Construction of Novel Aspartokinase Mutant A380I and Its Characterization by Molecular Dynamics Simulation


Abstract
1. Introduction
2. Results and Discussion
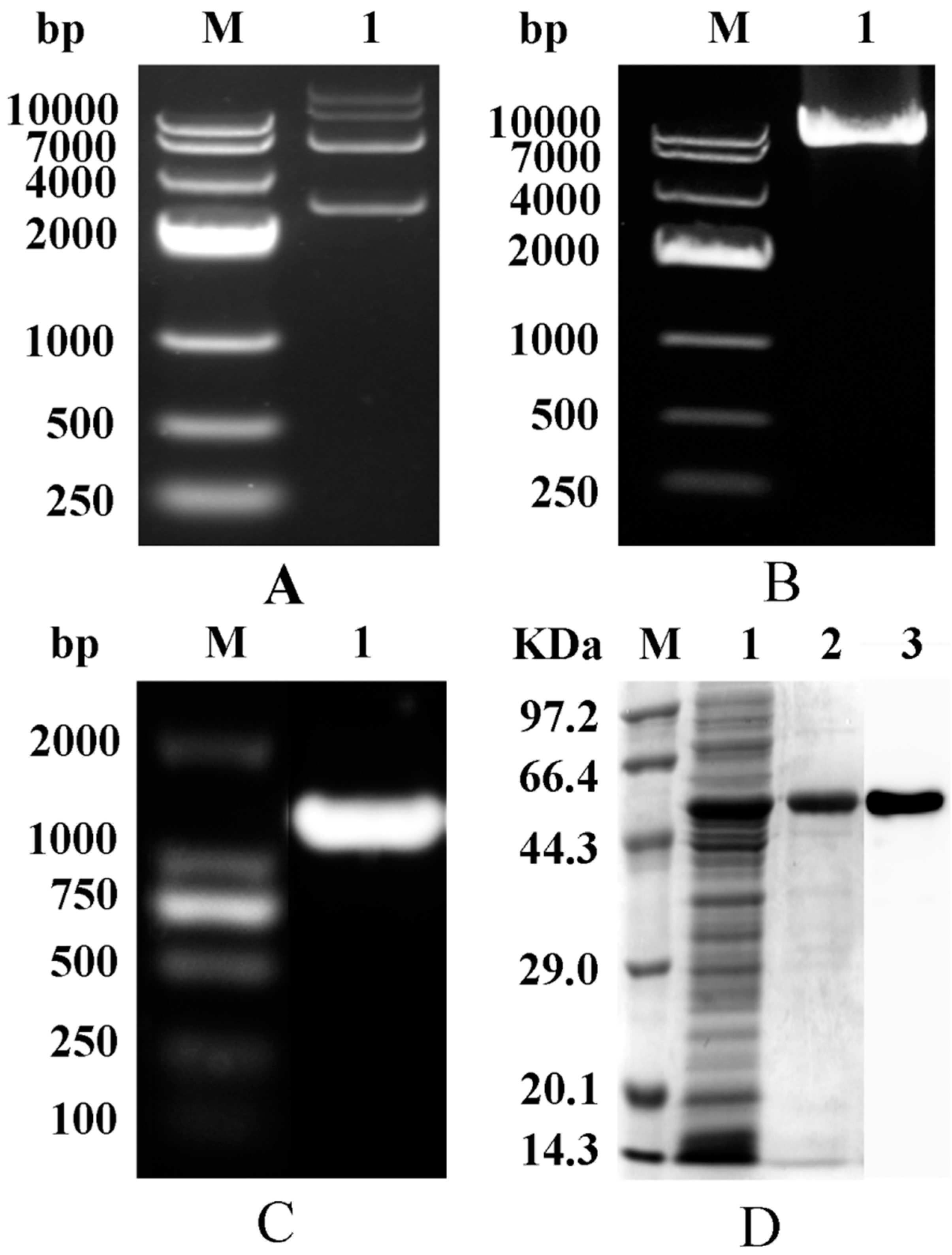
2.1. Construction and Purification of Mutant Strains
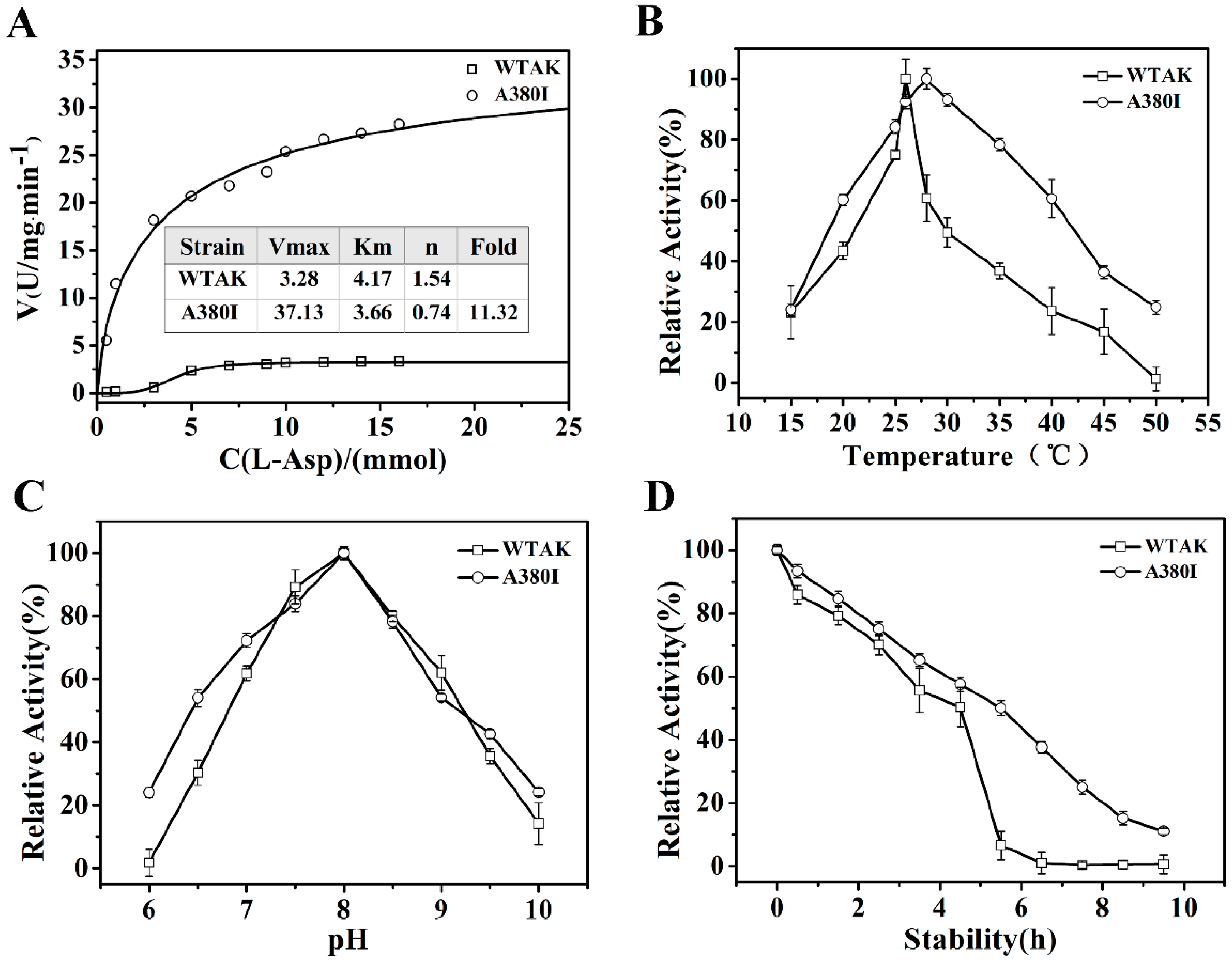
2.2. Dynamics and Enzymatic Properties
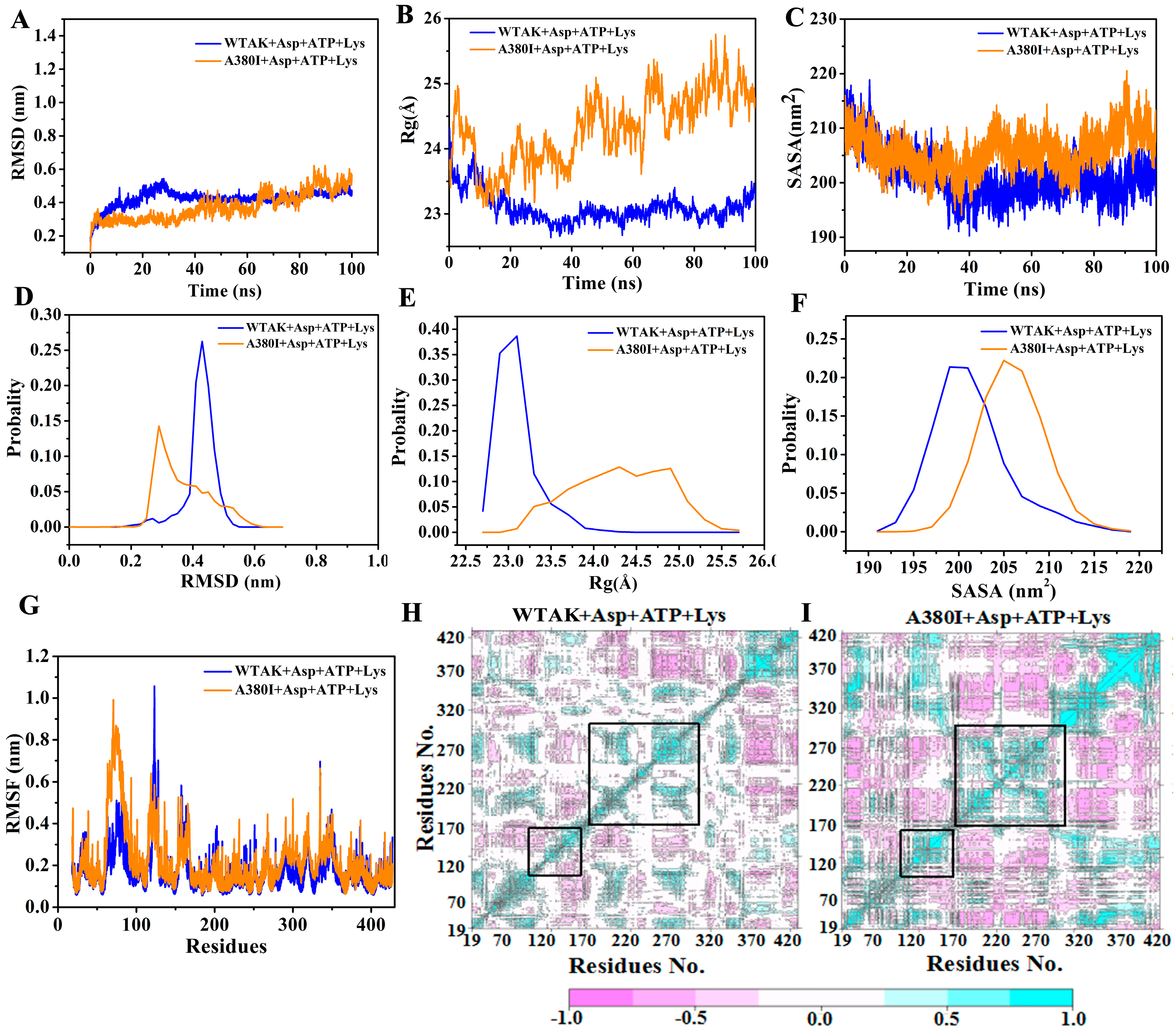
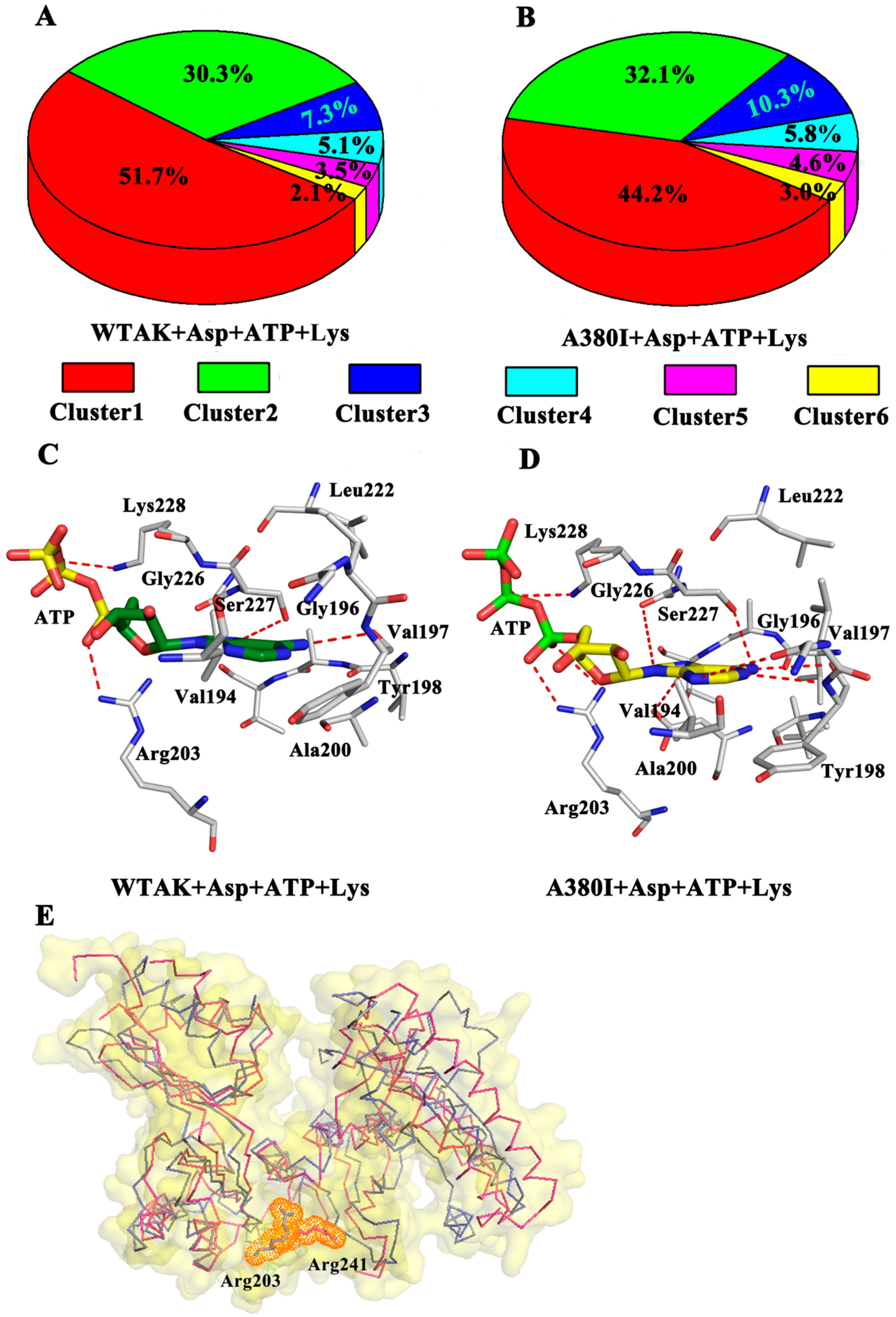
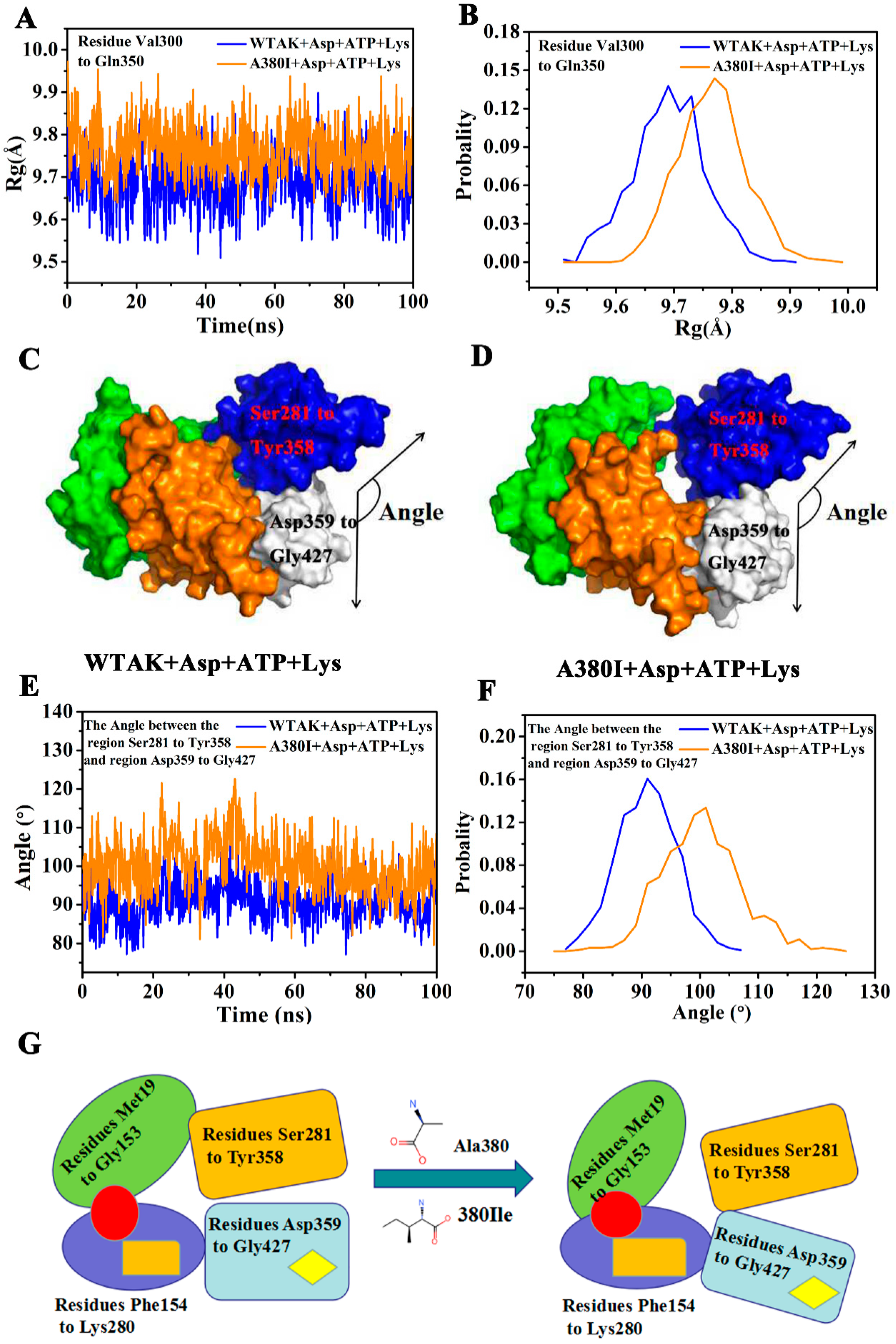
2.3. Analysis of MD Simulation
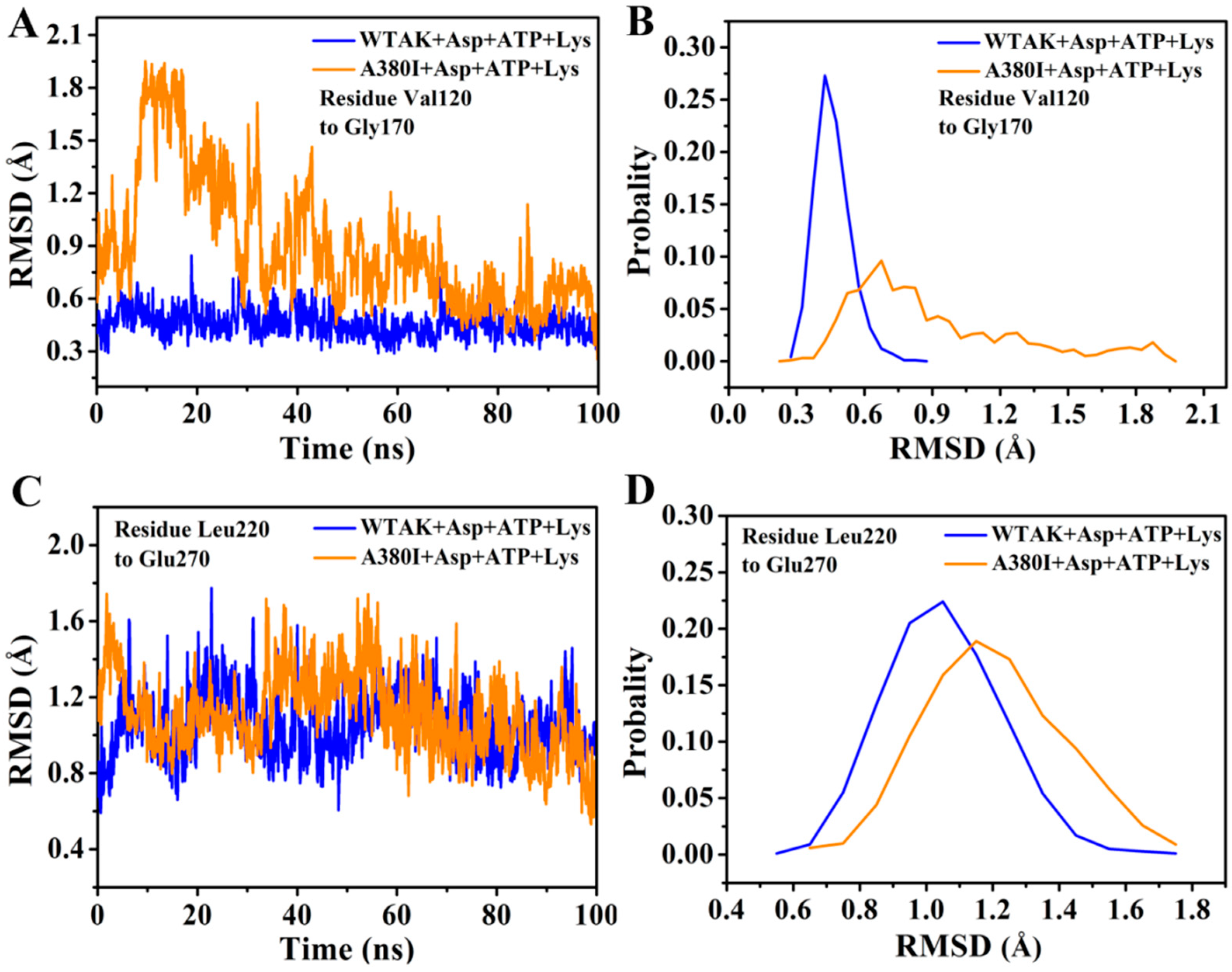
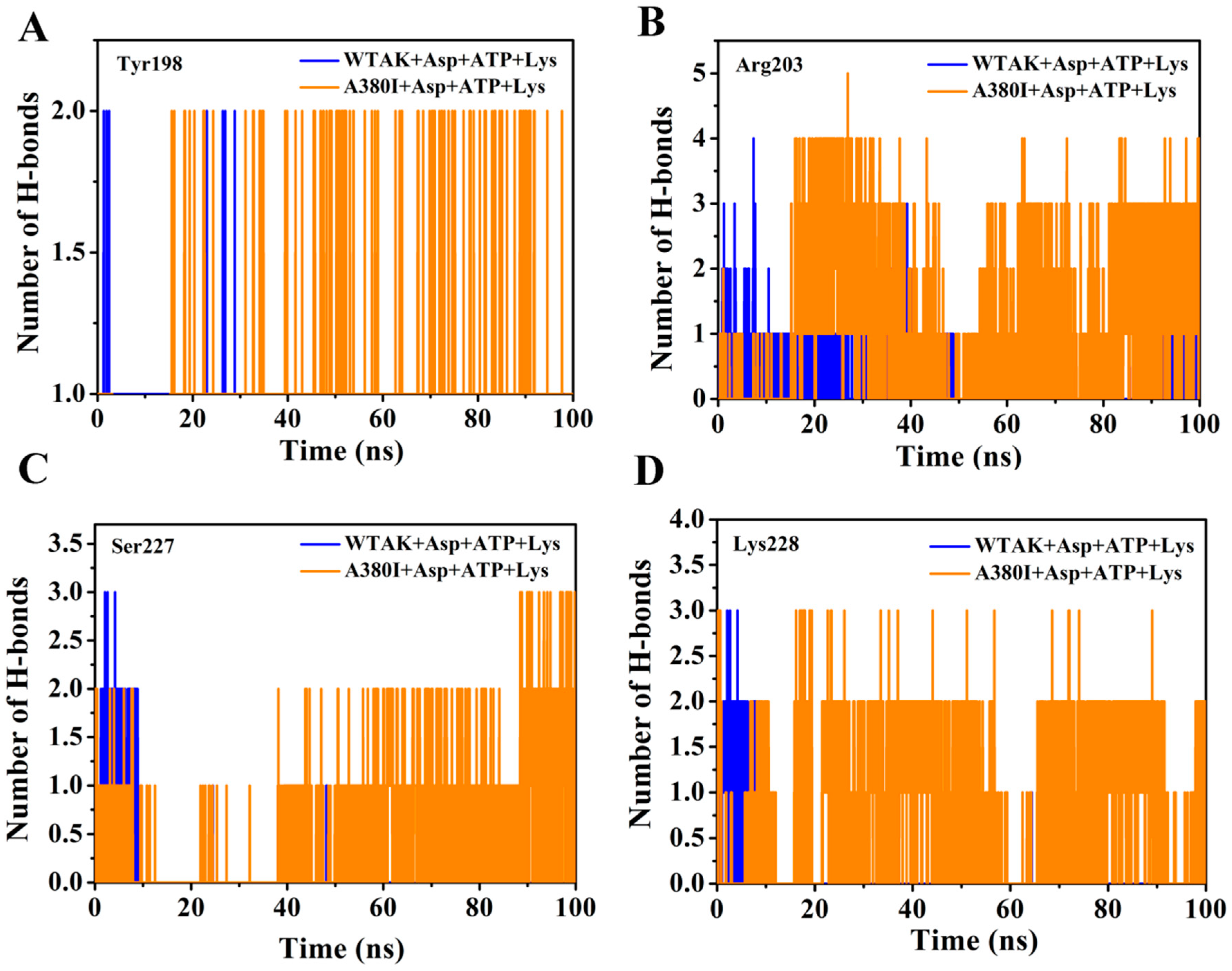
2.4. Mutations Affect the Flexibility of Active Site Residues
2.5. Mutation Generates an Open Conformation of the Protein
3. Materials and Methods
3.1. Experimental Materials
3.2. Test Method
3.2.1. Construction of Strain
3.2.2. Purification and Validation of Strain
3.2.3. Determination of Dynamics
3.2.4. Determination of Enzymatic Properties
3.2.5. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Faehnle, C.R.; Liu, X.Y.; Pavlovsky, A.; Viola, R.E. The initial step in the archaeal aspartate biosynthetic pathway catalyzed by a monofunctional aspartokinase. Acta Cryst. 2006, 62, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Rappert, S.; Sun, J.B.; Zeng, A.P. Integrating molecular dynamics and co-evolutionary analysis for reliable target prediction and deregulation of the allosteric inhibition of aspartokinase for amino acid production. J. Biotechnol. 