
Classical QSAR and Docking Simulation of 4-Pyridone Derivatives for Their Antimalarial Activity

 , ,
, ,
Abstract
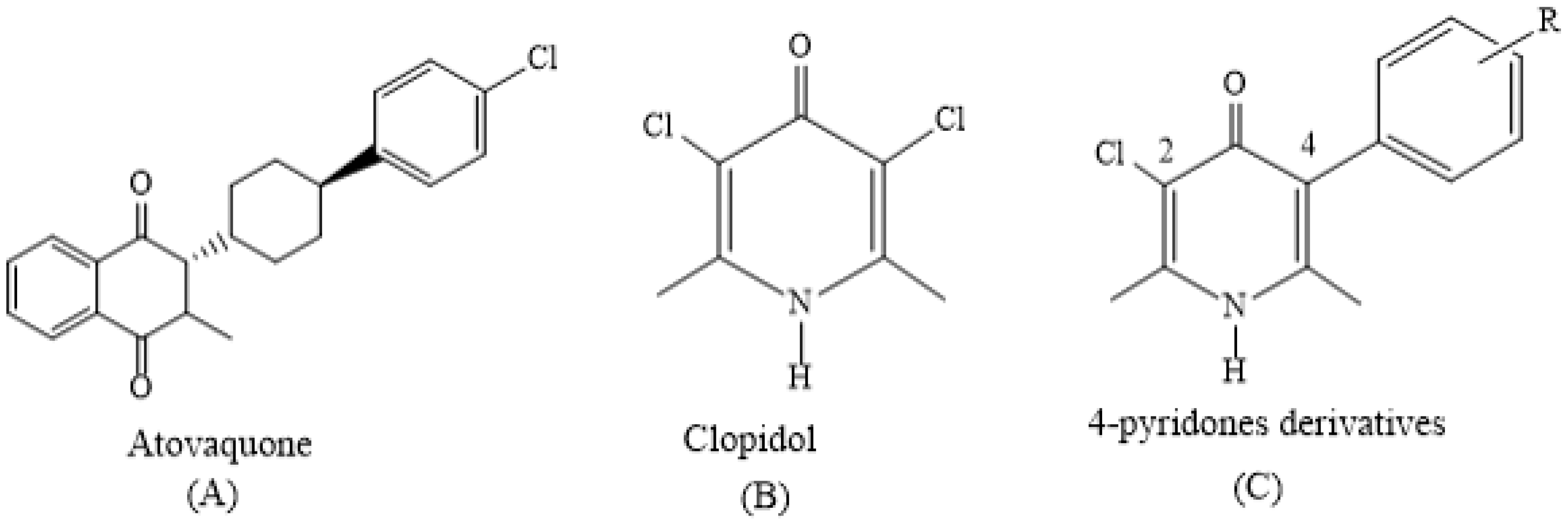
1. Introduction
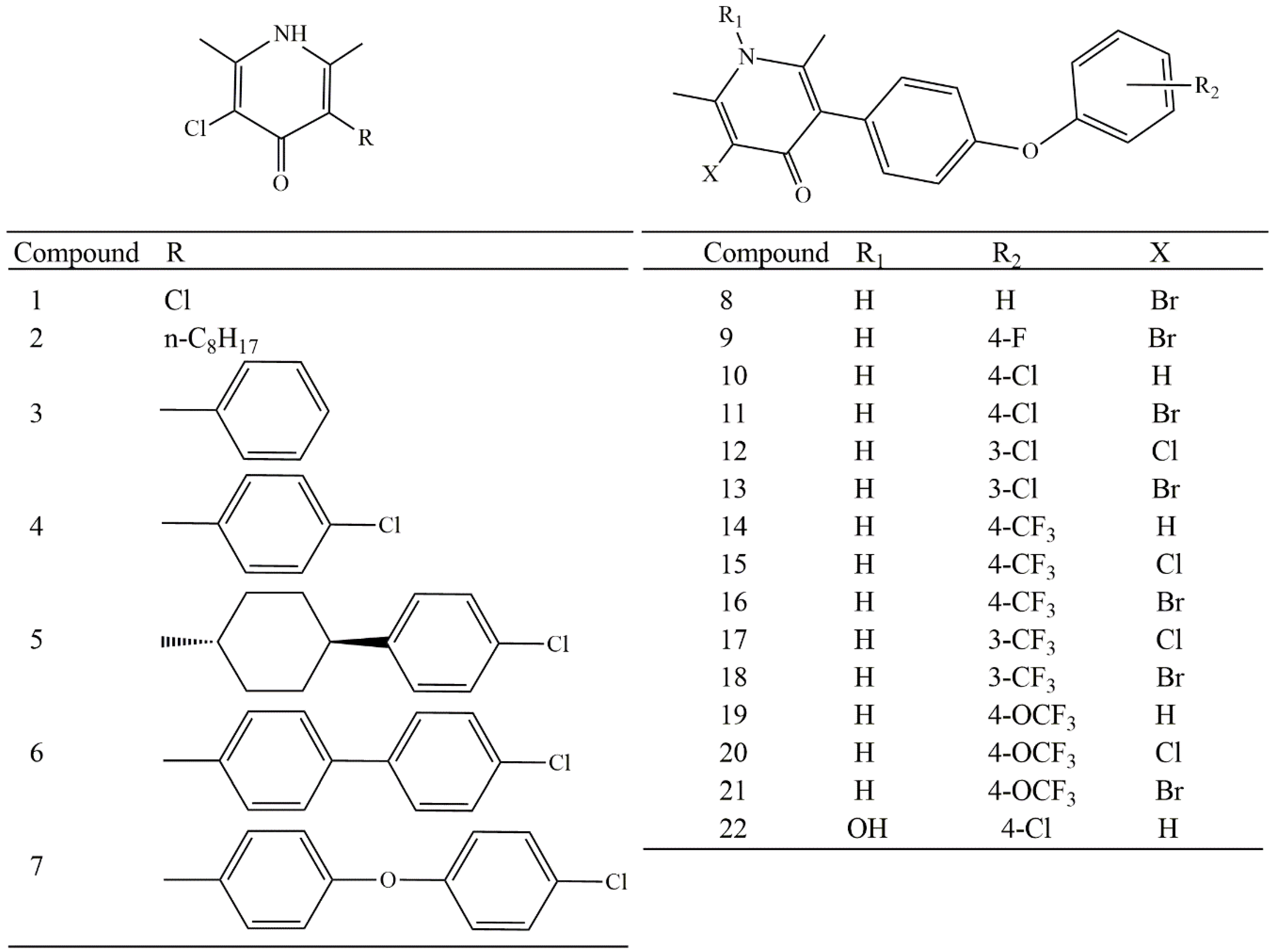
2. Results and Discussion
r = 0.950; r2 = 0.903; σ = 0.23; F = 40.233; σF = 0.0004; Q2 = 0.52
r = 0.960; r2 = 0.920; σ = 0.480; F = 37.66; σF = 0.003; Q2 = 0.54
r = 0.941 r2 = 0.886 σ = 0.38 F = 33.159; σF = 0.0006; Q2 = 0.92
r = 0.965; r2 = 0.930 σ = 0.380; F = 56.686 σF = 5.5 × 10−7; Q2 = 0.56
3. Materials and Methods
3.1. Statistical Analysis
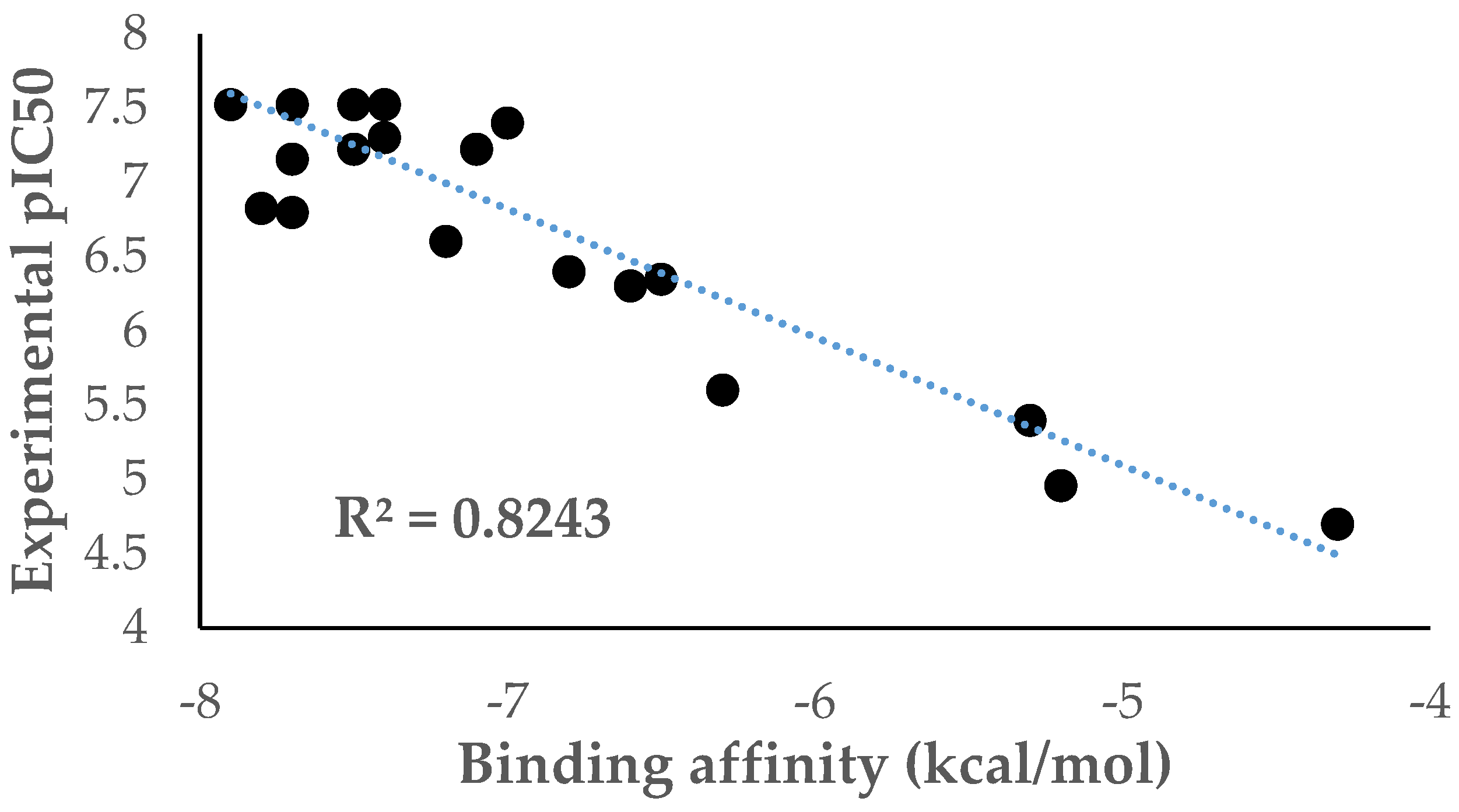
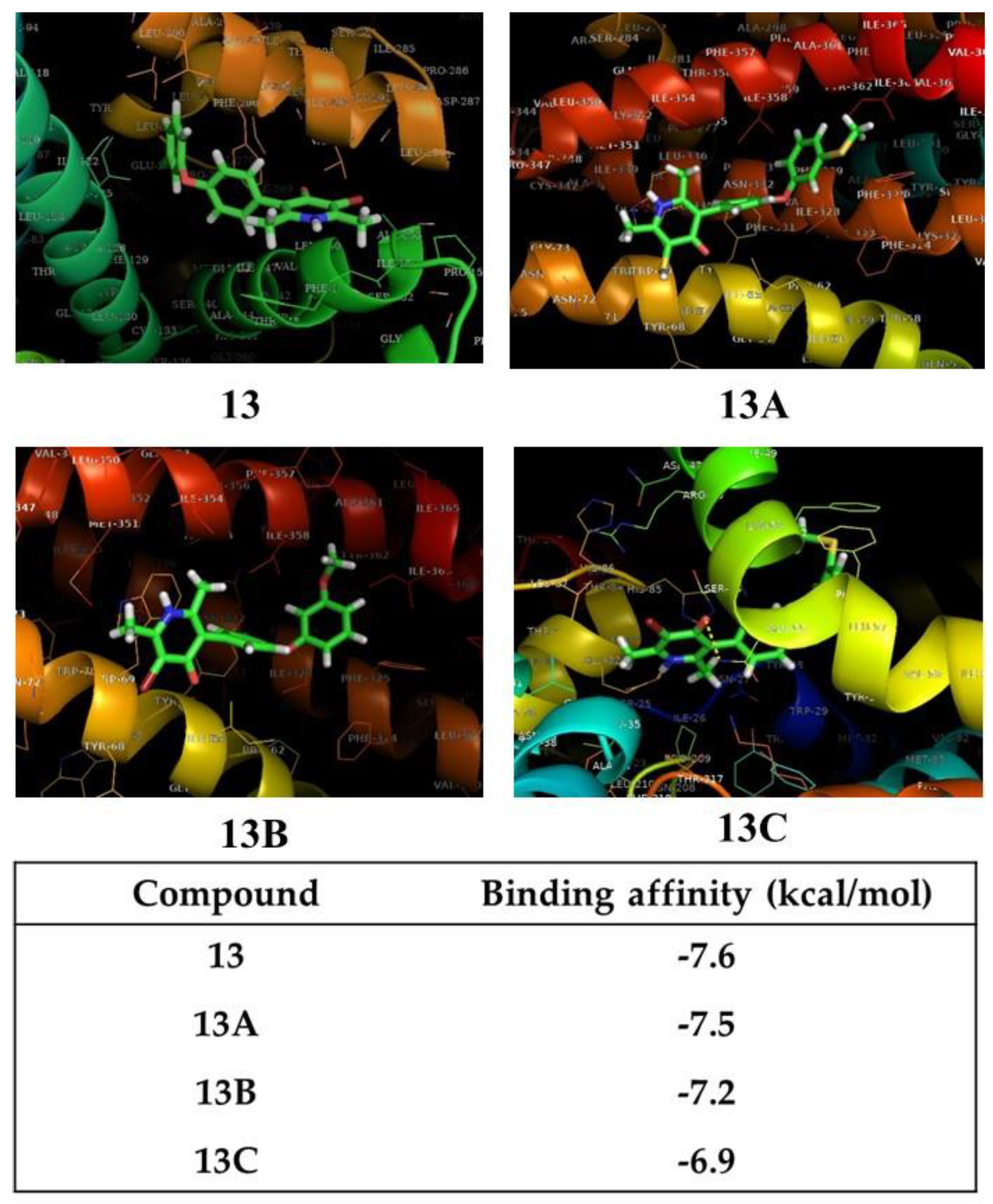
3.2. Docking Analysis
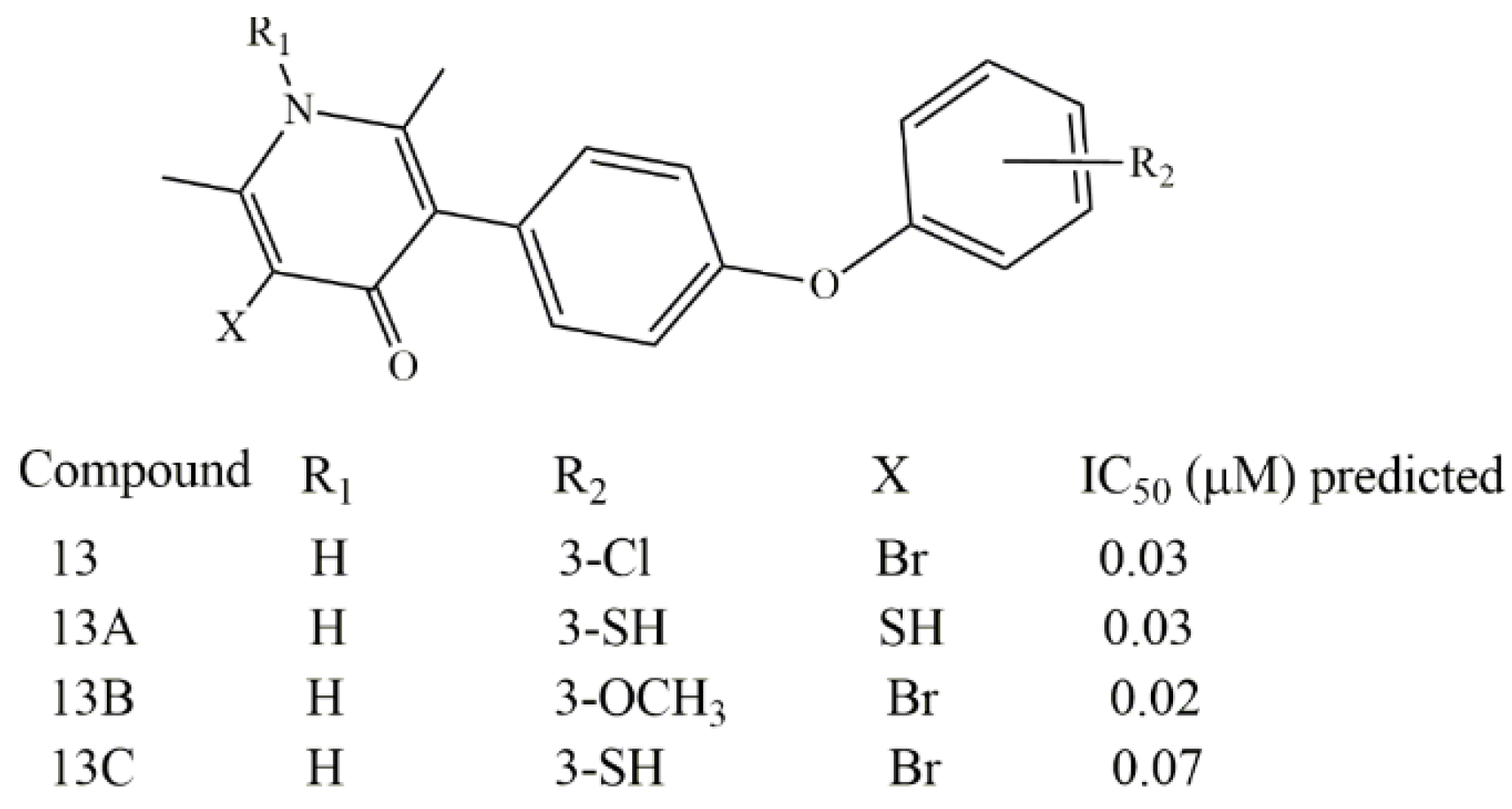
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Murray, C.J.; Rosenfeld, L.C.; Lim, S.S.; Andrews, K.G.; Foreman, K.J.; Haring, D.; Fullman, N.; Naghavi, M.; Lozano, R.; Lopez, A.D. Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet 2012, 379, 413–431. [Google Scholar] [CrossRef]
