Maritoclax Enhances TRAIL-Induced Apoptosis via CHOP-Mediated Upregulation of DR5 and miR-708-Mediated Downregulation of cFLIP


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
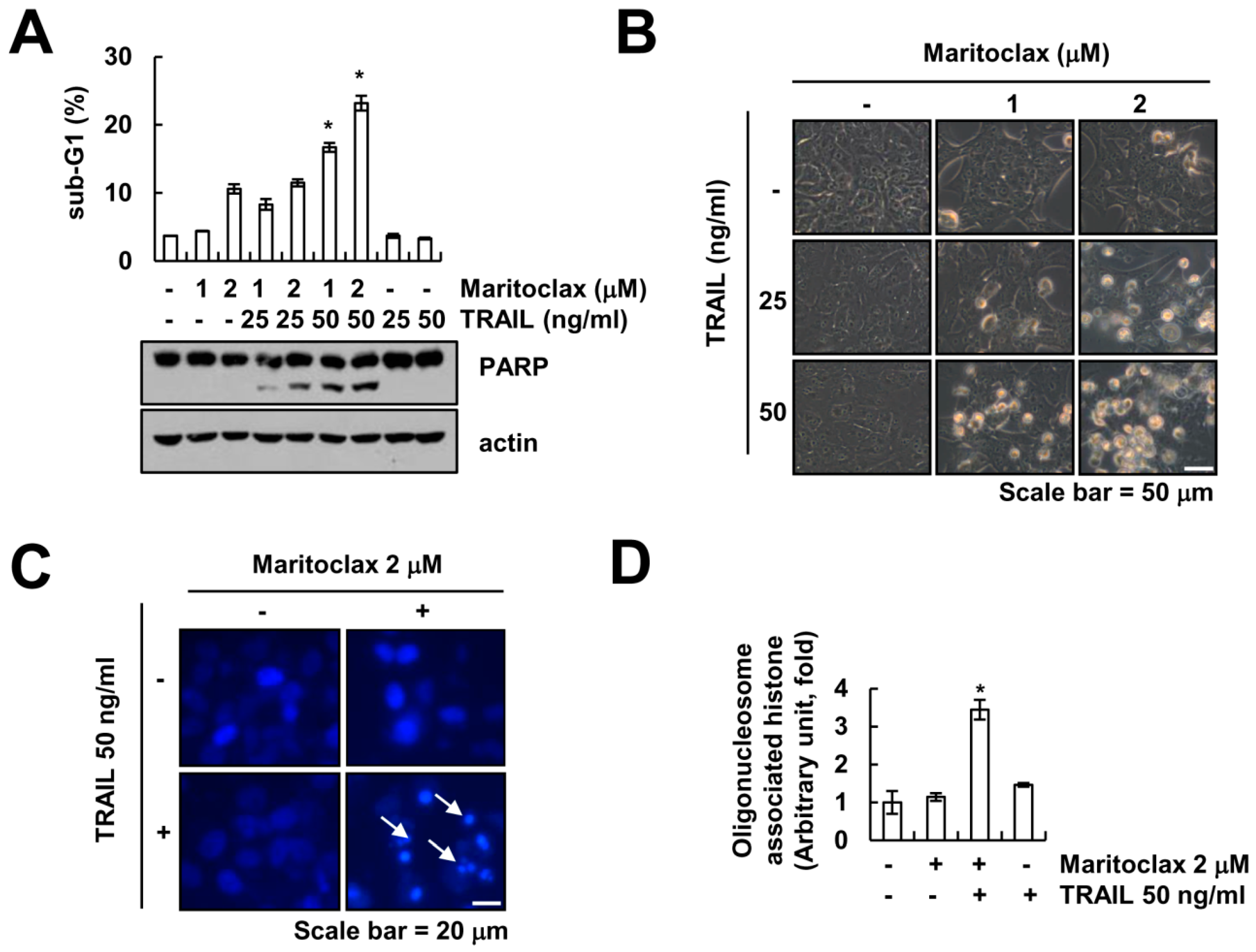
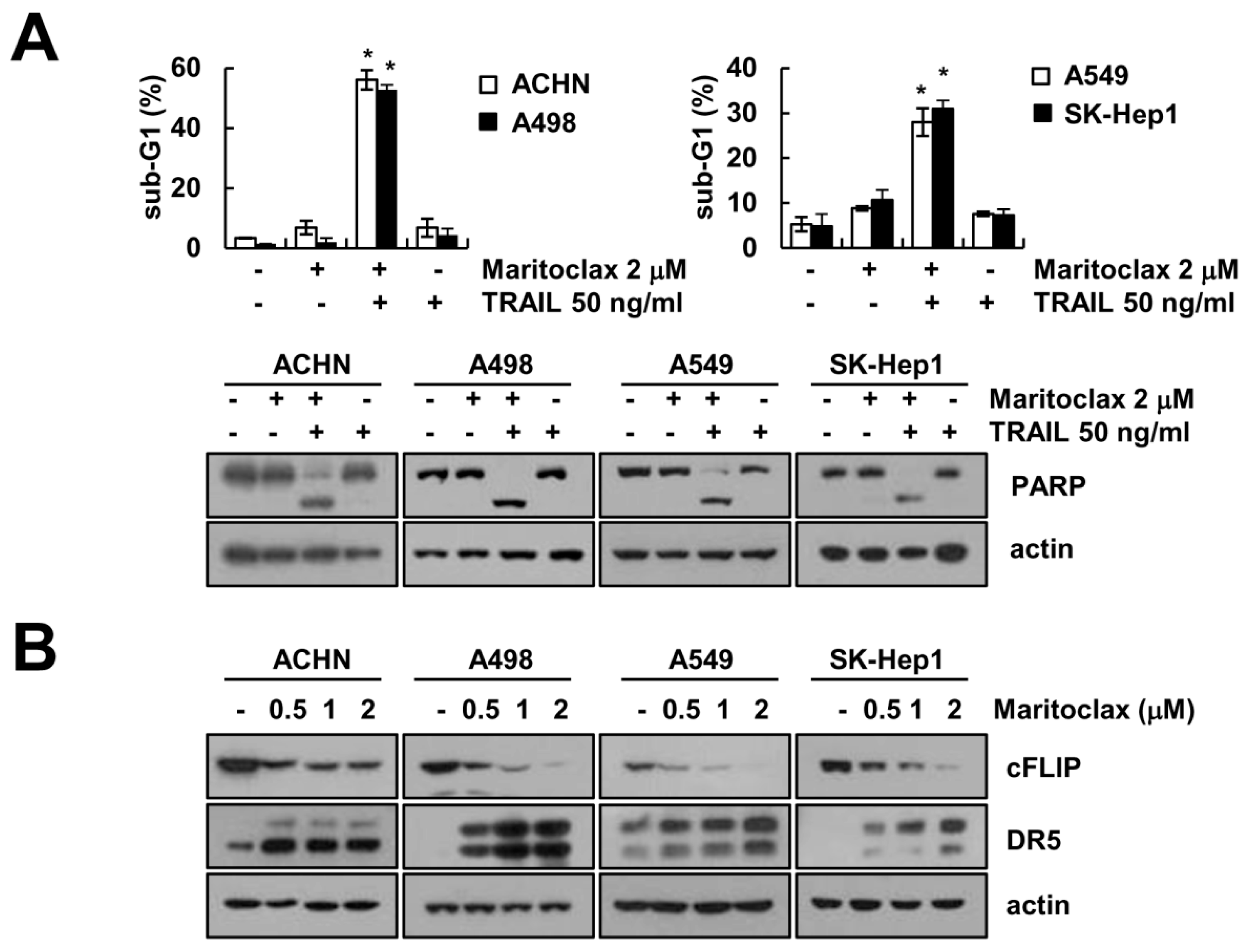
2.1. Effect of Maritoclax on TRAIL-Mediated Apoptosis in Human Renal Carcinoma Caki Cells
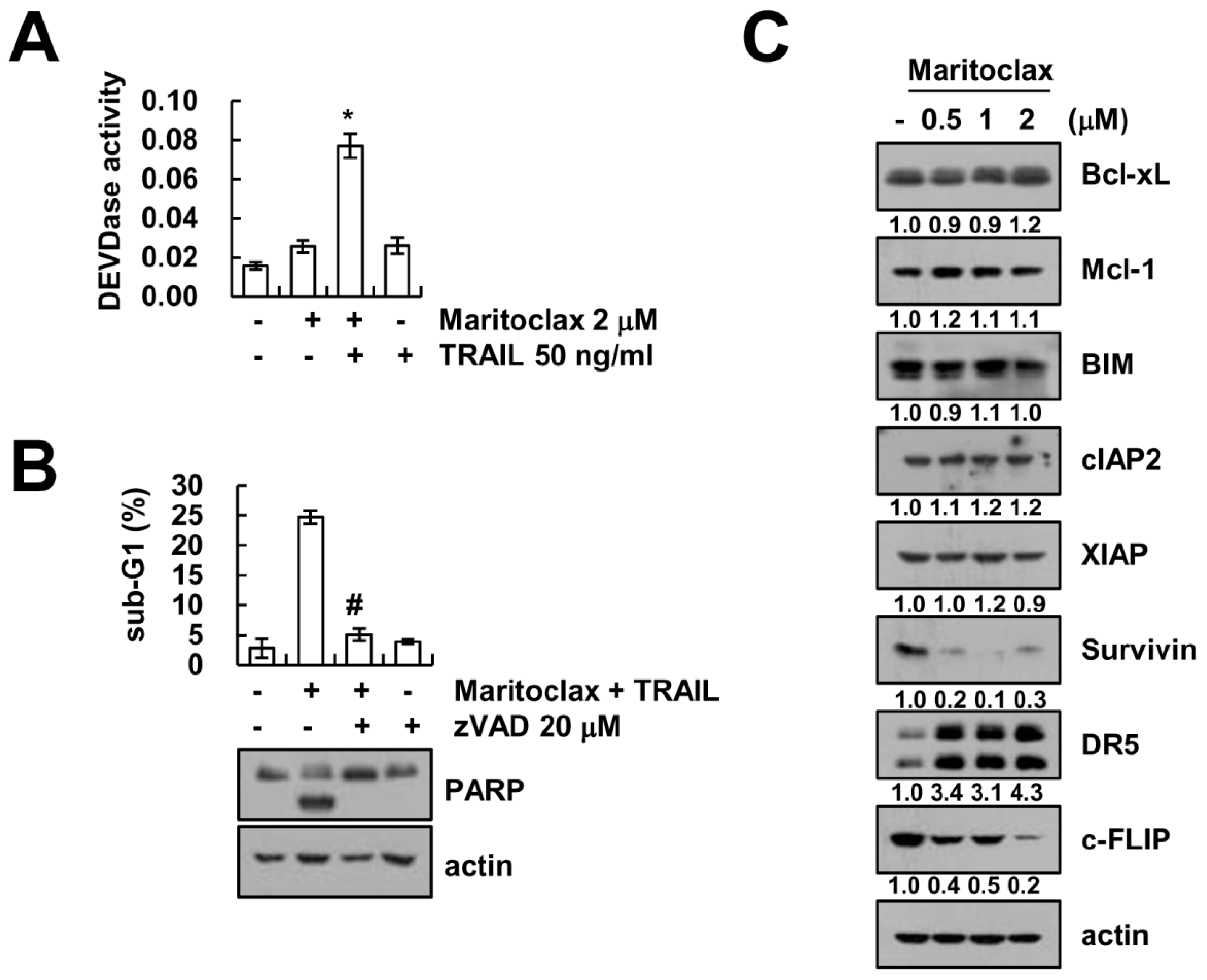
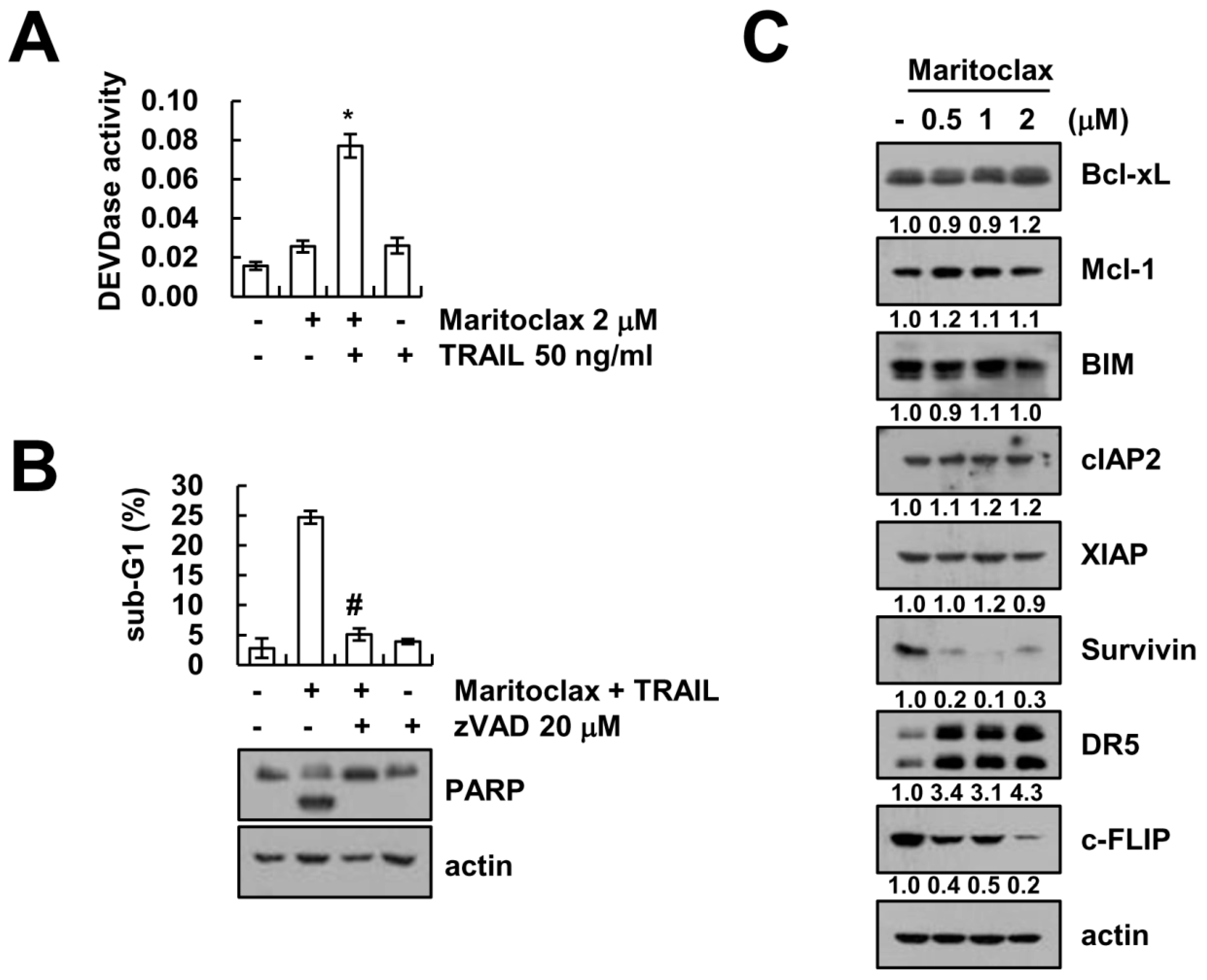
2.2. The Effect of Caspase Activation on Maritoclax Plus TRAIL-Induced Apoptosis
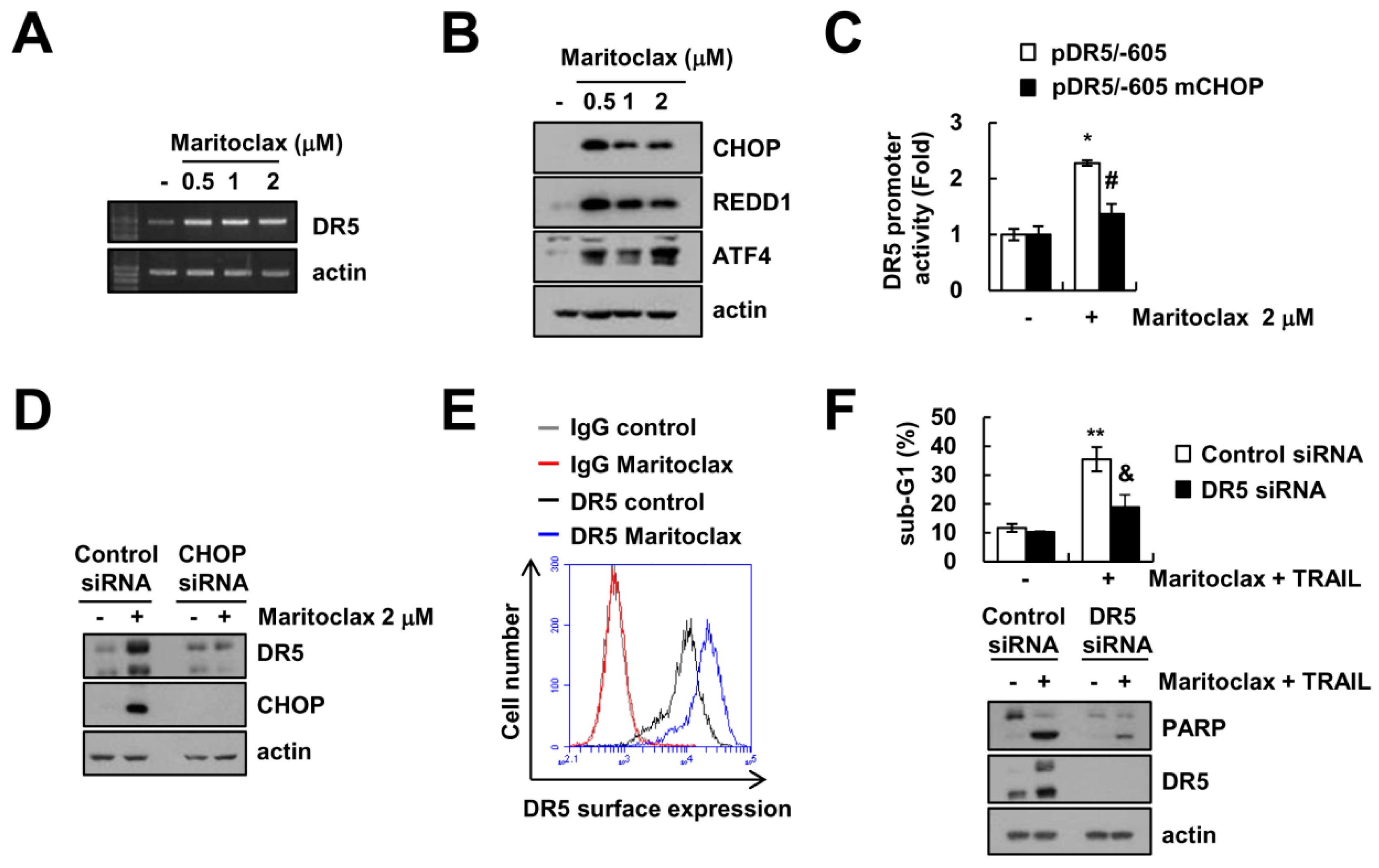
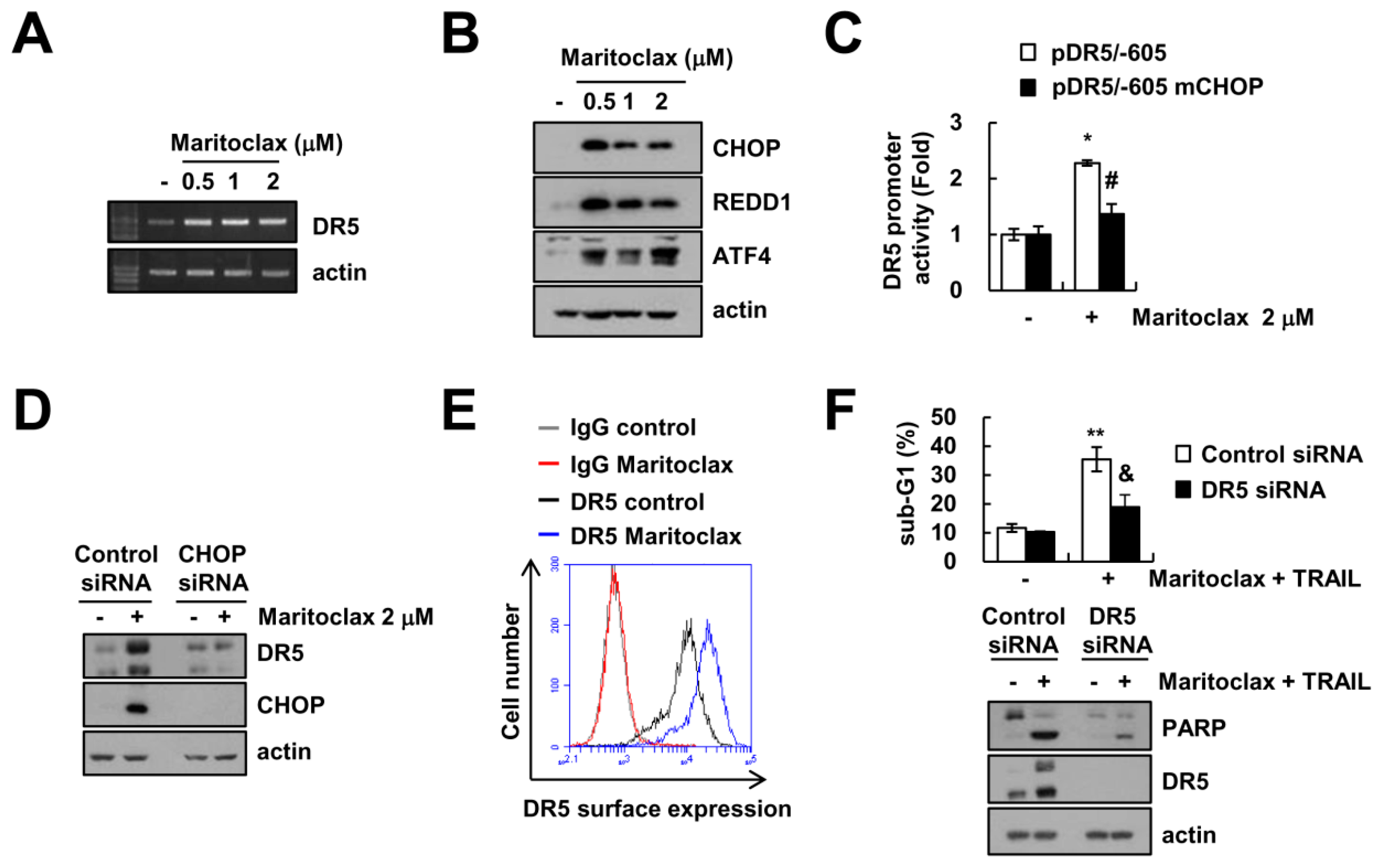
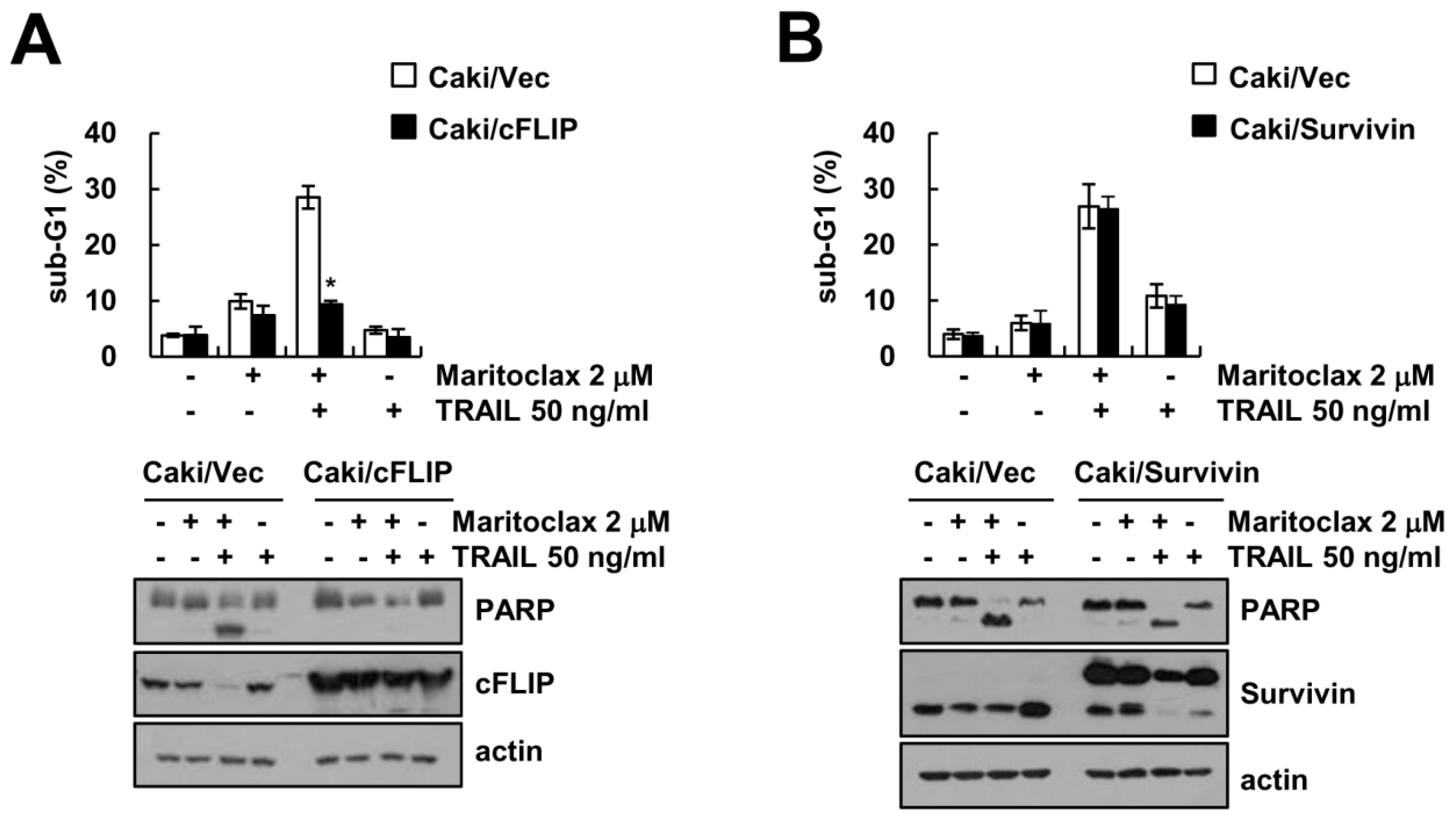
2.3. The Effect of DR5 Upregulation on Maritoclax Plus TRAIL-Induced Apoptosis
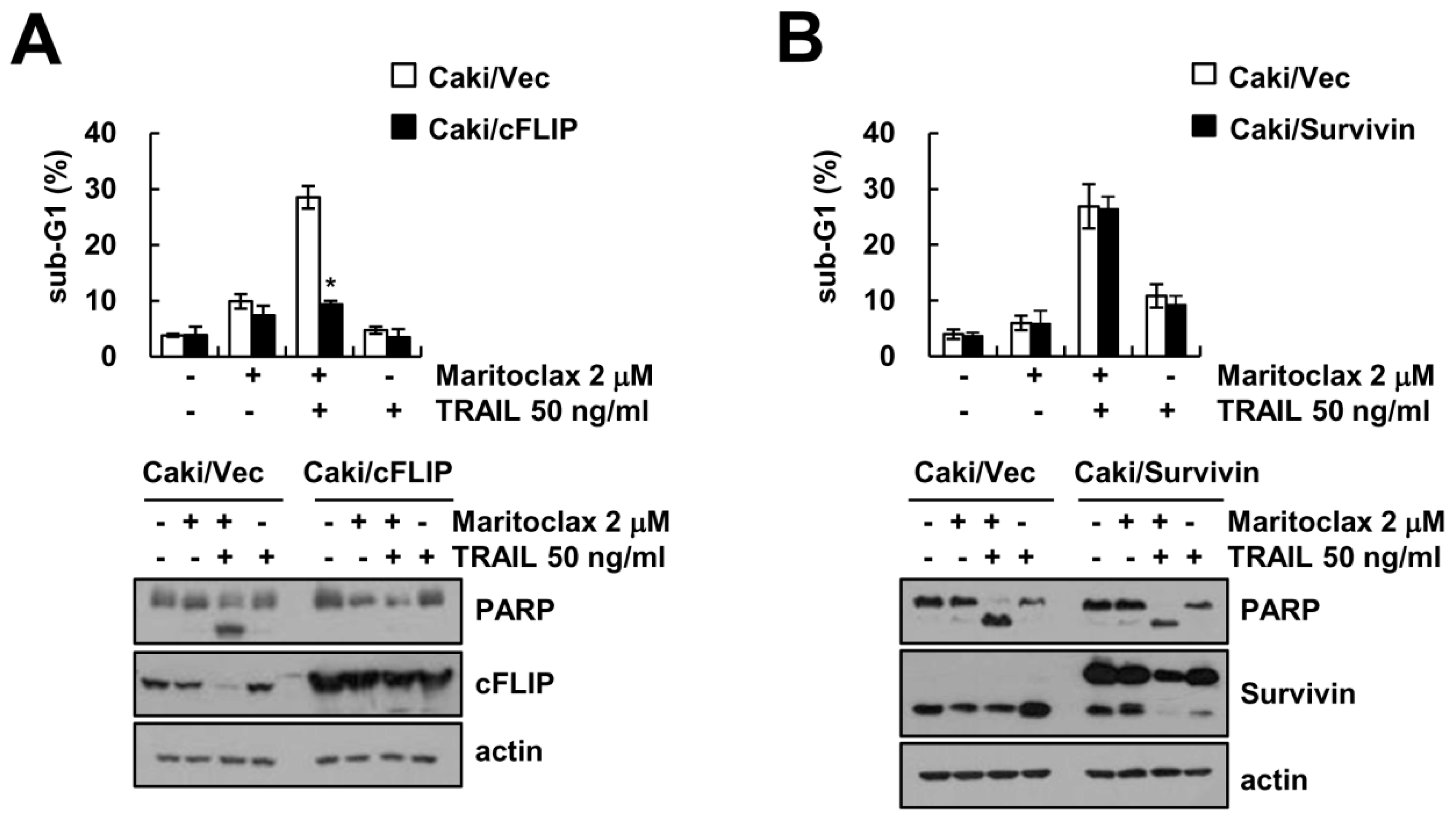
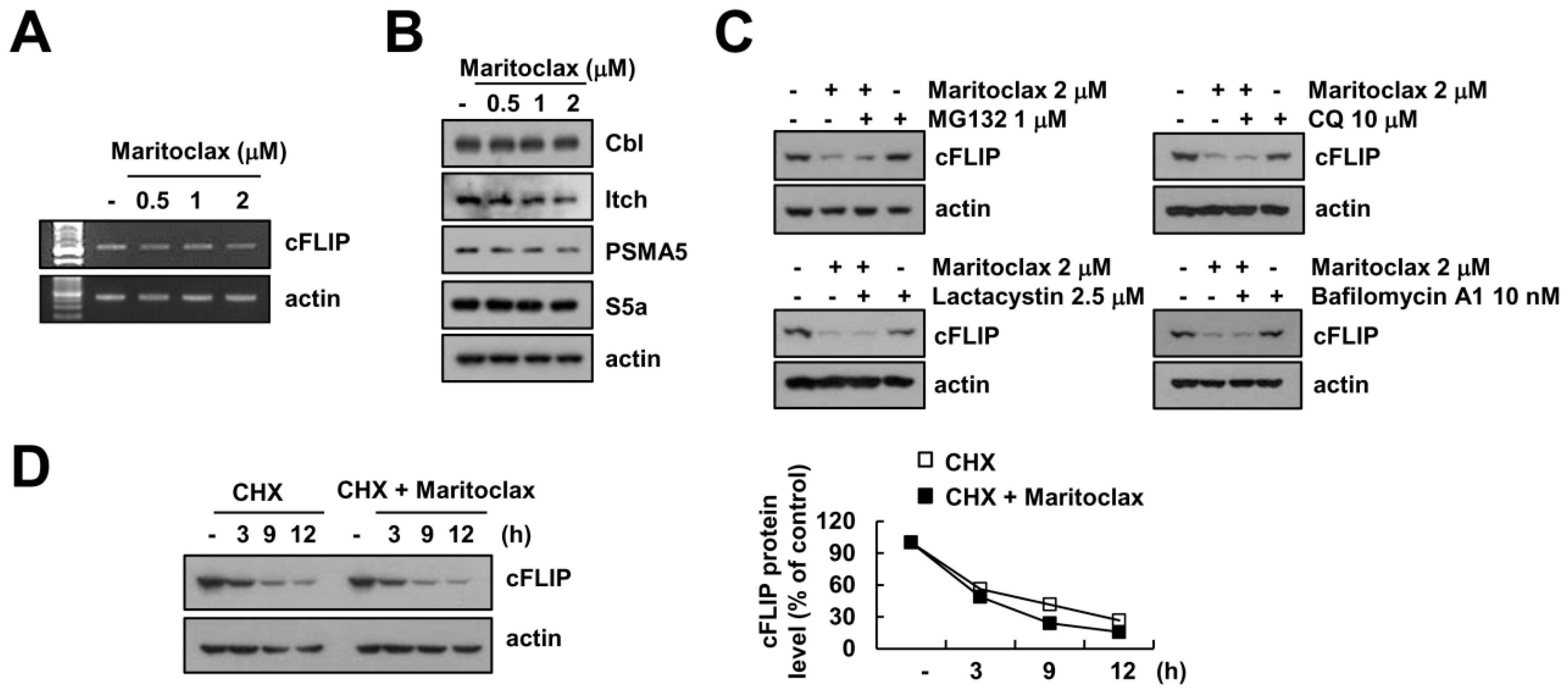
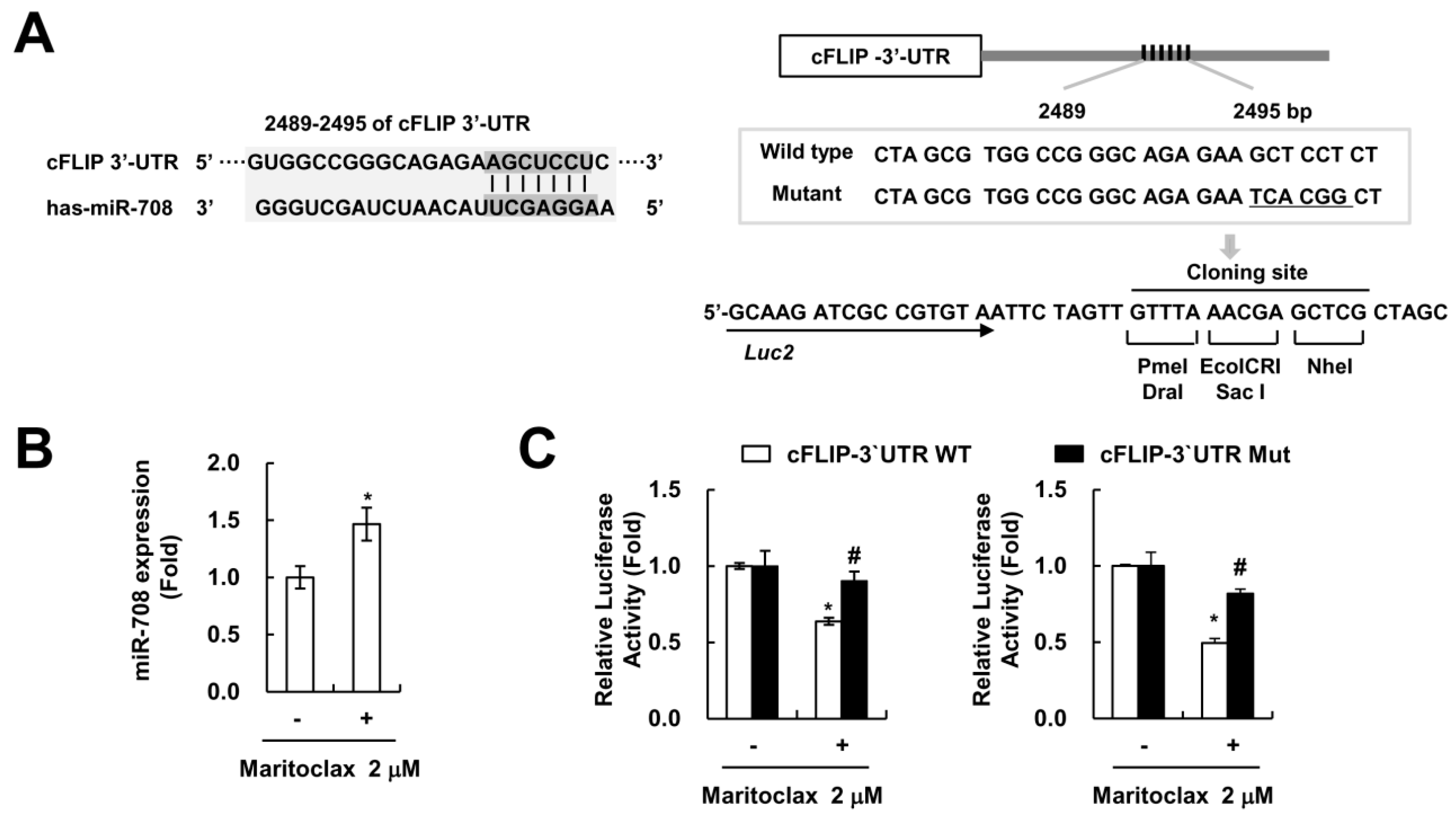
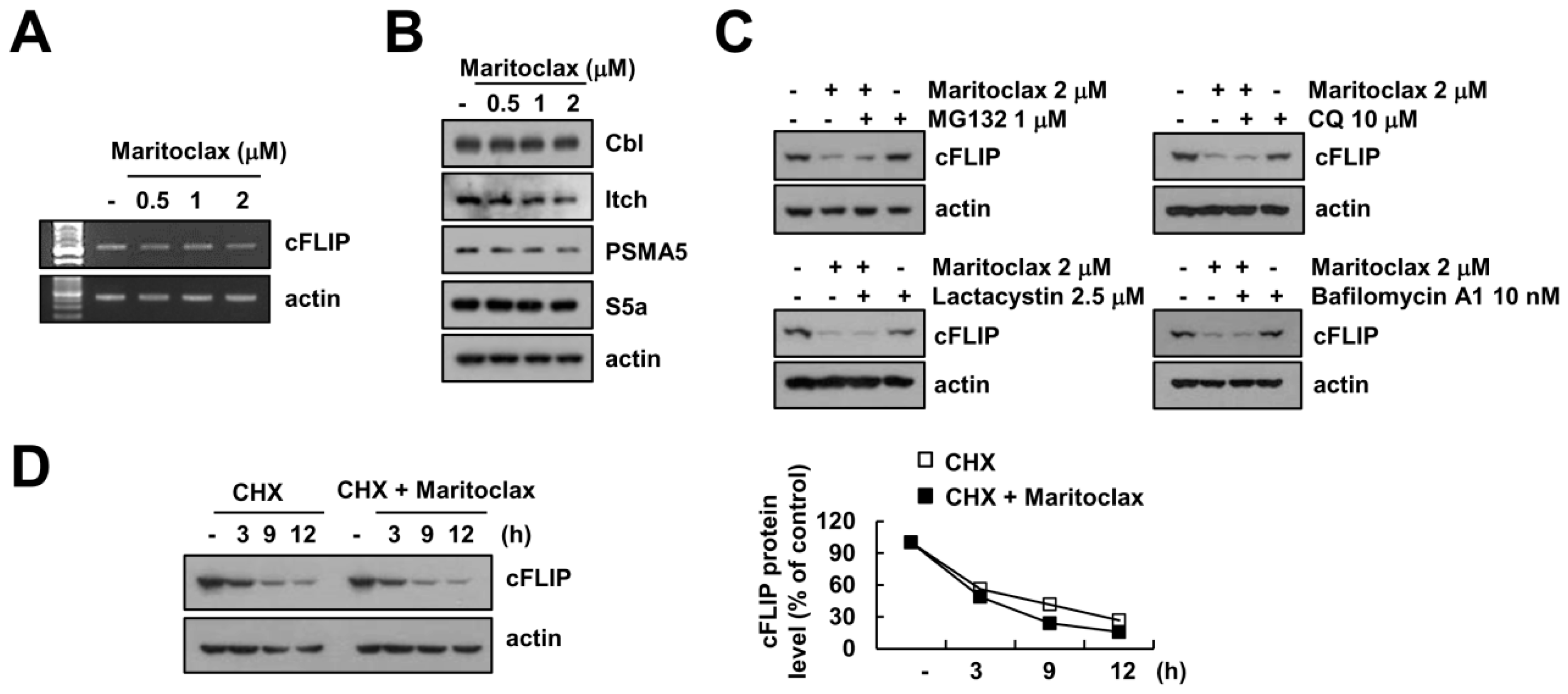
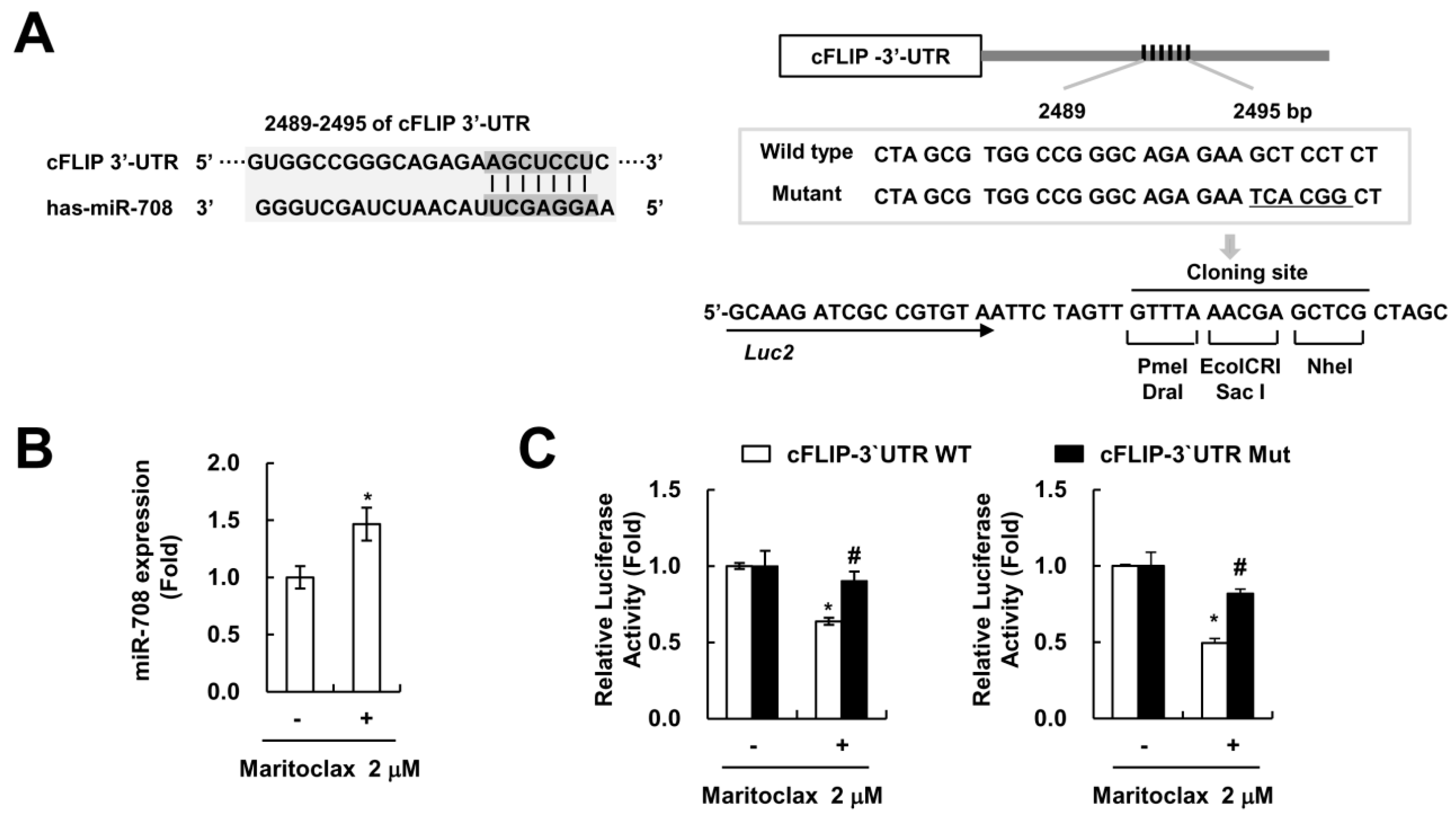
2.4. The Effect of cFLIP Downregulation on Maritoclax Plus TRAIL-Induced Apoptosis
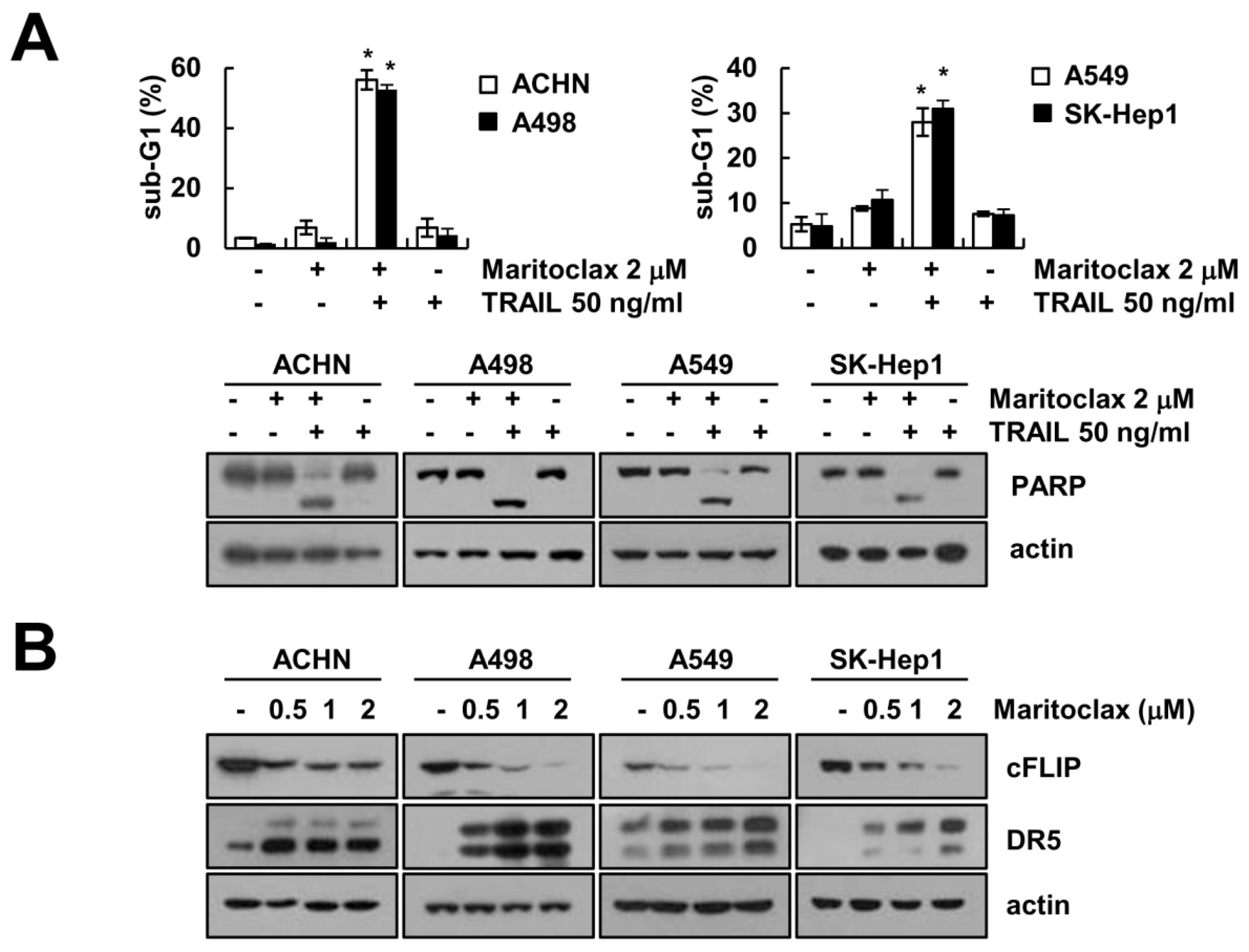
2.5. Effect of Combined Treatment with Maritoclax and TRAIL on Apoptosis in Other Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Materials
4.2. Western Blot Analysis and Flow Cytometry Analysis
4.3. Caspase Activity Assay, DNA Fragmentation Assay and DAPI Staining
4.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.6. Detection of DR5 on Cell Surface
