
Nutritional Regulators of Bcl-xL in the Brain


Abstract
1. Introduction
2. Soy and Soy Isoflavones
3. Ginseng and Ginsenosides
4. Omega-3 Fatty Acids
5. Resveratrol
6. Alcohol
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barclay, L.A.; Wales, T.E.; Garner, T.P.; Wachter, F.; Lee, S.; Guerra, R.M.; Stewart, M.L.; Braun, C.R.; Bird, G.H.; Gavathiotis, E.; et al. Inhibition of pro-apoptotic bax by a noncanonical interaction mechanism. Mol. Cell 2015, 57, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.C.S. The conserved N-terminal bh4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with ced-4. EMBO J. 1998, 17, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; La Rovere, R.; Karamanou, S.; Welkenhuyzen, K.; Ivanova, H.; Vandermarliere, E.; Di Martile, M.; Del Bufalo, D.; De Smedt, H.; Parys, J.B.; et al. A double point mutation at residues Ile14 and Val15 of Bcl-2 uncovers a role for the BH4 domain in both protein stability and function. FEBS J. 2018, 285, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.-P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; De Smedt, H.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The bh4 domain of Bcl-2 inhibits er calcium release and apoptosis by binding the regulatory and coupling domain of the ip3 receptor. Proc. Nat. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef] [PubMed]
- Westphal, D.; Kluck, R.M.; Dewson, G. Building blocks of the apoptotic pore: How bax and bak are activated and oligomerize during apoptosis. Cell Death Differ. 2013, 21, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Osen-Sand, A.; Nichols, A.; Eskes, R.; Montessuit, S.; Lauper, S.; Maundrell, K.; Antonsson, B.; Martinou, J.-C. Bid-induced conformational change of bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999, 144, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Gene. Dev. 2000, 14, 2060–2071. [Google Scholar] [PubMed]
- Wei, M.C. Proapoptotic bax and bak: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Moore, V.D.G.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic Bcl-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M. Structure of Bcl-xl-bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Jurgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Nettesheim, D.G.; Wang, Y.; Olejniczak, E.T.; Meadows, R.P.; Mack, J.; Swift, K.; Matayoshi, E.D.; Zhang, H.; Fesik, S.W.; et al. Rationale for Bcl-xl/bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000, 9, 2528–2534. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-xl and Bcl-2, displaces bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Chen, Y.B.; Aon, M.A.; Hsu, Y.T.; Soane, L.; Teng, X.; McCaffery, J.M.; Cheng, W.-C.; Qi, B.; Li, H.; Alavian, K.N.; et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 2011, 195, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Chevance, S.; Del Conte, R.; Lalli, D.; Turano, P. The anti-apoptotic Bcl-x(L.) protein, a new piece in the puzzle of cytochrome c interactome. PLoS ONE 2011, 6, e18329. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Pandey, P.; Schofield, L.; Israels, S.; Roncinske, R.; Yoshida, K.; Bharti, A.; Yuan, Z.-M.; Saxena, S.; Weichselbaum, R.; et al. Role for Bcl-xl as an inhibitor of cytosolic cytochrome c accumulation in dna damage-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 6939–6942. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Swahari, V.; Plestant, C.; Smith, I.; McCoy, E.; Smith, S.; Moy, S.S.; Anton, E.S.; Deshmukh, M. Bcl-xl is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J. Neurosci. 2016, 36, 5448–5461. [Google Scholar] [CrossRef] [PubMed]