- Bueno, J.M.; Manzano, P.; García, M.C.; Chicharro, J.; Puente, M.; Lorenzo, M.; García, A.; Ferrer, S.; Gómez, R.M.; Fraile, M.T.; et al. Potent antimalarial 4-pyridones with improved physico-chemical properties. Bioorg. Med. Chem. Lett. 2011, 21, 5214–5218. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Shahid, M.; Kasam, V.; Ziegler, W.; Hofmann-Apitius, M. In silico drug discovery approaches on grid computing infrastructures. Curr. Clin. Pharm. 2010, 5, 37–46. [Google Scholar] [CrossRef]
- Barton, V.; Fisher, N.; Biagini, G.A.; Ward, S.A.; O’Neill, P.M. Inhibiting plasmodium cytochrome bc1: A complex issue. Curr. Opin. Chem. Biol. 2010, 14, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Mather, M.; Henry, K.; Vaidya, A. Mitochondrial drug targets in apicomplexan parasites. Curr. Drug Targets 2007, 8, 49–60. [Google Scholar] [CrossRef]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Mi-Ichi, F. Parasite mitochondria as a target of chemotherapy: inhibitory effect of licochalcone a on the plasmodium falciparum respiratory chain. Ann. N. Y. Acad. Sci. 2005, 1056, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Horta, P.; Kuş, N.; Henriques, M.S.C.; Paixão, J.A.; Coelho, L.; Nogueira, F.; O’Neill, P.M.; Fausto, R.; Cristiano, M.L.S. Quinolone–hydroxyquinoline tautomerism in quinolone 3-esters. Preserving the 4-oxoquinoline structure to retain antimalarial activity. J. Org. Chem. 2015, 80, 12244–12257. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; McSurdy-Freed, J.; Moorthy, G.S.; Hugger, E.; Bambal, R.; Han, C.; Ferrer, S.; Gargallo, D.; Davis, C.B. Preclinical drug metabolism and pharmacokinetic evaluation of GW844520, a novel anti-malarial mitochondrial electron transport inhibitor. J. Pharm. Sci. 2006, 95, 2657–2672. [Google Scholar] [CrossRef] [PubMed]
- Yeates, C.L.; Batchelor, J.F.; Capon, E.C.; Cheesman, N.J.; Fry, M.; Hudson, A.T.; Pudney, M.; Trimming, H.; Woolven, J.; Bueno, J.M.; et al. Synthesis and structure–activity relationships of 4-pyridones as potential antimalarials. J. Med. Chem. 2008, 51, 2845–2852. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Ojha, P.K. Advances in quantitative structure–activity relationship models of antimalarials. Expert Opin. Drug Discov. 2010, 5, 751–778. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Maiti, M.K.; Jha, T. Predictive comparative QSAR modeling of 4–pyridones as potent antimalarials. Int. Electron. J. Mol. Des. 2010, 9, 1–19. [Google Scholar]
- Shaik, B.; Kaushal, T.; Agrawal, V.K. Quantitative structure activity relationship studies on a series of 4-pyridones as antimalerial agents. J. Indian Chem. Soc. 2016, 93, 871–876. [Google Scholar]
- McQuarrie, D.A. Statistical Mec.; Calif University Science Books: Sausalito, CA, USA, 2000; ISBN 978-1-891389-15-3. [Google Scholar]
- Winkler, D.A. The role of quantitative structure-activity relationships (QSAR) in biomolecular discovery. Brief. Bioinform. 2002, 3, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.C.; Márquez, E.A.; Mora, J.R. Molecular modeling studies of bromopyrrole alkaloids as potential antimalarial compounds: A DFT approach. Med. Chem. Res. 2018, 27, 844–856. [Google Scholar] [CrossRef]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methods on more than 96,000 compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Dunn, W.J. Linear relationships between lipophilic character and biological activity of drugs. J. Pharm. Sci. 1972, 61, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Chemical hardness and the electronic chemical potential. Inorg. Chim. Acta 1992, 198–200, 781–786. [Google Scholar] [CrossRef]