4.7. Real-Time Quantitative RT-PCR for Detection of miR-708
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of interest
Abbreviations
References
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Cretney, E.; Shanker, A.; Yagita, H.; Smyth, M.J.; Sayers, T.J. TNF-related apoptosis-inducing ligand as a therapeutic agent in autoimmunity and cancer. Immunol. Cell Biol. 2006, 84, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.D.; Jacobsen, K.M.; Li, W.; Shanker, A.; Sayers, T.J. Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol. Cancer Res. 2010, 8, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Strater, J.; Hinz, U.; Walczak, H.; Mechtersheimer, G.; Koretz, K.; Herfarth, C.; Moller, P.; Lehnert, T. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clin. Cancer Res. 2002, 8, 3734–3740. [Google Scholar] [PubMed]
- Ichikawa, K.; Liu, W.; Zhao, L.; Wang, Z.; Liu, D.; Ohtsuka, T.; Zhang, H.; Mountz, J.D.; Koopman, W.J.; Kimberly, R.P.; et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat. Med. 2001, 7, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Halpin-McCormick, A.; Sessler, T.; Samali, A.; Szegezdi, E. Resistance to TRAIL in non-transformed cells is due to multiple redundant pathways. Cell Death Dis. 2013, 4, e702. [Google Scholar] [CrossRef] [PubMed]