2011, 154, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Min, W.H.; Li, H.Y.; Li, H.M.; Liu, C.L.; Liu, J.S. Characterization of Aspartate Kinase from Corynebacterium pekinense and the Critical Site of Arg169. Int. J. Mol. Sci. 2015, 16, 28270–28284. [Google Scholar] [CrossRef]
- Dong, X.Y.; Zhao, Y.; Zhao, J.X.; Wang, X.Y. Characterization of aspartate kinase and homoserine dehydrogenase from Corynebacterium glutamicum IWJ001 and systematic investigation of l-isoleucine biosynthesis. J. Ind. Microbiol. Biotechnol. 2016, 43, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Galili, G. Regulation of Lysine and Threonine Synthesis. Plant cell 1995, 7, 899–906. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Richaud, C.; Mazat, J.P.; Felenbok, B.; Patte, J.C. The role of lysine and leucine binding on the catalytical and structural properties of aspartokinase III of Escherichia coli K. 12. Eur. J. Biochem. 1974, 48, 147–156. [Google Scholar] [CrossRef]
- Curien, G.; Laurencin, M.; Robert-Genthon, M.; Dumas, R. Allosteric monofunctional aspartate kinases from Arabidopsis. FEBS J. 2007, 274, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Viemon, C.; Curien, G.; Dumas, R. Mechanism of control of Arabidopsis thaliana aspartate kinase-homoserine dehydrogenase by threonine. J. Biol. Chem 2003, 278, 5361–5366. [Google Scholar] [CrossRef]