- Ofengeim, D.; Chen, Y.; Miyawaki, T.; Li, H.; Sacchetti, S.; Flannery, R.J.; Alavian, K.N.; Pontarelli, F.; Roelofs, B.A.; Hickman, J.A.; et al. N-terminally cleaved Bcl-xl mediates ischemia-induced neuronal death. Nat. Neurosci. 2012, 15, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.S.; Kulikov, A.V.; Kondaurova, E.M.; Tsybko, A.S.; Kulikova, E.A.; Krasnov, I.B.; Shenkman, B.S.; Sychev, V.N.; Bazhenova, E.Y.; Sinyakova, N.A.; et al. Effect of actual long-term spaceflight on bdnf, trkb, p75, bax and Bcl-xl genes expression in mouse brain regions. Neuroscience 2015, 284, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Liu, J.Q.; Nakano, Y.; Ueno, S.; Ohmori, S.; Fueta, Y.; Ishidao, T.; Kunugita, N.; Yamashita, U.; Hori, H. 1-bp inhibits nf-κb activity and Bcl-xl expression in astrocytes in vitro and reduces Bcl-xl expression in the brains of rats in vivo. NeuroToxicology 2007, 28, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-A.; Licznerski, P.; Alavian, K.N.; Shanabrough, M.; Jonas, E.A. Bcl-xl is necessary for neurite outgrowth in hippocampal neurons. Antioxid. Redox 2015, 22, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kannagi, M.; Ferrante, R.J.; Kowall, N.W.; Ryu, H. Activation of ets-2 by oxidative stress induces Bcl-xl expression and accounts for glial survival in amyotrophic lateral sclerosis. FASEB J. 2009, 23, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Mai, J.K.; Zapata, J.M.; Ashwell, K.W.; Schendel, S.L.; Reed, J.C.; Krajewski, S. Dynamics of expression of apoptosis-regulatory proteins bid, Bcl-2, Bcl-x, bax and bak during development of murine nervous system. Cell Death Differ. 2002, 9, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xl regulates metabolic efficiency of neurons through interaction with the mitochondrial f1fo atp synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Alavian, K.N.; Lazrove, E.; Mehta, N.; Jones, A.; Zhang, P.; Licznerski, P.; Graham, M.; Uo, T.; Guo, J.; et al. A Bcl-xl–drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat. Cell Biol. 2013, 15, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, Y.; Jones, A.F.; Sanger, R.H.; Collis, L.P.; Flannery, R.; McNay, E.C.; Yu, T.; Schwarzenbacher, R.; Bossy, B.; et al. Bcl-xl induces drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E. BCL-xL regulates synaptic plasticity. Mol. Interv. 2006, 6, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.; Porter, G.A.; Beutner, G.; Mnatsakanyan, N.; Park, H.A.; Mehta, N.; Chen, R.; Alavian, K.N. The mitochondrial permeability transition pore: Molecular structure and function in health and disease. In Molecular Basis for Mitochondrial Signaling; Rostovtseva, T.K., Ed.; Springer: New York, NY, USA, 2017; pp. 69–105. [Google Scholar]
- Park, H.A.; Jonas, E. Mitochondrial regulators of synaptic plasticity in the ischemic brain. In Synaptic Plasticity; Heinbockel, T., Ed.; INTECH: London, UK, 2017; pp. 39–67. [Google Scholar]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R. In vivo delivery of a Bcl-xl fusion protein containing the tat protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Fujihara, H.; Yao, J.; Qi, S.; Li, H.; Shimoji, K. Different expression patterns of Bcl-2, Bcl-xl, and Bax proteins after sublethal forebrain ischemia in C57Black/Crj6 mouse striatum. Stroke A J. Cerebr. Circ. 2003, 34, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Eguchi, Y.; Kosaka, H.; Kamiike, W.; Matsuda, H.; Tsujimoto, Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xl. Nature 1995, 374, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Graham, S.H.; Nakayama, M.; Zhu, R.L.; Jin, K.; Stetler, R.A.; Simon, R.P. Apoptosis repressor genes Bcl-2 and Bcl-x-long are expressed in the rat brain following global ischemia. J. Cereb. Blood Flow Metab. 1997, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Allegrini, P.R.; Rupalla, K.; Sauer, D.; Oltersdorf, T.; McGregor, A.L.; Bischoff, S.; Böttiger, B.W.; van der Putten, H. Neuron-specific transgene expression of Bcl-xl but not Bcl-2 genes reduced lesion size after permanent middle cerebral artery occlusion in mice. Neurosci. Lett. 1999, 268, 119–122. [Google Scholar] [CrossRef]
- Kilic, E.; Dietz, G.P.; Hermann, D.M.; Bahr, M. Intravenous TAT-Bcl-Xl is protective after middle cerebral artery occlusion in mice. Ann Neurol. 2002, 52, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Nesic, O.; Ye, Z.; Rea, H.; Westlund, K.N.; Xu, G.Y.; McAdoo, D.; Hulsebosch, C.E.; Perez-Polo, J.R. Bcl-xL expression after contusion to the rat spinal cord. J. Neurotrauma 2001, 18, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Nesic-Taylor, O.; Cittelly, D.; Ye, Z.; Xu, G.Y.; Unabia, G.; Lee, J.C.; Svrakic, N.M.; Liu, X.H.; Youle, R.J.; Wood, T.G.; et al. Exogenous Bcl-xl fusion protein spares neurons after spinal cord injury. J. Neurosci. Res. 2005, 79, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Kim, B.G.; Hwang, D.H.; Kim, H.M.; Kim, S.U. Overexpression of Bcl-xlin human neural stem cells promotes graft survival and functional recovery following transplantation in spinal cord injury. J. Neurosci. Res. 2009, 87, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Peng, X.; Insolera, R.; Fink, D.J.; Mata, M. IL-10 promotes neuronal survival following spinal cord injury. Exp Neurol. 