- Geserick, P.; Drewniok, C.; Hupe, M.; Haas, T.L.; Diessenbacher, P.; Sprick, M.R.; Schon, M.P.; Henkler, F.; Gollnick, H.; Walczak, H.; et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- and CD95L-mediated apoptosis. Oncogene 2008, 27, 3211–3220. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, A.I.; Partanen, J.I.; Hau, A.; Klefstrom, J. c-Myc primed mitochondria determine cellular sensitivity to TRAIL-induced apoptosis. EMBO J. 2007, 26, 1055–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.H.; Bellail, A.; Tse, M.C.; Yong, V.W.; Hao, C. Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. J. Neurosci. 2006, 26, 3299–3308. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Smith, F.; Wang, Q.; Wang, X.; Evers, B.M. Assessment of differential gene expression patterns in human colon cancers. Ann. Surg. 2000, 232, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Ganten, T.M.; Koschny, R.; Haas, T.L.; Sykora, J.; Li-Weber, M.; Herzer, K.; Walczak, H. Proteasome inhibition sensitizes hepatocellular carcinoma cells, but not human hepatocytes, to TRAIL. Hepatology 2005, 42, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2004, 12, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Gu, J.; Lin, T.; Huang, X.; Roth, J.A.; Fang, B. Mechanisms involved in development of resistance to adenovirus-mediated proapoptotic gene therapy in DLD1 human colon cancer cell line. Gene Ther. 2002, 9, 1262–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, H.; Bouchon, A.; Stahl, H.; Krammer, P.H. Tumor necrosis factor-related apoptosis-inducing ligand retains its apoptosis-inducing capacity on Bcl-2- or Bcl-xL-overexpressing chemotherapy-resistant tumor cells. Cancer Res. 2000, 60, 3051–3057. [Google Scholar] [PubMed]