- Curien, G.; Ravanel, S.; Robert, M.; Dumas, R. Identification of six novel allosteric effectors of Arabidopsis thaliana aspartate kinase-homoserine dehydrogenase isoforms. Physiological context sets the specificity. J. Biol. Chem. 2005, 280, 41178–41183. [Google Scholar] [CrossRef]
- Angeles, T.S.; Viola, R.E. The kinetic mechanisms of the bifunctional enzyme aspartokinase-homoserine dehydrogenase I from Escherichia coli. Arch. Biochem. Biophys. 1990, 283, 96–101. [Google Scholar] [CrossRef]
- Veron, M.; Guillou, Y.; Cohen, G.N. Isolation of the aspartokinase domain of bifunctional aspartokinase I-homoserine dehydrogenase I. from E.coli K12. FEBS Lett. 1985, 181, 381–384. [Google Scholar] [CrossRef]
- Dong, X.; Quinn, P.J.; Wang, X. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for the production of l-threonine. Biotechnol. Adv. 2011, 29, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tomita, T.; Kurihara, T.; Fushinobu, S.; Kuzuyama, T.; Nishiyama, M. Structural Insight into concerted inhibition of α2β2-type aspartate kinase from Corynebacterium glutamicum. J. Mol. Biol. 2007, 368, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, J.; Cremer, J.; Bachmann, B.; Eggeling, L.; Sahm, H.; Pühler, A. Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum. Mol. Microbiol. 2010, 5, 1197–1204. [Google Scholar] [CrossRef]
