Combining a QSAR Approach and Structural Analysis to Derive an SAR Map of Lyn Kinase Inhibition


Abstract

1. Introduction
2. Materials and Methods
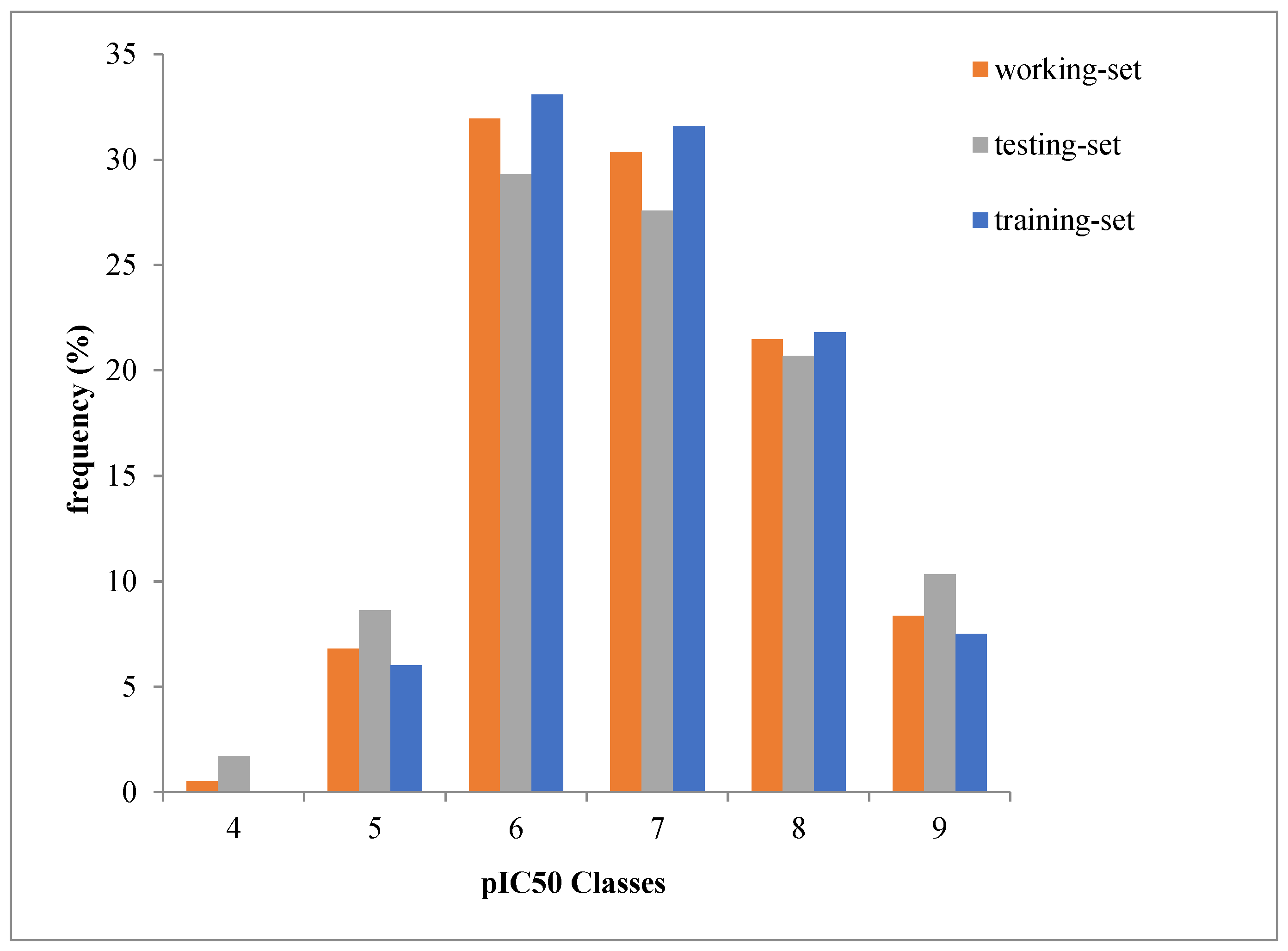
2.1. Dataset Source and Preparation
2.2. Calculation of Molecular Descriptors
2.3. Diversity Analysis
2.4. Descriptors Selection
2.5. Model Development and Validation
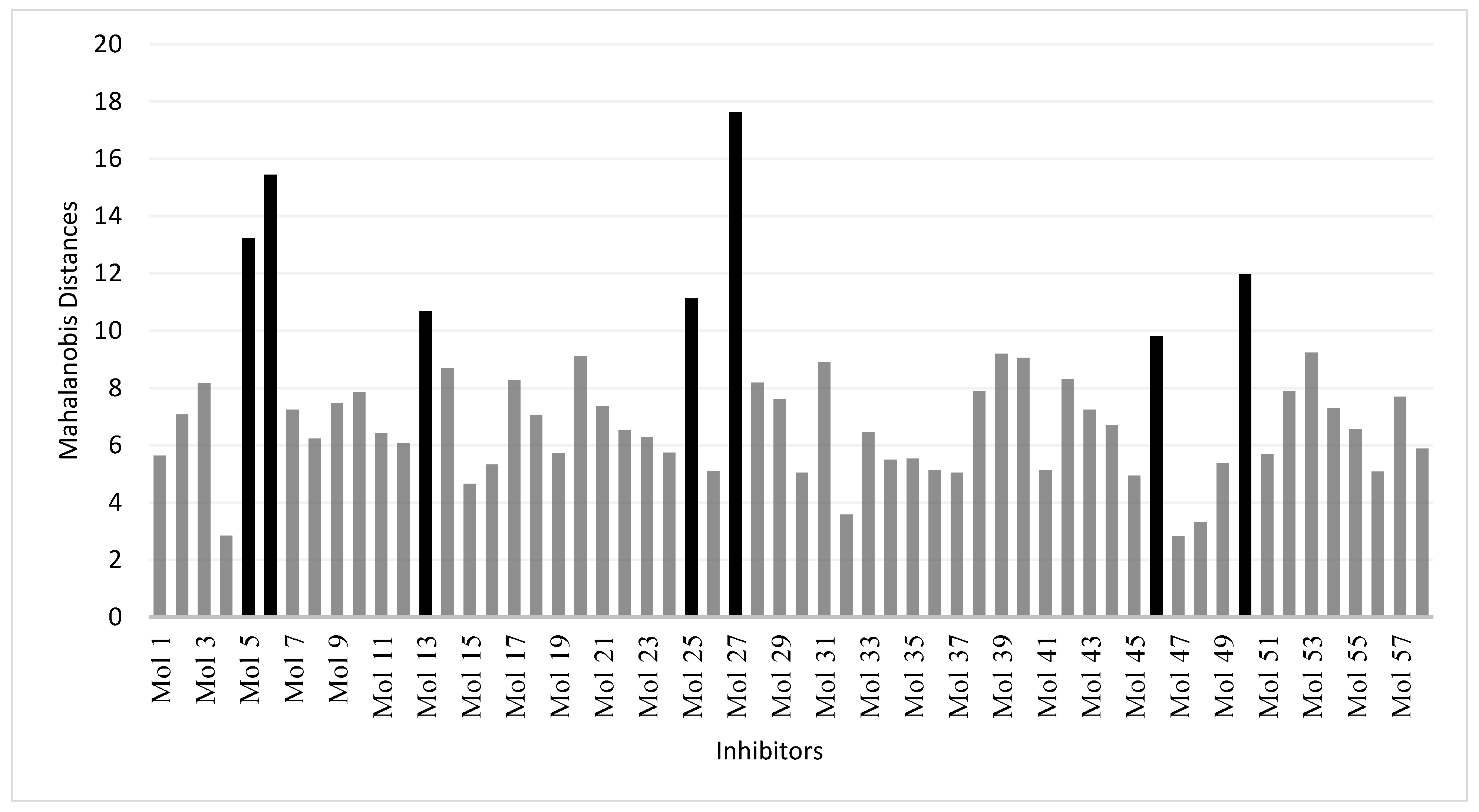
2.6. Domain of Applicability of the Models
3. Results and Discussion
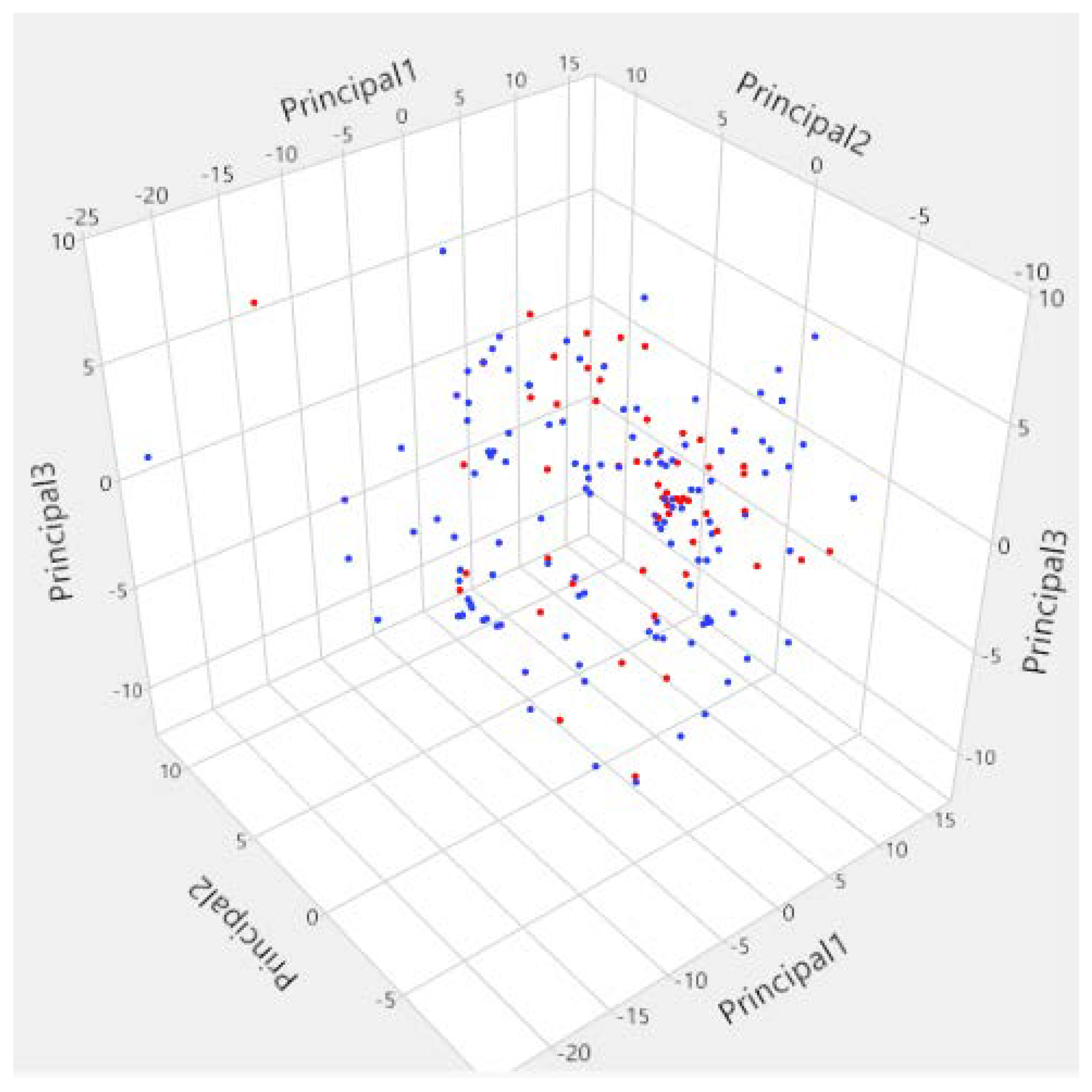
3.1. Diversity Analysis
3.2. Descriptors Pertinence
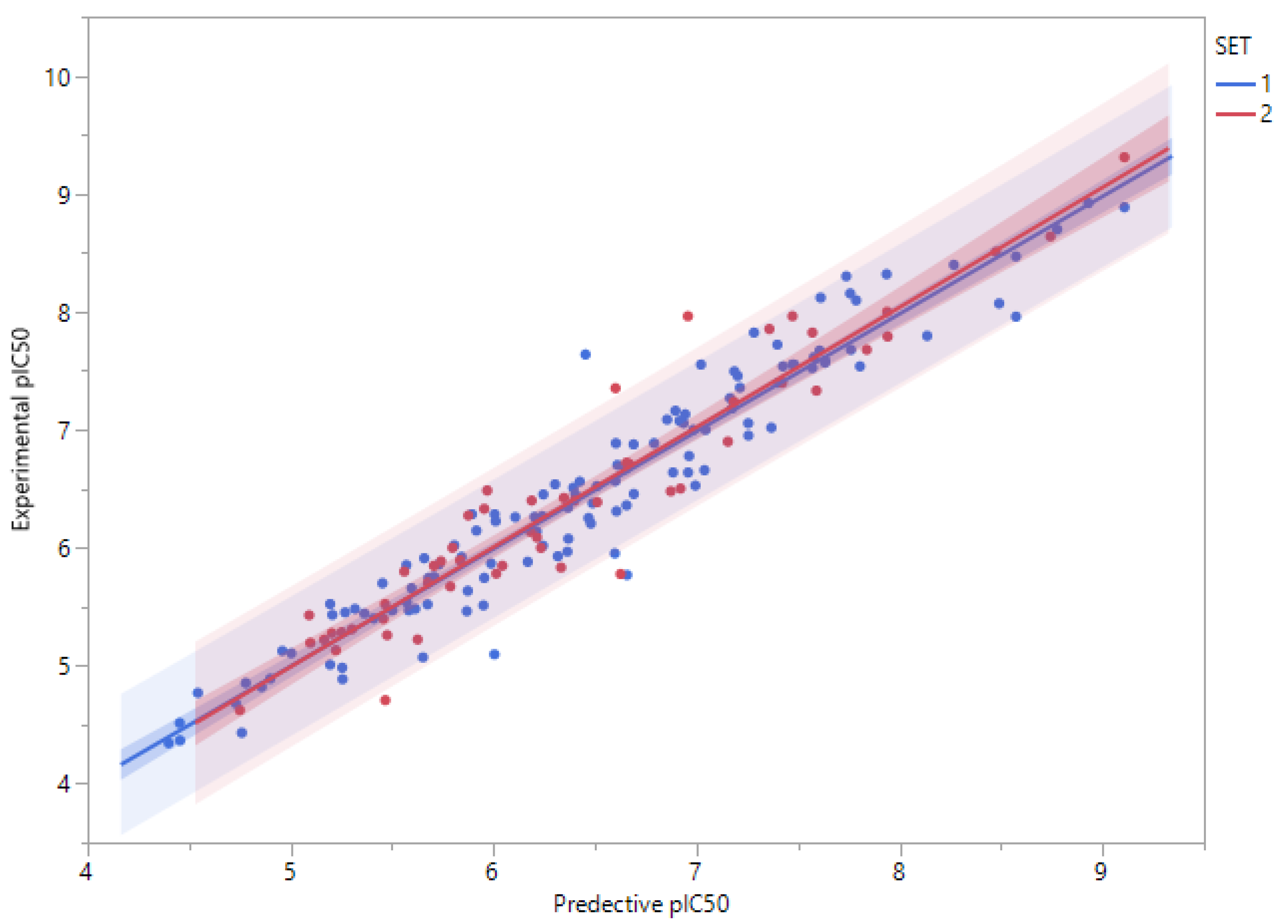
3.3. QSAR Model Derivation and Validation
3.4. Applicability Domains of QSAR Models
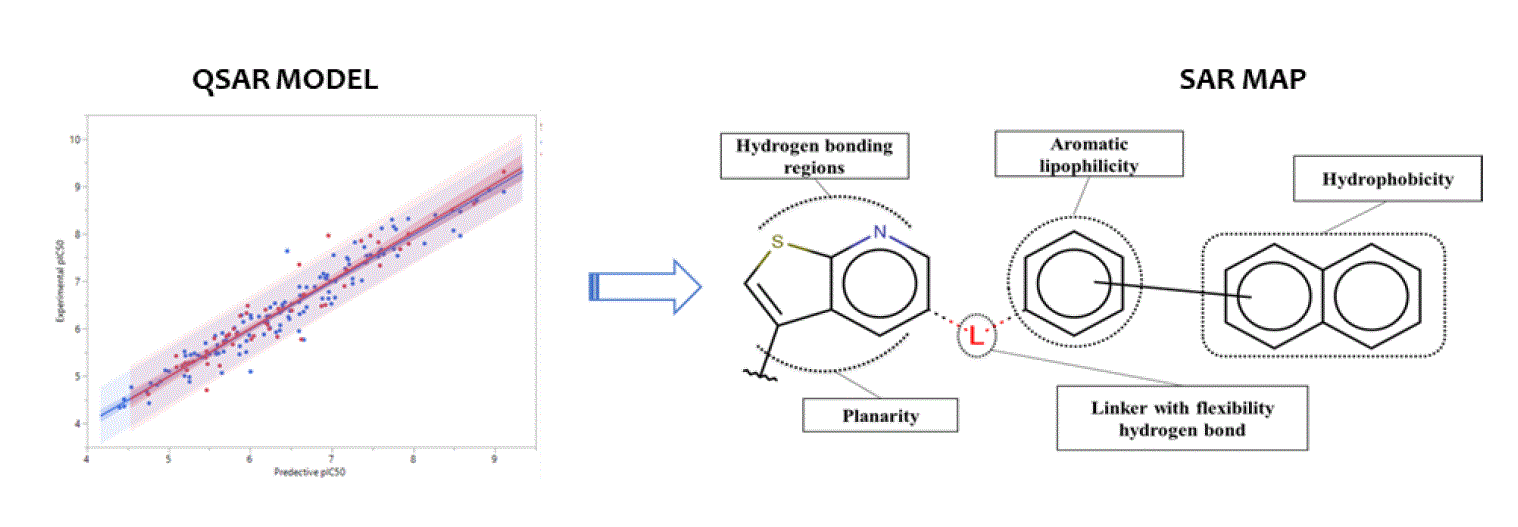
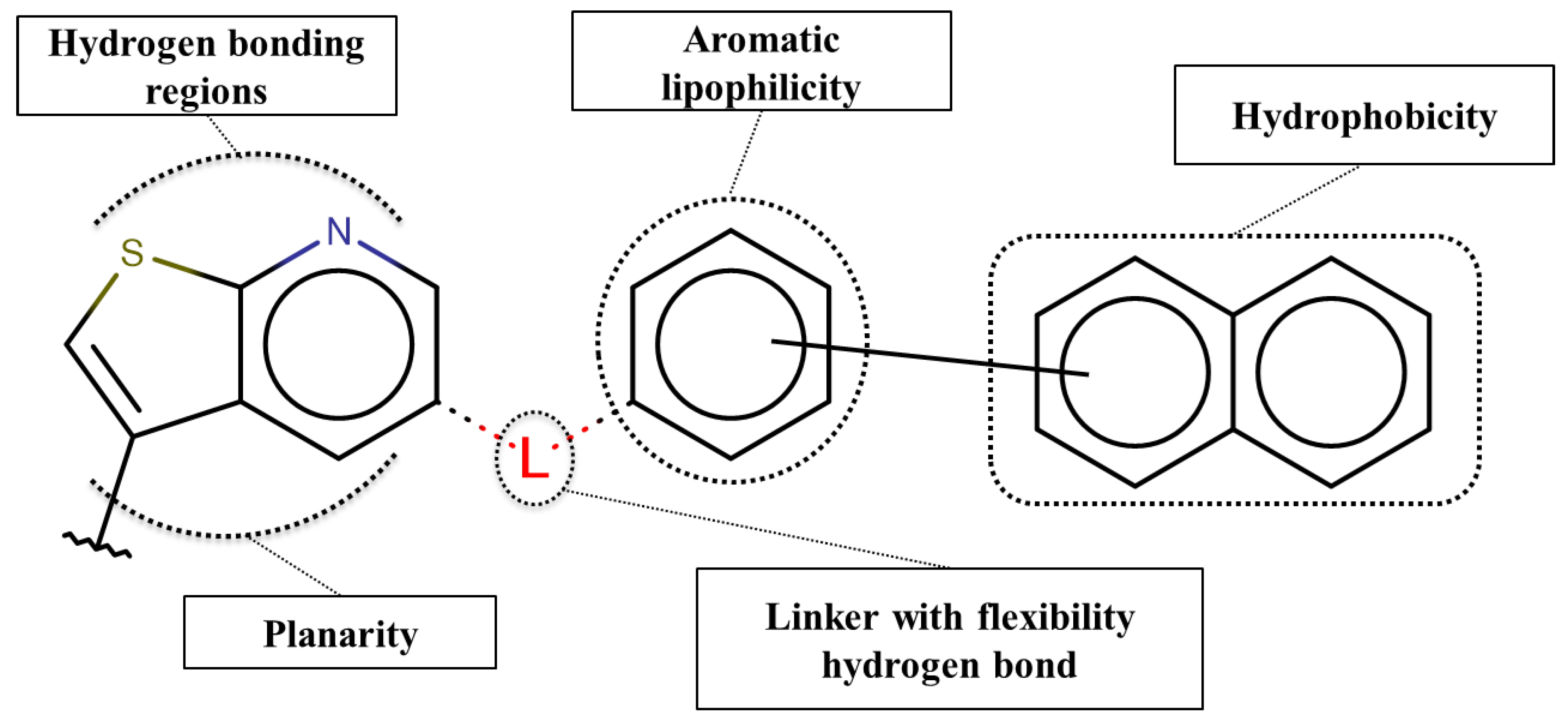
3.5. Structure-Activity Relationship Map Derivation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Merlin, J.L. Les inhibiteurs de tyrosine kinase en oncologie. Lett. Pharmacol. 2008, 22, 51–62. [Google Scholar]
- van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef] [PubMed]
- Zámečníkova, A. Novel approaches to the development of tyrosine kinase inhibitors and their role in the fight against cancer. Expert Opin. Drug Discov. 