- Kelly, M.M.; Hoel, B.D.; Voelkel-Johnson, C. Doxorubicin pretreatment sensitizes prostate cancer cell lines to TRAIL induced apoptosis which correlates with the loss of c-FLIP expression. Cancer Biol. Ther. 2002, 1, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.P.; Zisman, A.; Bonavida, B. Synergy is achieved by complementation with Apo2L/TRAIL and actinomycin D in Apo2L/TRAIL-mediated apoptosis of prostate cancer cells: Role of XIAP in resistance. Prostate 2002, 53, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; McDonald, E.R., 3rd; Dicker, D.T.; El-Deiry, W.S. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J. Biol. Chem. 2004, 279, 35829–35839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol. Cancer Res. 2008, 6, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Haste, N.M.; Hughes, C.C.; Tran, D.N.; Fenical, W.; Jensen, P.R.; Nizet, V.; Hensler, M.E. Pharmacological properties of the marine natural product marinopyrrole A against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Li, R.; Sung, S.S.; Wu, H.; Liu, Y.; Manieri, W.; Krishnegowda, G.; Awwad, A.; Dewey, A.; Liu, X.; Amin, S.; et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Liu, Q.; Gowda, K.; Barth, B.M.; Claxton, D.; Amin, S.; Loughran, T.P., Jr.; Wang, H.G. Maritoclax induces apoptosis in acute myeloid leukemia cells with elevated Mcl-1 expression. Cancer Biol. Ther. 2014, 15, 107710–107786. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Poornima, P.; Milani, M.; Gowda, K.; Amin, S.; Wang, H.G.; Cohen, G.M. Maritoclax and dinaciclib inhibit MCL-1 activity and induce apoptosis in both a MCL-1-dependent and -independent manner. Oncotarget 2015, 6, 12668–12681. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, J.M.; Alford, S.E.; Hughes, C.C.; Fenical, W.; Chambers, T.C. Purported Mcl-1 inhibitor marinopyrrole A fails to show selective cytotoxicity for Mcl-1-dependent cell lines. Cell Death Dis. 2013, 4, e880. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; Hamilton, S.R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Bedi, G.C.; Engstrom, L.W.; Zeng, Q.; Mookerjee, B.; Gelinas, C.; Fuchs, E.J.; Bedi, A. Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-kappaB. Nat. Cell Biol. 2001, 3, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Yoshida, T.; Nakata, S.; Horinaka, M.; Wakada, M.; Mizutani, Y.; Miki, T.; Sakai, T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005, 65, 6364–6370. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.R.; Min, K.J.; Woo, S.M.; Choe, M.; Choi, K.S.; Lee, Y.K.; Yoon, G.; Kwon, T.K. Inhibition of Cathepsin S Induces Mitochondrial ROS That Sensitizes TRAIL-Mediated Apoptosis Through p53-Mediated Downregulation of Bcl-2 and c-FLIP. Antioxid. Redox Signal. 