- Muehlbauer, G.J.; Somers, D.A.; Matthews, B.F.; Gengenbach, B.G. Molecular genetics of the maize (Zea mays L.) aspartate kinase-homoserine dehydrogenase gene family. Plant. Physiol. 1994, 106, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Ren, J.; Lockyer, M.; Hawkins, A.R.; Stammers, D.K. Structures of R- and T-state Escherichia coli aspartokinase III Mechanisms of the allosteric transition and inhibition by lysine. J. Biol. Chem. 2006, 281, 31544–31552. [Google Scholar] [CrossRef] [PubMed]
- Schuldt, L.; Suchowersky, R.; Veith, K.; Mueller-Dieckmann, J.; Weiss, M.S. Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of the regulatory domain of aspartokinase (Rv3709c) from Mycobacterium tuberculosis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 380–385. [Google Scholar] [CrossRef]
- Chen, Z.; Meyer, W.Q.; Rappert, S.; Sun, J.B.; Zeng, A.P. Coevolutionary Analysis Enabled Rational Deregulation of Allosteric Enzyme Inhibition in Corynebacterium glutamicum for Lysine Production. Appl. Environ. Microbiol. 2011, 77, 4352–4360. [Google Scholar] [CrossRef]
- Robin, A.Y.; Cobessi, D.; Curien, G.; Robert-Genthon, M.; Ferrer, J.L.; Dumas, R. A New Mode of Dimerization of Allosteric Enzymes with ACT Domains Revealed by the Crystal Structure of the Aspartate Kinase from Cyanobacteria. J. Mol. Biol. 2010, 399, 283–293. [Google Scholar] [CrossRef]
- Manjasetty, B.A.; Chance, M.R.; Burley, S.K.; Panjikar, S.; Almo, S.C. Crystal structure of Clostridium acetobutylicum Aspartate kinase (CaAK): An important allosteric enzyme for amino acids production. Biotechnol. Rep. 2014, 3, 73–85. [Google Scholar] [CrossRef]