2014, 9, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Lieu, C.; Kopetz, S. The SRC family of protein tyrosine kinases: A new and promising target for colorectal cancer therapy. Clin. Colorectal Cancer 2010, 9, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Prabhu, K.S.; Achkar, I.W.; Kuttikrishnan, S.; Shyam, S.; Khan, A.Q.; Merhi, M.; Dermime, S.; Uddin, S. Role of Non Receptor Tyrosine Kinases in Hematological Malignances and its Targeting by Natural Products. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Benati, D.; Baldari, C.T. SRC family kinases as potential therapeutic targets for malignancies and immunological disorders. Curr. Med. Chem. 2008, 15, 1154–1165. [Google Scholar] [CrossRef]
- Engen, J.R.; Wales, T.E.; Hochrein, J.M.; Meyn, M.A.; Banu Ozkan, S.; Bahar, I.; Smithgall, T.E. Structure and dynamic regulation of Src-family kinases. Cell. Mol. Life Sci. CMLS 2008, 65, 3058–3073. [Google Scholar] [CrossRef] [PubMed]
- Liu, D. LYN, a Key Gene from Bioinformatics Analysis, Contributes to Development and Progression of Esophageal Adenocarcinoma. Med. Sci. Monit. Basic Res. 2015, 21, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg-Furmanov, M.; Stein, I.; Pikarsky, E.; Rubin, H.; Kasem, S.; Wygoda, M.; Weinstein, I.; Reuveni, H.; Ben-Sasson, S.A. Lyn is a target gene for prostate cancer: Sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004, 64, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Zardan, A.; Nip, K.M.; Thaper, D.; Toren, P.; Vahid, S.; Beraldi, E.; Fazli, L.; Lamoureux, F.; Gust, K.M.; Cox, M.E.; et al. Lyn tyrosine kinase regulates androgen receptor expression and activity in castrate-resistant prostate cancer. Oncogenesis 2014, 3, e115. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zagozdzon, R.; Avraham, R.; Avraham, H.K. CHK negatively regulates Lyn kinase and suppresses pancreatic cancer cell invasion. Int. J. Oncol. 2006, 29, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hao, X.; Ouyang, X.; Dong, X.; Yang, Y.; Yu, T.; Hu, J.; Hu, L. Tyrosine kinase LYN is an oncotarget in human cervical cancer: A quantitative proteomic based study. Oncotarget 2016, 7, 75468–75481. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Bocanegra, M.; Kwon, M.J.; Shin, Y.K.; Nam, S.J.; Yang, J.H.; Kao, J.; Godwin, A.K.; Pollack, J.R. LYN is a mediator of epithelial-mesenchymal transition and target of dasatinib in breast cancer. Cancer Res. 2010, 70, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, K.; Baumann, C.; Szabadkai, I.; Orfi, L.; Kéri, G.; Ullrich, A.; Torka, R. Combined inhibition of AXL, Lyn and p130Cas kinases block migration of triple negative breast cancer cells. Cancer Biol. Ther. 2014, 15, 1571–1582. [Google Scholar] [CrossRef]
