Recognition of Protein Pupylation Sites by Adopting Resampling Approach


Abstract
1. Introduction
2. Results and Discussion
2.1. Pearson Correlation Coefficient for Feature Selection
2.2. Combination of KPCA and FUS for Training Set Balancing
2.3. Predictive Performance Improvement
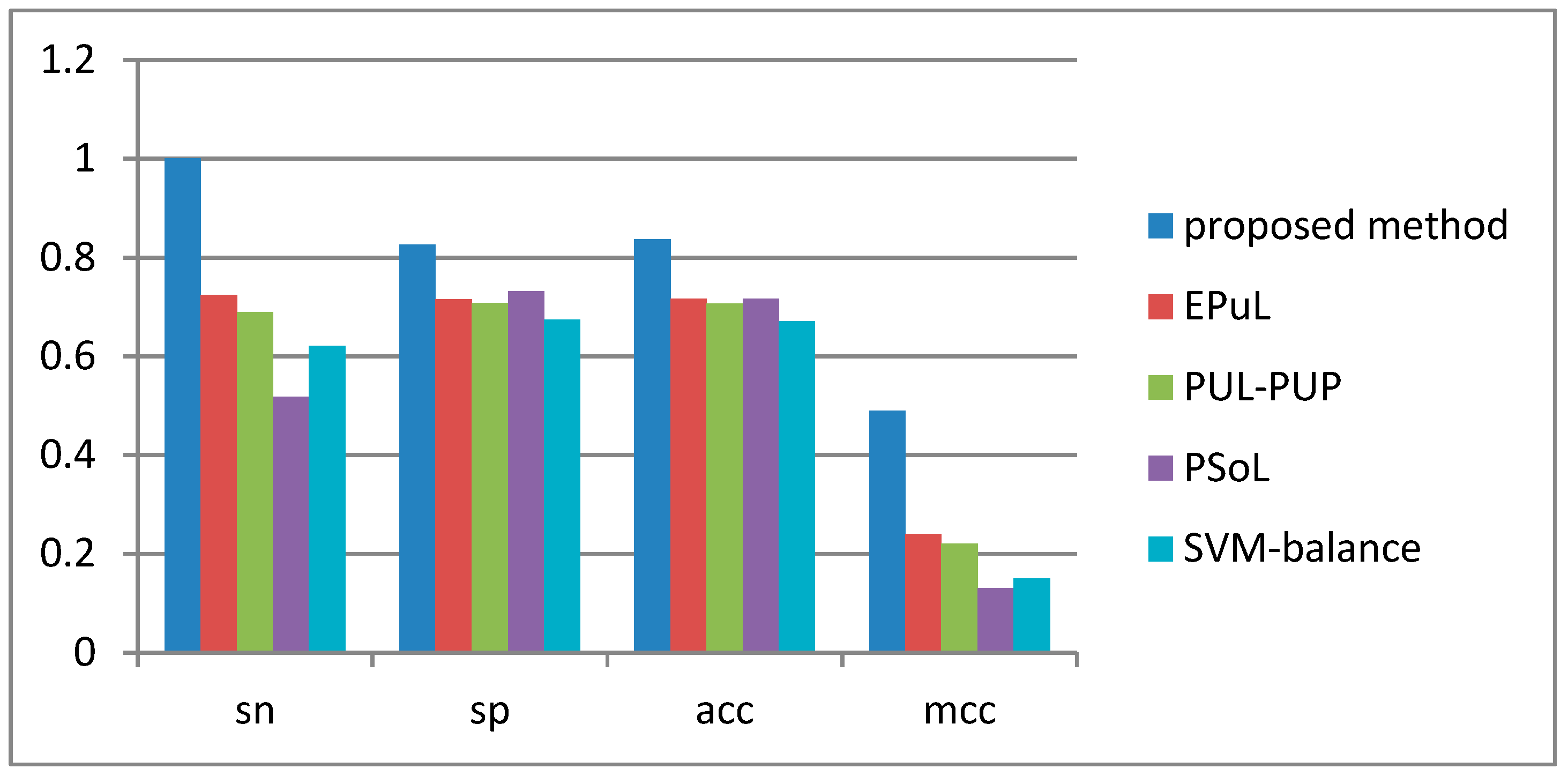
2.4. Comparison between the Proposed Method and Other Prediction Methods
2.5. Performance on the Independent Test Dataset
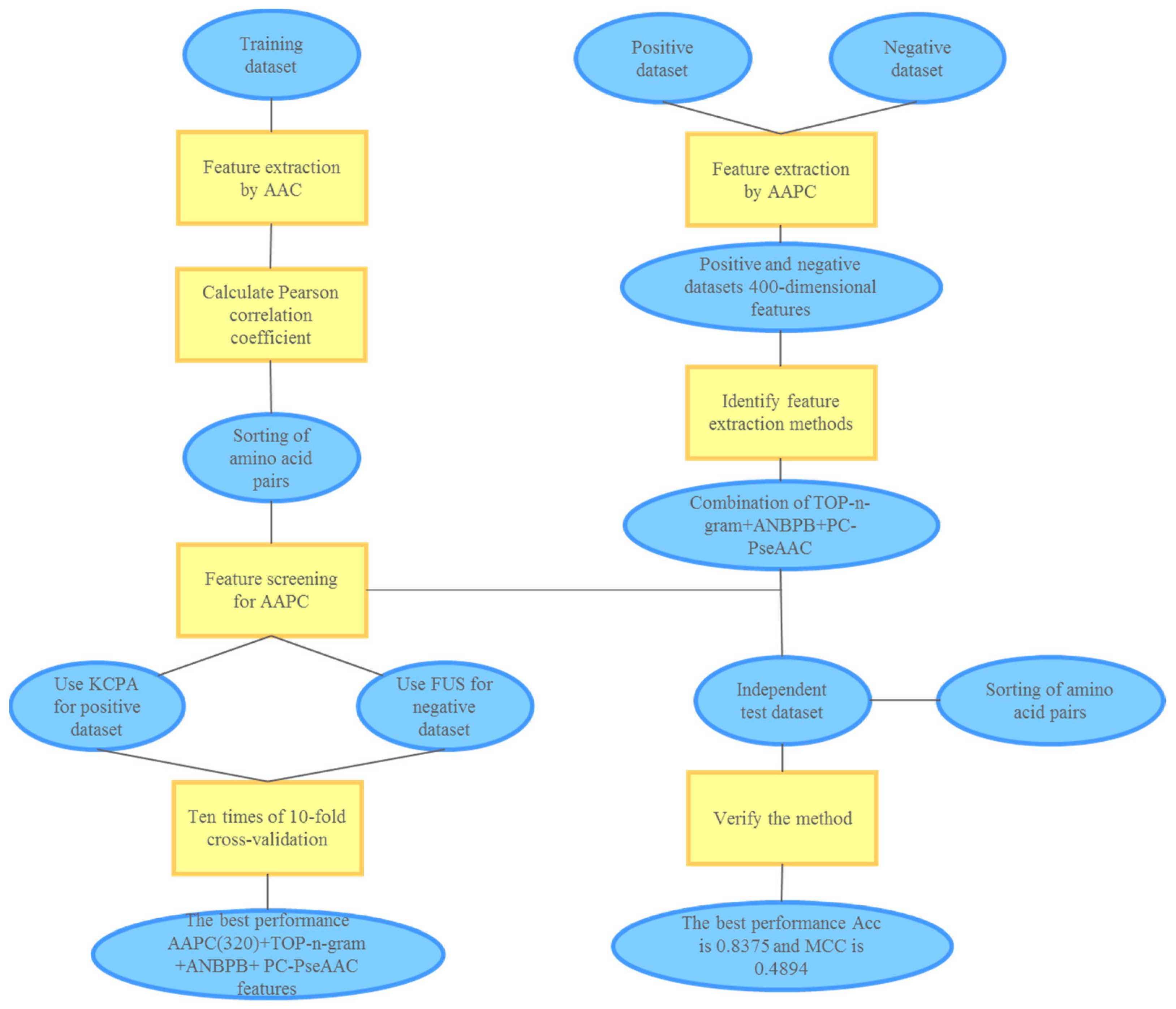
3. Materials and Methods
3.1. Formatting of Mathematical Components
3.2. Information from the Protein Peptide Sequence
3.2.1. Amino Acid Composition (AAC)
3.2.2. Amino Acid Pair Composition
3.2.3. Adapted Normal Distribution bi-profile Bayes
3.2.4. TOP-n-gram
3.2.5. Parallel Correlation Pseudo Amino Acid Composition
3.3. Pearson Correlation Coefficient
3.4. Processing of Positive and Negative Datasets
3.4.1. KPCA Oversampling Technique
3.4.2. Fuzzy Undersampling Method
3.5. SVMs Implementation and Parameter Selectiont
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Pup | prokaryotic ubiquitin-like protein |
| KPCA | k-means principal component analysis oversampling technique |
| FUS | fuzzy undersampling method |
| Sn | sensitivity |
| Sp | specificity |
| Acc | accuracy |
| MCC | Matthews correlation coefficient |
| CKSAAP | composition of k-spaced amino acid pairs feature |
| pbCKSAAP | profile-based composition of k-spaced amino acid pairs |
| AAC | extracting amino acid composition |
| M. smegmatis | Mycobacterium smegmatis |
| M. tuberculosis | Mycobacterium tuberculosis |
| E. coli | Escherichia coli |
| AAPC | amino acid pair composition |
| ANBPB | adapted normal distribution bi-profile Bayes |
| BPB | bi-profile Bayes |
| PC-PseAAC | parallel correlation pseudo amino acid composition |
| PCA | principal component analysis |
| SVM | support vector machine |
| RBF | radial basis kernel function |
References
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 2007, 100, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell. Bio. 2005, 6, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Austin, M.C.; Schmidt, R.; Kurland, B.F.; Vaezi, A.; Hayes, D.N.; Mendez, E.; Parvathaneni, U.; Chai, X.; Sampath, S.; et al. ERCC1 is a prognostic biomarker in locally advanced head and neck cancer: Results from a randomised, phase II trial. Br. J. Cancer 2013, 109, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Falzon, M.; Blackhall, F.; Spicer, J.; Nicolson, M.; Chaudhuri, A.; Middleton, G.; Ahmed, S.; Hicks, J.; Crosse, B. Randomized Prospective Biomarker Trial of ERCC1 for Comparing Platinum and Nonplatinum Therapy in Advanced Non–Small-Cell Lung Cancer: ERCC1 Trial (ET). J. Clin. Oncol. 