- Han, G.; Hu, X.; Qin, T.; Li, Y.; Wang, X. Metabolic engineering of Corynebacterium glutamicum ATCC13032 to produce S-adenosyl-l-methionine. Enzym. Microb. Technol. 2016, 83, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.K.; Chen, G.J.; Wang, L.S. Structural and dynamic evolution of the amphipathic N-terminus diversifies enzyme thermostability in the glycoside hydrolase family 12. Phys. Chem. Chem. Phys. 2016, 18, 21340–21350. [Google Scholar] [CrossRef] [PubMed]
- Eisold, A.; Labudde, D. Detailed Analysis of 17β-Estradiol-Aptamer Interactions: A Molecular Dynamics Simulation Study. Molecules 2018, 23, 1690. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.S.; Zhao, L.; Jin, H.Y.; Shan, N.; Han, W.W.; Wang, S.; Shan, Y.M. Binding modes of phosphotriesterase-like lactonase complexed with -nonanoic lactone and paraoxon using molecular dynamics simulations. J. Biomol. Struct. Dyn. 2017, 35, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Tu, T.; Wang, X.Y.; Wang, Y.; Ma, R.; Su, X.Y.; Xie, X.M.; Yao, B.; Luo, H.Y. Enhancing the catalytic activity of a novel GH5 cellulase GtCel5 from Gloeophyllum trabeum CBS 900.73 by site-directed mutagenesis on loop 6. Biotechnol. Biofuels 2018, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tomita, T.; Kuzuyama, T.; Nishiyama, M. Mechanism of concerted inhibition of α2β2-type hetero-oligomeric aspartate kinase from Corynebacterium glutamicum. J. Biol. Chem. 2010, 285, 27477–27486. [Google Scholar] [CrossRef] [PubMed]