- Mukhomorov, V.K. Biological activity of chemical compounds and their molecular Structure-information approach. J. Chem. Eng. Chem. Res. 2014, 1, 54–65. [Google Scholar]
- Cárdenas-Jirón, G.I.; Gutiérrez-Oliva, S.; Melin, J.; Toro-Labbé, A. Relations between potential energy, electronic chemical potential, and hardness profiles. J. Phys. Chem. A 1997, 101, 4621–4627. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K.; Kyle, D.E.; Vennerstrom, J.L. Structural analysis of chloroquine resistance reversal by imipramine analogs. Antimicrob. Agents Chem. 2001, 45, 2655–2657. [Google Scholar] [CrossRef]
- Jiménez Villalobos, T.P.; Gaitán Ibarra, R.; Montalvo Acosta, J.J. 2D, 3D-QSAR and molecular docking of 4(1H)-quinolones analogues with antimalarial activities. J. Mol. Graph. Model. 2013, 46, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Egan, T.J. Interactions of quinoline antimalarials with hematin in solution. J. Inorg. Biochem. 2006, 100, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Mammino, L.; Bilonda, M.K. Computational study of naphthylisoquinoline alkaloids with antimalarial activity from dioncophyllaceae and ancistrodaceae in vacuo. Theor. Chem. Acc. 2016, 135. [Google Scholar] [CrossRef]
- Young, R.C.; Durant, G.J.; Emmett, J.C.; Ganellin, C.R.; Graham, M.J.; Mitchell, R.C.; Prain, H.D.; Roantree, M.L. Dipole moment in relation to H2 receptor histamine antagonist activity for cimetidine analogues. J. Med. Chem. 1986, 29, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Hill, P.; Lange, B.B.; Meshnick, S.R.; Meunier, B.; Trumpower, B.L. Molecular basis for atovaquone resistance in modeled in parasites and pathogenic fungi. Trends Parasitol. 2007, 23, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Pacák, P. Molar refractivity and interactions in solutions I. Molar refractivity of some monovalent ions in aqueous and dimethyl sulfoxide solutions. Chem. Pap. 1989, 1989. 43, 289–500. [Google Scholar]
- Warhurst, D.C. Antimalarial drug discovery: Development of inhibitors of dihydrofolate reductase active in drug resistance. Drug Discov. Today 1998, 3, 538–546. [Google Scholar] [CrossRef]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 09 Citation Gaussian.com. Available online: http://gaussian.com/g09citation/ (accessed on 12 October 2018).