- Li, Y.; Xiong, L.; Gong, J. Lyn kinase enhanced hepatic fibrosis by modulating the activation of hepatic stellate cells. Am. J. Transl. Res. 2017, 9, 2865–2877. [Google Scholar]
- Contri, A.; Brunati, A.M.; Trentin, L.; Cabrelle, A.; Miorin, M.; Cesaro, L.; Pinna, L.A.; Zambello, R.; Semenzato, G.; Donella-Deana, A. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J. Clin. Invest. 2005, 115, 369–378. [Google Scholar] [CrossRef]
- Oncogenic Association of the Cbp/PAG Adaptor Protein with the Lyn Tyrosine Kinase in Human B-NHL Rafts. Available online: http://www.bloodjournal.org/content/111/4/2310/tab-figures-only?sso-checked=true (accessed on 9 April 2018).
- Almamun, M.; Levinson, B.T.; van Swaay, A.C.; Johnson, N.T.; McKay, S.D.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Integrated methylome and transcriptome analysis reveals novel regulatory elements in pediatric acute lymphoblastic leukemia. Epigenetics 2015, 10, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Dong, F.; Zhou, Q. Triptonide acts as a novel potent anti-lymphoma agent with low toxicity mainly through inhibition of proto-oncogene Lyn transcription and suppression of Lyn signal pathway. Toxicol. Lett. 2017, 278, 9–17. [Google Scholar] [CrossRef]