2009, 220, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Kadota, R.; Koda, M.; Kawabe, J.; Hashimoto, M.; Nishio, Y.; Mannoji, C.; Miyashita, T.; Furuya, T.; Okawa, A.; Takahashi, K.; et al. Granulocyte colony-stimulating factor (g-csf) protects oligpdendrocyte and promotes hindlimb functional recovery after spinal cord injury in rats. PLoS ONE 2012, 7, e50391. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: Relevance to Alzheimer’s disease. J. Neurochem. 1999, 72, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Keil, U.; Bonert, A.; Marques, C.A.; Scherping, I.; Weyermann, J.; Strosznajder, J.B.; Müller-Spahn, F.; Haass, C.; Czech, C.; Pradier, L.; et al. Amyloid β-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J. Biol. Chem. 2004, 279, 50310–50320. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W.E. Mitochondrial dysfunction: An early event in alzheimer pathology accumulates with age in ad transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Vohra, B.P.S.; Sasaki, Y.; Miller, B.R.; Chang, J.; DiAntonio, A.; Milbrandt, J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J. Neurosci. 2010, 30, 13729–13738. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Mouatt-Prigent, A.; Vila, M.; Abbas, N.; Perier, C.; Faucheux, B.A.; Vyas, S.; Hirsch, E.C. Increased expression and redistribution of the antiapoptotic molecule Bcl-xL in Parkinson’s disease. Neurobiol. Dis. 2002, 10, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Liste, I. The generation of dopaminergic neurons by human neural stem cells is enhanced by Bcl-xl, both in vitro and in vivo. J. Neurosci. 2004, 24, 10786–10795. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.-W. Enhanced in vitro midbrain dopamine neuron differentiation, dopaminergic function, neurite outgrowth, and 1-methyl-4-phenylpyridium resistance in mouse embryonic stem cells overexpressing Bcl-xl. J. Neurosci. 2004, 24, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, C.; Courtois, E.; Jensen, P.; Jørgensen, J.R.; Zimmer, J.; Martínez-Serrano, A.; Meyer, M. Enhanced dopaminergic differentiation of human neural stem cells by synergistic effect of Bcl-xland reduced oxygen tension. J. Neurochem. 2009, 110, 1908–1920. [Google Scholar] [CrossRef] [PubMed]
- Courtois, E.T.; Castillo, C.G.; Seiz, E.G.; Ramos, M.; Bueno, C.; Liste, I.; Martinez-Serrano, A. In vitro and in vivo enhanced generation of human a9 dopamine neurons from neural stem cells by Bcl-xl. J. Biol. Chem. 2010, 285, 9881–9897. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Fu, K.; Wang, D.; Mu, C.; Wang, G. Oxidized dj-1 interacts with the mitochondrial protein Bcl-xl. J. Biol. Chem. 2011, 286, 35308–35317. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Ku, H.H.; Tsai, T.H.; Lin, H.L.; Chen, L.H.; Chien, C.S.; Ho, L.L.-T.; Lee, C.-H.; Chang, Y.-L. Moclobemide upregulated Bcl-2 expression and induced neural stem cell differentiation into serotoninergic neuron via extracellular-regulated kinase pathway. Br. J. Pharmacol. 2006, 148, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Kosten, T.A.; Galloway, M.P.; Duman, R.S.; Russell, D.S.; D’Sa, C. Repeated unpredictable stress and antidepressants differentially regulate expression of the Bcl-2 family of apoptotic genes in rat cortical, hippocampal, and limbic brain structures. Neuropsychopharmacology 2007, 33, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Shishkina, G.T.; Kalinina, T.S.; Berezova, I.V.; Dygalo, N.N. Stress-induced activation of the brainstem Bcl-xl gene expression in rats treated with fluoxetine: Correlations with serotonin metabolism and depressive-like behavior. Neuropharmacology 2012, 62, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, G.; Zhang, Y.; Su, X.; Hang, C. The potential role