- Gill, P.M.W.; Johnson, B.G.; Pople, J.A.; Frisch, M.J. The performance of the Becke—Lee—Yang—Parr (B—LYP) density functional theory with various basis sets. Chem. Phys. Lett. 1992, 197, 499–505. [Google Scholar] [CrossRef]
- Marquez, E.; Domínguez, R.M.; Mora, J.R.; Córdova, T.; Chuchani, G. Experimental and theoretical studies of the homogeneous, unimolecular gas-phase elimination kinetics of trimethyl orthovalerate and trimethyl orthochloroacetate. J. Phys. Chem. A 2010, 114, 4203–4209. [Google Scholar] [CrossRef] [PubMed]
- Lezama, J.; Márquez, E.; Mora, J.R.; Córdova, T.; Chuchani, G. Theoretical calculations on the mechanisms of the gas phase elimination kinetics of chlorocyclohexane, 3-chlorocyclohexene and 4-chlorocyclohexene. J. Mol. Struct.: THEOCHEM 2009, 916, 17–22. [Google Scholar] [CrossRef]
- ChemAxon—Software Solutions and Services for Chemistry & Biology. Available online: https://chemaxon.com/search (accessed on 12 October 2018).
- Biostatistics: A Foundation for Analysis in the Health Sciences, 10th Edition. Available online: https://www.wiley.com/enus/Biostatistics%3A+A+Foundation+for+Analysis+in+the+Health+Sciences%2C+10th+Edition-p-9781118302798 (accessed on 12 October 2018).
- Golub, G.H.; Heath, M.; Wahba, G. Generalized cross-validation as a method for choosing a good ridge parameter. Technometrics 1979, 21, 215. [Google Scholar] [CrossRef]
- Sodero, A.C.R.; Abrahim-Vieira, B.; Torres, P.H.M.; Pascutti, P.G.; Garcia, C.R.; Ferreira, V.F.; Rocha, D.R. da; Ferreira, S.B.; Silva, F.P., Jr. Insights into cytochrome bc1 complex binding mode of antimalarial 2-hydroxy-1,4-naphthoquinones through molecular modelling. Memórias do Instituto Oswaldo Cruz 2017, 112, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.A.; Fisher, N.; Berry, N.; Stocks, P.A.; Meunier, B.; Williams, D.P.; Bonar-Law, R.; Bray, P.G.; Owen, A.; O’Neill, P.M.; et al. Acridinediones: Selective and potent inhibitors of the malaria parasite mitochondrial bc1 complex. Mol. Pharmacol. 2008, 73, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Birth, D.; Kao, W.-C.; Hunte, C. Structural analysis of atovaquone-inhibite8d cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Lange, B.B.; Merbitz-Zahradnik, T.; Zwicker, K.; Hill, P.; Meunier, B.; Pálsdóttir, H.; Hunte, C.; Meshnick, S.; Trumpower, B.L. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J. Biol. Chem. 2003, 278, 31312–31318. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Meshnick, S.R.; Trumpower, B.L. Modeling the molecular basis of atovaquone resistance in parasites and pathogenic fungi. Trends. Parasitol. 2007, 23, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Lange, C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001, 20, 6591–6600. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | pIC50 | s | µe | µ | q2 | q7 | G | MR | ClogP |
|---|---|---|---|---|---|---|---|---|---|
| Clopidol | 4.70 | 3.32 | −0.15 | 8.85 | 0.04 | −0.65 | −1321.26 | 49.35 | 1.22 |