- Kim, A.; Seong, K.M.; Kang, H.J.; Park, S.; Lee, S.-S. Inhibition of Lyn is a promising treatment for mantle cell lymphoma with bortezomib resistance. Oncotarget 2015, 6, 38225–38238. [Google Scholar] [CrossRef] [PubMed]
- Ptasznik, A.; Nakata, Y.; Kalota, A.; Emerson, S.G.; Gewirtz, A.M. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat. Med. 2004, 10, 1187–1189. [Google Scholar] [CrossRef]
- Gioia, R.; Trégoat, C.; Dumas, P.Y.; Lagarde, V.; Prouzet-Mauléon, V.; Desplat, V.; Sirvent, A.; Praloran, V.; Lippert, E.; Villacreces, A.; et al. CBL controls a tyrosine kinase network involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid leukaemia. J. Pathol. 2015, 237, 14–24. [Google Scholar] [CrossRef] [PubMed]
- He, W.Q.; Gu, J.W.; Li, C.Y.; Kuang, Y.Q.; Kong, B.; Cheng, L.; Zhang, J.H.; Cheng, J.M.; Ma, Y. The PPI network and clusters analysis in glioblastoma. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4784–4790. [Google Scholar]
- González, M.P.; Terán, C.; Saíz-Urra, L.; Teijeira, M. Variable selection methods in QSAR: An overview. Curr. Top. Med. Chem. 2008, 8, 1606–1627. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Arodz, T.; Gálvez, J. Computational methods in developing quantitative structure-activity relationships (QSAR): A review. Comb. Chem. High Throughput Screen. 2006, 9, 213–228. [Google Scholar] [CrossRef]