2017, 27, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tay, K.H.; Dong, L.; Thorne, R.F.; Jiang, C.C.; Yang, E.; Tseng, H.Y.; Liu, H.; Christopherson, R.; Hersey, P.; et al. Cystatin B inhibition of TRAIL-induced apoptosis is associated with the protection of FLIP(L) from degradation by the E3 ligase itch in human melanoma cells. Cell Death Differ. 2010, 17, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.A.; Kim, S.W.; Nam, J.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Kwon, T.K.; Lee, T.J. Inhibition of c-FLIPL expression by miRNA-708 increases the sensitivity of renal cancer cells to anti-cancer drugs. Oncotarget 2016, 7, 31832–31846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.K.; Gowda, K.; Doi, K.; Sharma, A.K.; Wang, H.G.; Amin, S. Proteasomal degradation of Mcl-1 by maritoclax induces apoptosis and enhances the efficacy of ABT-737 in melanoma cells. PLoS ONE 2013, 8, e78570. [Google Scholar] [CrossRef] [PubMed]
- Mert, U.; Sanlioglu, A.D. Intracellular localization of DR5 and related regulatory pathways as a mechanism of resistance to TRAIL in cancer. Cell. Mol. Life Sci. 2017, 74, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhu, H.H.; Zhou, L.F.; Wu, S.S.; Wang, J.; Chen, Z. Inhibition of c-FLIP expression by miR-512-3p contributes to taxol-induced