- Thongekkaew, J.; Ikeda, H.; Masaki, K.; Iefuji, H. Fusion of cellulose binding domain from Trichoderma reesei CBHI to Cryptococcus sp S-2 cellulase enhances its binding affinity and its cellulolytic activity to insoluble cellulosic substrates. Enzym. Microb. Technol. 2013, 52, 241–246. [Google Scholar] [CrossRef]
- Wu, X.Y.; Tian, Z.N.; Jiang, X.K.; Zhang, Q.; Wang, L.S. Enhancement in catalytic activity of Aspergillus niger XynB by selective site-directed mutagenesis of active site amino acids. Appl. Microbiol. Biotechnol. 2018, 102, 249–260. [Google Scholar] [CrossRef]
- Li, C.C.; Yang, M.J.; Liu, L.; Li, T.; Peng, C.T.; He, L.H.; Song, Y.J.; Zhu, Y.B.; Shen, Y.L.; Yang, J.; et al. Mechanistic insights into the allosteric regulation of Pseudomonas aeruginosa aspartate kinase. Biochem. J. 2018, 475, 1107–1119. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Hess, B.; van der Vegt, N.F.A. Hydration thermodynamic properties of amino acid analogues: A systematic comparison of biomolecular force fields and water models. J. Phys. Chem. B 2006, 110, 17616–17626. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals: A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 4, 1463–1472. [Google Scholar] [CrossRef]
- Wang, J.; Gao, D.; Yu, X.; Li, W.; Qi, Q. Evolution of a chimeric aspartate kinase for L-lysine production using a synthetic RNA device. Appl. Microbiol. Biotechnol. 2015, 99, 8527–8536. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | WT-AK | A380I | ||||||