- Puri, M.; Solanki, A.; Padawer, T.; Tipparaju, S.M.; Moreno, W.A.; Pathak, Y. Chapter 1–Introduction to Artificial Neural Network (ANN) as a Predictive Tool for Drug Design, Discovery, Delivery, and Disposition: Basic Concepts and Modeling. In Artificial Neural Network for Drug Design, Delivery and Disposition; Academic Press: Boston, MA, USA, 2016; pp. 3–13. ISBN 978-0-12-801559-9. [Google Scholar]
- Todeschini, R.; Consonni, V.; Gramatica, P. Chemometrics in QSAR. In Comprehensive Chemometrics; Brown, S.D., Tauler, R., Walczak, B., Eds.; Elsevier: Oxford, UK, 2009; Volume 4, pp. 129–172. ISBN 978-0-444-52701-1. [Google Scholar]
- Agrafiotis, D.K.; Shemanarev, M.; Connolly, P.J.; Farnum, M.; Lobanov, V.S. SAR Maps: A New SAR Visualization Technique for Medicinal Chemists. J. Med. Chem. 2007, 50, 5926–5937. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Chapter 5-Analysis of the Content of SAR Databases. In Ligand Efficiency Indices for Drug Discovery; Academic Press: San Diego, CA, USA, 2013; pp. 67–79. ISBN 978-0-12-404635-1. [Google Scholar]
- Molecular Operating Environment (MOE), 2008.10; Chemical Computing Group ULC: Montreal, QC, Canada, 2008.