apoptosis in hepatocellular carcinoma cells. Oncol. Rep. 2010, 23, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Zhong, Y.; Chen, Y.; Du, J.; Ju, X.; Zhao, C.; Zhang, G.; Zhang, L.; Liu, K.; Yang, N.; et al. Hsa-miR-1246, hsa-miR-320a and hsa-miR-196b-5p inhibitors can reduce the cytotoxicity of Ebola virus glycoprotein in vitro. Sci. China Life Sci. 2014, 57, 959–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, S.; Yamamura, S.; Majid, S.; Shahryari, V.; Hirata, H.; Tanaka, Y.; Dahiya, R. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res. 2011, 71, 6208–6219. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Valk-Lingbeek, M.E.; Bruggeman, S.W.; van Lohuizen, M. Stem cells and cancer; the polycomb connection. Cell 2004, 118, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Kim, T.H.; Kim, D.E.; Min, K.J.; Kwon, T.K. NOX4-mediated ROS production induces apoptotic cell death via down-regulation of c-FLIP and Mcl-1 expression in combined treatment with thioridazine and curcumin. Redox Biol. 2017, 13, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Kwon, Y.J.; Chun, Y.J. CYP1B1 Activates Wnt/beta-Catenin Signaling through Suppression of Herc5-Mediated ISGylation for Protein Degradation on beta-Catenin in HeLa Cells. Toxicol. Res. 2017, 33, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Shin, D.Y. Repression of the F-box protein Skp2 is essential for actin damage-induced tetraploid G1 arrest. BMB Rep. 2017, 50, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, M.