2017, 35, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Faridounnia, M.; Wienk, H.; Kovacic, L.; Folkers, G.E.; Nicolass, G.J.; Kaptein, R.; Jan, H.J.; Boelens, R. The Cerebro-oculo-facio-skeletal Syndrome Point Mutation F231L in the ERCC1 DNA Repair Protein Causes Dissociation of the ERCC1-XPF Complex. J. Biol. Chem. 2015, 33, 20541–20555. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.J.; Mintseris, J.; Ferreyra, J.; Steven, P.G.; Heran, D.K. Ubiquitin-Like Protein Involved in the Proteasome Pathway of Mycobacterium tuberculosis. Science. 2008, 5904, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Perez-Oliva, A.B.; Lachaud, C.; Szyniarowski, P.; Muñoz, I.; Macartney, T.; Hickson, I.; Rouse, J.; Alessi, D.R. USP45 deubiquitylase controls ERCC1-XPF endonuclease-mediated DNA damage responses. EMBO J. 2015, 34, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gong, F. The emerging role of deubiquitination in nucleotide excision repair. DNA Repair 2016, 43, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Cuijk, L.V.; Van Belle, G.J.; Turkyilmaz, Y.; Poulsen, S.L.; Janssens, R.C.; Theil, F.A.; Sabatella, M. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat. Commun. 2015, 6, 7499. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.W. PupDB: A database of pupylated proteins. BMC Bioinf. 2012, 1186, 1471–2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, Q.; Cao, J.; Gao, X.; Ren, J.; Xue, Y. GPS-PUP: Computational prediction of pupylation sites in prokaryotic proteins. Mol. Biosystems. 2011, 7, 2737–2740. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.W. Prediction of pupylation sites using the composition of k-spaced amino acid pairs. J. Theor. Biol. 2013, 336, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.W.; Dai, J.Y.; Ning, Q.; Ma, Z.Q.; Yin, M.H.; Sun, P.P. Position-Specific Analysis and Prediction of Protein Pupylation Sites Based on Multiple Features. Biomed Res. Int. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qiu, J.D.; Shi, S.P.; Suo, S.B.; Liang, R.P. Systematic Analysis and Prediction of Pupylation Sites in Prokaryotic Proteins. PLoS ONE 2013, 8, e74002. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Zhou, Y.; Lu, X.T.; Li, J.Y.; Song, J.N.; Zhang, Z.D. Computational Identification of Protein Pupylation Sites by Using Profile-Based Composition of k-Spaced Amino Acid Pairs. PLoS ONE 2015, 10, e0129635. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Cao, J.Z. Positive-Unlabeled learning for pupylation sites prediction. Biomed. Res. Int. 2016, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.G.; Bao, L.L.; Zhao, X.S.; Zhao, X.W.; Sangaiah, A.K.; Wang, G.G.; Ma, Z.Q. EPuL: An Enhanced Positive-Unlabeled Learning Algorithm for the Prediction of Pupylation Sites. Molecules 2017, 22, 1463. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, X.; Lin, L.; Dong, Q.; Wang, X. A discriminative method for protein remote homology detection and fold recognition combining Top-n-grams and latent semantic analysis. BMC Bioinform. 2008, 9, 510. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Z.; Liu, T.; Wang, Z.P. O-GlcNAcPRED: A sensitive predictor to capture protein O-GlcNAcylation sites. Mol. Biosyst. 2013, 9, 2909–2913. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins: Structure Function Bioinform 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Z.; Zuo, Y. Computional prediction of protein O-GlcNAc modification. Methods Mol. Biol. 2018, 1754, 235–246. [Google Scholar] [PubMed]
- Hosseinzadeh, M.; Eftekhari, M. Using Fuzzy Undersampling and Fuzzy PCA to Improve Imbalanced Classification through Rotation Forest Algorithm. CSSE Int. Symp. Cmpt. Sci. Software Eng. 2015, 1–7. [Google Scholar]
- Kruskal, W.H. Ordinal measures of association. Journal of the American Statistical Association 1958, 53, 814–861. [Google Scholar] [CrossRef]
- Lee Rodgers, J.; Nicewander, W.A. Thirteen ways to look at the correlation coefficient. Am. Stat. 1988, 42, 59. [Google Scholar] [CrossRef]
- Liu, B.; Wu, H. Pse-in-One 2.0: A web server for generating comprehensive modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2017, 10, 4236. [Google Scholar]
- Wang, C.; Ding, C.; Meraz, R.F.; Holbrook, S.R. PSoL: A positive sample only learning algorithm for finding non-coding RNA genes. Bioinformatics 2006, 22, 2590. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.M.; Weng, S.L.; Lu, C.T.; Cheng, T.L.; Chang, T.H.; Weng, T.Y.; Lee, T.Y. SOHSite: Incorporating evolutionary information and physicochemical properties to identify protein S-sulfenylation sites. BMC Genomics 2016, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, J.; Wu, L.Y. iSulf-Cys: Prediction of S-sulfenylation Sites in Proteins with Physicochemical Properties of Amino Acids. PLoS ONE 2016, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Song, J.N.; Thomson, B.A. Cascleave: Towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 2010, 26, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, J.; Wu, L.Y.; Chou, K.C. iSNO-PseAAC: Predict Cysteine S-Nitrosylation Sites in Proteins by Incorporating Position Specific Amino Acid Propensity into Pseudo Amino Acid Composition. PLoS ONE 2013, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.Y.; Liu, F.L.; Wang, X.L.; Chen, J.J.; Chou, K.C. Identification of Real MicroRNA Precursors with a Pseudo Structure Status Composition Approach. PLoS ONE 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Sokal, R.R.; Thomson, B.A. Population structure inferred by local spatial autocorrelation: An example from an Amerindian tribal population. Am. J. Phys. Anthropol. 2006, 129, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Deng, E.Z.; Ding, H.; Chen, W.; Chou, K.C. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961–12972. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.S.; Eskin, E.; Cohen, A.; Weston, J.; Noble, W.S.S. Mismatch string kernels for discriminative protein classification. Bioinformatics 2004, 20, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Z.; Zuo, Y.; Zou, Q. O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 2018, 34, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Si, J.N.; Yan, R.X.; Wang, C.; Zhang, Z.D.; Su, X.D. TIM-Finder: A new method for identifying TIM-barrel proteins. BMC Struct. Biol. 2009, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.L.; Li, J.T.; Shu, Y.M.; Peng, J.H. SAR Image Denoising via Clustering-Based Principal Component Analysis. IEEE T. Geosci. Remote 2014, 52, 6858–6869. [Google Scholar]
- Yan, R.X.; Si, J.N.; Wang, C.; Zhang, Z.D. DescFold: A web server for protein fold recognition. BMC Bioinform. 2009, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.J.; Wan, S.X.; He, Y.; Zou, Q. Classification of Small GTPases with Hybrid Protein Features and Advanced Machine Learning Techniques. Curr. Bioinform. 2018, 13, 492–500. [Google Scholar] [CrossRef]
- Liao, Z.J.; Li, D.P.; Wang, X.R.; Li, L.S.; Zou, Q. Cancer Diagnosis Through IsomiR Expression with Machine Learning Method. Curr. Bioinform. 2018, 13, 57–63. [Google Scholar] [CrossRef]