- Afifi, A.; May, S.; Clark, V.A. Practical Multivariate Analysis, 5th ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2011; ISBN 978-1-4398-1680-6. [Google Scholar]
- JMP®, version 14.0.1; SAS Institute Inc.: Cary, NC, USA, 1989.
- Nelder, J.A.; Wedderburn, R.W.M. Generalized Linear Models. J. Roy. Stat. Soc. Ser. Gen. 1972, 135, 370–384. [Google Scholar] [CrossRef]
- Yasri, A.; Hartsough, D. Toward an optimal procedure for variable selection and QSAR model building. J. Chem. Inf. Comput. Sci. 2001, 41, 1218–1227. [Google Scholar] [CrossRef]
- Warren, R.; Smith, R.F.; Cybenko, A.K. Use of Mahalanobis Distance for Detecting Outliers and Outlier Clusters in Markedly Non-Normal Data: A Vehicular Traffic Example; SRA International, Inc.: Dayton, OH, USA, 2011. [Google Scholar]
- QuaSAR-Descriptor. Available online: http://www.cadaster.eu/sites/cadaster.eu/files/challenge/descr.htm (accessed on 3 August 2018).
- Horio, T.; Hamasaki, T.; Inoue, T.; Wakayama, T.; Itou, S.; Naito, H.; Asaki, T.; Hayase, H.; Niwa, T. Structural factors contributing to the Abl/Lyn dual inhibitory activity of 3-substituted benzamide derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 2712–2717. [Google Scholar] [CrossRef]
- Kim, K.H.; Maderna, A.; Schnute, M.E.; Hegen, M.; Mohan, S.; Miyashiro, J.; Lin, L.; Li, E.; Keegan, S.; Lussier, J.; et al. Imidazo[1,5-a]quinoxalines as irreversible BTK inhibitors for the treatment of rheumatoid arthritis. Bioorg. Med. Chem. Lett. 2011, 21, 6258–6263. [Google Scholar] [CrossRef]
- Goldberg, D.R.; Hao, M.-H.; Qian, K.C.; Swinamer, A.D.; Gao, D.A.; Xiong, Z.; Sarko, C.; Berry, A.; Lord, J.; Magolda, R.L.; et al. Discovery and Optimization of p38 Inhibitors via Computer-Assisted Drug Design. J. Med. Chem. 2007, 50, 4016–4026. [Google Scholar] [CrossRef]
Sample Availability: Data (sdf file and pIC50 values) are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | |
|---|---|
| 38 Molecules |  |
| 33 Molecules |  |
| 32 Molecules |  |
| 12 Molecules |  |
| 12 Molecules |  |
| 26 Molecules |  |
| Categories of Descriptors | Definition | Categories of Descriptors | Definition | |
|---|---|---|---|---|
| Physico-Chemical Properties | LogP (o/w) | Log of the octanol/water partition coefficient (including implicit hydrogens) | MR | Molecular refractivity (including implicit hydrogens) |
| logS | Log of the aqueous solubility (mol/L) | TPSA | Polar surface area (Å2) calculated using group contributions to approximate the polar surface area from connection table information only | |
| apol | Sum of the atomic polarizabilities (including implicit hydrogens) | SlogP_VSA0 SlogP_VSA1 SlogP_VSA3 SlogP_VSA5 SlogP_VSA6 | Subdivided logP Surface Areas are descriptors based on an approximate accessible van der Waals surface area (in Å2) calculation for each atom along with its contribution to logP property | |
| Atom and Bond Counts | PEOE_RPC_− | Relative negative/positive partial charge: the smallest negative qi divided by the sum of the negative qi. Q_RPC−/Q_RPC+ is identical to RPC−/RPC+ which has been retained for compatibility | PEOE_VSA_0 | Sum of vi (a der Waals surface area of atom i) where qi (partial charge of atom i) is in the range (−0.05, 0.00) |
| PEOE_RPC_+ | PEOE_VSA_FPOS | Fractional positive van der Waals surface area. This is the sum of the vi such that qiis non-negative divided by the total surface area. The vi are calculated using a connection table approximation | ||
| Atom and Bond Counts | b_double | Number of double bonds. Aromatic bonds are not considered to be double bonds | lip_acc | The number of O and N atoms |
| a_ICM | Atom information content (mean). This is the entropy of the element distribution in the molecule (including implicit hydrogens but not lone pair pseudo-atoms) | lip_don | The number of OH and NH atoms | |