|---|---|---|---|---|---|---|---|---|
| Inhibitors | Relative Activity (%) Concentration (mM) | |||||||
| 0.2 | 1 | 5 | 10 | 0.2 | 1 | 5 | 10 | |
| Control | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Lys | 93.87 ± 1.54 | 91.13 ± 2.69 | 88.33 ± 1.77 | 85.43 ± 1.52 | 94.91 ± 0.66 | 95.47 ± 0.69 | 96.27 ± 0.96 | 112.41 ± 1.18 |
| Thr | 95.02 ± 2.65 | 94.42 ± 0.45 | 89.37 ± 0.91 | 89.76 ± 2.90 | 84.49 ± 1.66 | 94.61 ± 0.83 | 95.48 ± 2.55 | 97.92 ± 1.04 |
| Met | 92.01 ± 2.40 | 83.11 ± 1.46 | 72.66 ± 0.88 | 73.7 2± 1.65 | 90.57 ± 0.82 | 95.73 ± 0.96 | 97.82 ± 0.78 | 98.35 ± 0.79 |
| Lys + Thr | 71.01 ± 3.65 | 59.26 ± 2.35 | 40.02 ± 1.45 | 34.77 ± 0.97 | 90.04 ± 0.94 | 96.94 ± 1.46 | 102.07 ± 3.12 | 104.48 ± 0.67 |
| Lys + Met | 80.31 ± 0.84 | 75.67 ± 1.31 | 72.97 ± 0.68 | 69.65 ± 1.56 | 90.51 ± 1.05 | 93.29 ± 2.18 | 99.12 ± 2.13 | 104.50 ± 1.36 |
| Thr + Met | 84.76 ± 1.59 | 81.66 ± 0.88 | 78.86 ± 1.35 | 70.91 ± 1.00 | 71.87 ± 2.02 | 85.19 ± 2.75 | 94.07 ± 0.091 | 97.46 ± 1.85 |
| Lys + Thr + Met | 76.51 ± 1.03 | 60.06 ± 1.88 | 49.16 ± 2.61 | 30.51 ± 0.78 | 84.17 ± 1.25 | 99.73 ± 1.95 | 105.01 ± 1.53 | 99.79 ± 2.13 |
| Hydrogen Bonds Occupancies | WTAK | A380I | |
|---|---|---|---|
| Donor | Accepter | ||
| Lys228:NZ | ATP:O3 | 47.49% | 56.26% |
| Arg203:NH1 | ATP:O | 38.34% | 51.78% |
| Arg203:NH1 | ATP:O1 | 43.11% | 49.55% |
| Arg203:NH2 | ATP:P | 44.01% | 63.27% |