-Y.; Min, K.-j.; Woo, S.M.; Seo, S.U.; Choi, Y.H.; Kim, S.H.; Kim, D.E.; Lee, T.-J.; Kim, S.; Park, J.-W.; et al. Maritoclax Enhances TRAIL-Induced Apoptosis via CHOP-Mediated Upregulation of DR5 and miR-708-Mediated Downregulation of cFLIP. Molecules 2018, 23, 3030. https://doi.org/10.3390/molecules23113030
Jeon M-Y, Min K-j, Woo SM, Seo SU, Choi YH, Kim SH, Kim DE, Lee T-J, Kim S, Park J-W, et al. Maritoclax Enhances TRAIL-Induced Apoptosis via CHOP-Mediated Upregulation of DR5 and miR-708-Mediated Downregulation of cFLIP. Molecules. 2018; 23(11):3030. https://doi.org/10.3390/molecules23113030
Chicago/Turabian StyleJeon, Mi-Yeon, Kyoung-jin Min, Seon Min Woo, Seung Un Seo, Yung Hyun Choi, Sang Hyun Kim, Dong Eun Kim, Tae-Jin Lee, Shin Kim, Jong-Wook Park, and et al. 2018. "Maritoclax Enhances TRAIL-Induced Apoptosis via CHOP-Mediated Upregulation of DR5 and miR-708-Mediated Downregulation of cFLIP" Molecules 23, no. 11: 3030. https://doi.org/10.3390/molecules23113030
APA StyleJeon, M.-Y., Min, K.-j., Woo, S. M., Seo, S. U., Choi, Y. H., Kim, S. H., Kim, D. E., Lee, T.-J., Kim, S., Park, J.-W., & Kwon, T. K. (2018). Maritoclax Enhances TRAIL-Induced Apoptosis via CHOP-Mediated Upregulation of DR5 and miR-708-Mediated Downregulation of cFLIP. Molecules, 23(11), 3030. https://doi.org/10.3390/molecules23113030