- Li, D.P.; Ju, Y.; Zou, Q. Protein Folds Prediction with Hierarchical Structured SVM. Curr. Proteomics 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines. ACM T. Intel. Syst. Tec. 2011, 2, 3. [Google Scholar] [CrossRef]
- Ju, Z.; Cao, J.Z.; Gu, H. Predicting lysine phosphoglycerylation with fuzzy SVM by incorporating k-spaced amino acid pairs into Chou’s general PseAAC. Theor. Biol. 2016, 397, 145–150. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



{kind=link}
{kind=link}
| Method | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| Without resampling | 0 | 100 | 92.50 | NaN |
| KPCA oversampling | 25.54 | 99.87 | 92.67 | 0.33 |
| KPCA oversampling and FUS undersampling | 81.42 | 76.44 | 78.10 | 0.55 |
| Feature | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| AAPC(320) | 81.42 | 76.44 | 78.1 | 0.55 |
| AAPC(320) + TOP-n-gram | 80.33 | 100 | 93.43 | 0.86 |
| AAPC(320) + ANBPB | 64.48 | 100 | 88.14 | 0.74 |
| AAPC(320) + PC-PseAAC | 84.15 | 72.60 | 76.46 | 0.54 |
| AAPC(320) + TOP-n-gram + ANBPB | 70.49 | 98.36 | 89.05 | 0.75 |
| AAPC(320) + TOP-n-gram + PC-PseAAC | 87.43 | 75.34 | 79.38 | 0.59 |
| AAPC(320) + ANBPB + PC-PseAAC | 75.41 | 100 | 91.79 | 0.82 |
| AAPC(320) + TOP-n-gram + ANBPB + PC-PseAAC | 94.54 | 100 | 98.18 | 0.96 |
| Method | Sn (%) | Sp (%) | Acc (%) | MCC | AUC |
|---|---|---|---|---|---|
| Proposed method | 90.53 | 99.8 | 95.09 | 0.91 | 0.96 |
| * EPuL | 84.21 | 95.45 | 90.24 | 0.81 | 0.93 |
| * PUL-PUP | 82.24 | 91.57 | 88.92 | 0.74 | 0.92 |
| * PSoL | 67.5 | 73.6 | 70.55 | 0.42 | 0.8 |
| * SVM balance | 76.71 | 63.65 | 69.88 | 0.4 | 0.77 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Chen, Y.; Li, T.; Jia, C. Recognition of Protein Pupylation Sites by Adopting Resampling Approach. Molecules 2018, 23, 3097. https://doi.org/10.3390/molecules23123097
Li T, Chen Y, Li T, Jia C. Recognition of Protein Pupylation Sites by Adopting Resampling Approach. Molecules. 2018; 23(12):3097. https://doi.org/10.3390/molecules23123097
Chicago/Turabian StyleLi, Tao, Yan Chen, Taoying Li, and Cangzhi Jia. 2018. "Recognition of Protein Pupylation Sites by Adopting Resampling Approach" Molecules 23, no. 12: 3097. https://doi.org/10.3390/molecules23123097
APA StyleLi, T., Chen, Y., Li, T., & Jia, C. (2018). Recognition of Protein Pupylation Sites by Adopting Resampling Approach. Molecules, 23(12), 3097. https://doi.org/10.3390/molecules23123097