| Arg203:NH2 | ATP:O1 | 48.00% | 57.76% |
| Lys228:NZ | ATP:O1 | 34.33% | 45.57% |
| Arg203:NH2 | ATP:O9 | 90.62% | 93.89% |
| Ser227:OG | ATP:N5 | 32.53% | |
| ATP:N5 | Tyr198:O | 42.48% | 54.35% |
| ATP:N5 | Ser227:OG | 31.74% | 39.14% |
| ATP:N5 | Ala200:O | 27.52% | 32.84% |
| Tyr198:N | ATP:N4 | 55.79% | 55.79% |
| Ser281:OG | ATP:O3 | 28.14% | 41.37% |
| ATP:C1 | GLY226:O | 45.38% | |
| Val194:CG1 | ATP:O11 | 34.21% | |
| Tyr198:CD1 | ATP:C10 | 31.97% | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.; Fang, L.; Liu, C.; Gao, Y.; Min, W. Construction of Novel Aspartokinase Mutant A380I and Its Characterization by Molecular Dynamics Simulation. Molecules 2018, 23, 3379. https://doi.org/10.3390/molecules23123379
Han C, Fang L, Liu C, Gao Y, Min W. Construction of Novel Aspartokinase Mutant A380I and Its Characterization by Molecular Dynamics Simulation. Molecules. 2018; 23(12):3379. https://doi.org/10.3390/molecules23123379
Chicago/Turabian StyleHan, Caijing, Li Fang, Chunlei Liu, Yunna Gao, and Weihong Min. 2018. "Construction of Novel Aspartokinase Mutant A380I and Its Characterization by Molecular Dynamics Simulation" Molecules 23, no. 12: 3379. https://doi.org/10.3390/molecules23123379
APA StyleHan, C., Fang, L., Liu, C., Gao, Y., & Min, W. (2018). Construction of Novel Aspartokinase Mutant A380I and Its Characterization by Molecular Dynamics Simulation. Molecules, 23(12), 3379. https://doi.org/10.3390/molecules23123379