| 2 | 5.40 | 3.52 | −0.14 | 7.80 | 0.04 | −0.66 | −1175.99 | 81.12 | 4.63 |
| 3 | 4.96 | 3.57 | −0.14 | 7.71 | 0.05 | −0.72 | −1092.66 | 69.01 | 1.78 |
| 4 | 5.60 | 3.50 | −0.14 | 8.70 | 0.05 | −0.73 | −1552.26 | 73.82 | 2.49 |
| 5 | 7.30 | 3.57 | −0.14 | 9.02 | 0.04 | −0.70 | −1786.81 | 99.52 | 5.17 |
| 6 | 6.40 | 3.58 | −0.13 | 8.74 | 0.07 | −0.77 | −1783.26 | 98.95 | 4.38 |
| 7 | 7.22 | 3.66 | −0.13 | 8.00 | 0.06 | −0.75 | −1858.47 | 100.06 | 4.59 |
| 8 | 6.82 | 3.76 | −0.13 | 7.58 | 0.02 | −0.76 | −3510.40 | 98.07 | 4.02 |
| 9 | 7.40 | 3.68 | −0.13 | 8.13 | 0.01 | −0.75 | −3609.65 | 98.29 | 4.17 |
| 10 | 6.60 | 3.73 | −0.13 | 8.45 | −0.33 | −0.76 | −1398.87 | 95.27 | 3.79 |
| 11 | 7.40 | 3.67 | −0.13 | 8.41 | 0.02 | −0.76 | −3970.00 | 102.87 | 4.74 |
| 12 | 7.52 | 3.63 | −0.13 | 9.70 | 0.06 | −0.75 | −1858.47 | 100.06 | 4.59 |
| 13 | 7.52 | 3.63 | −0.13 | 9.60 | 0.01 | −0.76 | −3970.00 | 102.87 | 4.74 |
| 14 | 6.30 | 3.43 | −0.14 | 11.19 | −0.34 | −0.75 | −1276.32 | 96.43 | 3.96 |
| 15 | 7.22 | 3.47 | −0.14 | 11.36 | 0.05 | −0.74 | −1735.92 | 101.23 | 4.76 |
| 16 | 7.52 | 3.52 | −0.13 | 11.09 | −0.01 | −0.75 | −3847.46 | 104.04 | 4.91 |
| 17 | 7.52 | 3.51 | −0.13 | 10.97 | 0.06 | −0.75 | −1735.92 | 101.23 | 4.76 |
| 18 | 7.52 | 3.63 | −0.13 | 9.01 | 0.01 | −0.76 | −3886.75 | 109.08 | 5.41 |
| 19 | 6.80 | 3.63 | −0.13 | 10.48 | −0.34 | −0.75 | −1351.54 | 97.00 | 4.11 |
| 20 | 7.52 | 3.57 | −0.13 | 10.52 | 0.05 | −0.74 | −1811.14 | 101.79 | 4.90 |
| 21 | 7.52 | 3.59 | −0.13 | 10.36 | 0.00 | −0.75 | −3922.67 | 104.61 | 5.05 |
| 22 | 5.66 | 3.65 | −0.13 | 8.67 | −0.34 | −0.35 | −1474.02 | 0.00 | 3.69 |
| Parameter | pIC50 | ClogP | MR | µ | µe |
|---|---|---|---|---|---|
| pIC50 | 1.000 | 0.850 | 0.726 | 0.427 | 0.647 |
| ClogP | 0.850 | 1.000 | 0.606 | 0.352 | 0.597 |
| MR | 0.726 | 0.606 | 1.000 | 0.301 | 0.217 |
| µ | 0.427 | 0.352 | 0.301 | 1.000 | −0.035 |
| µe | 0.647 | 0.597 | 0.217 | −0.035 | 1.000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Sumoza, M.; Alcázar, J.J.; Márquez, E.; Mora, J.R.; Lezama, J.; Puello, E. Classical QSAR and Docking Simulation of 4-Pyridone Derivatives for Their Antimalarial Activity. Molecules 2018, 23, 3166. https://doi.org/10.3390/molecules23123166
Flores-Sumoza M, Alcázar JJ, Márquez E, Mora JR, Lezama J, Puello E. Classical QSAR and Docking Simulation of 4-Pyridone Derivatives for Their Antimalarial Activity. Molecules. 2018; 23(12):3166. https://doi.org/10.3390/molecules23123166
Chicago/Turabian StyleFlores-Sumoza, Máryury, Jackson J. Alcázar, Edgar Márquez, José R. Mora, Jesús Lezama, and Esneyder Puello. 2018. "Classical QSAR and Docking Simulation of 4-Pyridone Derivatives for Their Antimalarial Activity" Molecules 23, no. 12: 3166. https://doi.org/10.3390/molecules23123166
APA StyleFlores-Sumoza, M., Alcázar, J. J., Márquez, E., Mora, J. R., Lezama, J., & Puello, E. (2018). Classical QSAR and Docking Simulation of 4-Pyridone Derivatives for Their Antimalarial Activity. Molecules, 23(12), 3166. https://doi.org/10.3390/molecules23123166