| b_count | Number of bonds (including implicit hydrogens) | lip_druglike | One if and only if Lipinski’s rules violation < 2 otherwise zero | |
| Pharmacophoric Features | a_acc, | Number of hydrogen bond acceptor atoms (not counting acidic atoms but counting atoms that are both hydrogen bond donors and acceptors such as -OH) | vsa_don, | Approximation to the sum of VDW surface areas of pure hydrogen bond donors (not counting basic atoms and atoms that are both hydrogen bond donors and acceptors such as -OH) (Å2) |
| a_don, | Number of hydrogen bond donor atoms (not counting basic atoms but counting atoms that are both hydrogen bond donors and acceptors such as -OH) | vsa_other | Approximation to the sum of VDW surface areas (Å2) of atoms typed as “other” | |
| Connectivity and Shape Indices | chi0 | Atomic connectivity index (order 0) | KierFlex | Kier molecular flexibility index |
| chil_C | Carbon connectivity index (order 1) | |||
| Adjacency and Distance Matrix Descriptors | VDistMa | If m is the sum of the distance matrix entries | BCUT_SLOGP_1 | The BCUT descriptors using atomic contribution to logP (using the Wildman and Crippen SlogP method) |
| WeinerPath | Wiener path number. | BCUT_SLOGP_3 | ||
| balabanJ | Balaban’s connectivity topological index | GCUT_SMR_1 | The GCUT descriptors using atomic contribution to molar refractivity (using the Wildman and Crippen SMR method) instead of partial charge | |
| BCUT_PEOE_3 | Adjacency and distance matrix descriptors. The BCUT descriptors are calculated from the eigenvalues of a modified adjacency matrix | GCUT_SMR_3 | ||
| BCUT_SMR_2 | The BCUT descriptors using atomic contribution to molar refractivity (using the Wildman and Crippen SMR method) instead of partial charge |
| RT2 | RMSET | Rv2 | RMSEV | |
|---|---|---|---|---|
| ANN 3-layers | 0.75 | 0.54 | 0.48 | 0.68 |
| ANN 4-layers | 0.81 | 0.47 | 0.71 | 0.51 |
| ANN 5-layers | 0.76 | 0.53 | 0.64 | 0.56 |
| ANN 6-layers | 0.84 | 0.43 | 0.78 | 0.46 |
| ANN 7-layers | 0.90 | 0.34 | 0.85 | 0.39 |
| ANN 8-layers | 0.86 | 0.40 | 0.62 | 0.66 |
| ANN 9-layers | 0.92 | 0.29 | 0.90 | 0.32 |
| ANN 10-layers | 0.90 | 0.34 | 0.78 | 0.47 |
| ANN 11-layers | 0.91 | 0.32 | 0.72 | 0.62 |
| ANN 12-layers | 0.88 | 0.37 | 0.82 | 0.40 |
| ANN 13-layers | 0.86 | 0.40 | 0.74 | 0.51 |
| RTs2 | RMSETs | |
|---|---|---|
| ANN 3-layers | 0.77 | 0.52 |
| ANN 4-layers | 0.82 | 0.46 |
| ANN 5-layers | 0.73 | 0.56 |
| ANN 6-layers | 0.79 | 0.50 |
| ANN 7-layers | 0.89 | 0.37 |
| ANN 8-layers | 0.87 | 0.40 |
| ANN 9-layers | 0.91 | 0.33 |
| ANN 10-layers | 0.89 | 0.36 |
| ANN 11-layers | 0.92 | 0.31 |
| ANN 12-layers | 0.91 | 0.33 |
| ANN 13-layers | 0.90 | 0.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naboulsi, I.; Aboulmouhajir, A.; Kouisni, L.; Bekkaoui, F.; Yasri, A. Combining a QSAR Approach and Structural Analysis to Derive an SAR Map of Lyn Kinase Inhibition. Molecules 2018, 23, 3271. https://doi.org/10.3390/molecules23123271
Naboulsi I, Aboulmouhajir A, Kouisni L, Bekkaoui F, Yasri A. Combining a QSAR Approach and Structural Analysis to Derive an SAR Map of Lyn Kinase Inhibition. Molecules. 2018; 23(12):3271. https://doi.org/10.3390/molecules23123271
Chicago/Turabian StyleNaboulsi, Imane, Aziz Aboulmouhajir, Lamfeddal Kouisni, Faouzi Bekkaoui, and Abdelaziz Yasri. 2018. "Combining a QSAR Approach and Structural Analysis to Derive an SAR Map of Lyn Kinase Inhibition" Molecules 23, no. 12: 3271. https://doi.org/10.3390/molecules23123271
APA StyleNaboulsi, I., Aboulmouhajir, A., Kouisni, L., Bekkaoui, F., & Yasri, A. (2018). Combining a QSAR Approach and Structural Analysis to Derive an SAR Map of Lyn Kinase Inhibition. Molecules, 23(12), 3271. https://doi.org/10.3390/molecules23123271