of jak2/stat3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhepo) after experimental traumatic brain injury of rats. Cytokine 2011, 56, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Lončarević-Vasiljković, N.; Milanović, D.; Pešić, V.; Tešić, V.; Brkić, M.; Lazić, D.; Avramović, V.; Kanazir, S. Dietary restriction suppresses apoptotic cell death, promotes Bcl-2 and Bcl-xl mrna expression and increases the Bcl-2/bax protein ratio in the rat cortex after cortical injury. Neurochem. Int. 2016, 96, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Pang, A.L.; Xiong, L.L.; Xia, Q.J.; Liu, F.; Wang, Y.C.; Liu, F.; Zhang, P.; Meng, B.-L.; Tan, S.; Wang, T.-H. Neural stem cell transplantation is associated with inhibition of apoptosis, Bcl-xL upregulation, and recovery of neurological function in a rat model of traumatic. brain injury. Cell Transpl. 2017, 26, 1262–1275. [Google Scholar] [CrossRef] [PubMed]
- Soler-Botija, C.; Ferrer, I.; Alvarez, J.L.; Baiget, M.; Tizzano, E.F. Downregulation of Bcl-2 proteins in type i spinal muscular atrophy motor neurons during fetal development. J. Neuropathol. Exp. Neurol. 2003, 62, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Tsai, M.S.; Ting, C.H.; Li, H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J. Mol. Med. 2008, 86, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Tsai, M.S.; Ting, C.H.; Wang, S.H.; Li, H. Restoring Bcl-x(L) levels benefits a mouse model of spinal muscular atrophy. Neurobiol. Dis. 2008, 31, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Garcera, A.; Mincheva, S.; Gou-Fabregas, M.; Caraballo-Miralles, V.; Lladó, J.; Comella, J.X.; Soler, R.M. A new model to study spinal muscular atrophy: Neurite degeneration and cell death is counteracted by Bcl-xl overexpression in motoneurons. Neurobiol. Dis. 2011, 42, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Anderton, R.S.; Price, L.L.; Turner, B.J.; Meloni, B.P.; Mitrpant, C.; Mastaglia, F.L.; Goh, C.; Wilton, S.D.; Boulos, S. Co-regulation of survival of motor neuron and Bcl-xl expression: Implications for neuroprotection in spinal muscular atrophy. Neuroscience 2012, 220, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kaufmann, J.A.; Sanchez-Ross, M.G.; Johnson, K.M. Mechanisms of N-methyl-d-aspartate-induced apoptosis in phencyclidine-treated cultured forebrain neurons. J. Pharmacol. Exp. Ther. 2000, 294, 287–295. [Google Scholar] [PubMed]
- Wang, C.; McInnis, J.; Ross-Sanchez, M.; Shinnick-Gallagher, P.; Wiley, J.; Johnson, K. Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: Implications for schizophrenia. Neuroscience 2001, 107, 535–550. [Google Scholar] [CrossRef]
- He, J.; Xu, H.; Yang, Y.; Rajakumar, D.; Li, X.; Li, X.-M. The effects of chronic administration of quetiapine on the phencyclidine-induced reference memory impairment and decrease of Bcl-xl/Bax ratio in the posterior cingulate cortex in rats. Behav. Brain Res. 2006, 168, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J.; Cheng, E.H.-Y.; Karp, C.L.; Kirsch, D.G.; Ueno, K.; Takahashi, A.; Kastan, M.B.; Griffin, D.E.; Earnshaw, W.C.; Veliuona, M.A.; et al. Modulation of cell death by Bcl-xl through caspase interaction. Proc. Natl. Acad. Sci. USA 1998, 95, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, T.; Mashiko, T.; Ofengeim, D.; Flannery, R.J.; Noh, K.-M.; Fujisawa, S.; Bonanni, L.; Bennett, M.V.L.; Zukin, R.S.; Jonas, E.A. Ischemic preconditioning blocks bad translocation, Bcl-xl cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4892–4897. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yokota, H.; Jover, T.; Cappuccio, I.; Calderone, A.; Simionescu, M.; Bennett, M.V.; Zukin, R.S. Ischemic preconditioning: Neuronal survival in the face of caspase-3 activation. J. Neurosci. 2004, 24, 2750–2759. [Google Scholar] [CrossRef] [PubMed]
- Tornero, D.; Posadas, I.; Ceña, V. Bcl-xl blocks a mitochondrial inner membrane channel and prevents ca2+ overload-mediated cell death. PLoS ONE 2011, 6, e20423. [Google Scholar] [CrossRef] [PubMed]
- Park, H.A.; Jonas, E.A. ΔN-Bcl-xL, a therapeutic target for neuroprotection. Neural Regen Res. 2017, 12, 1791–1794. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.A.; Hickman, J.A.; Chachar, M.; Polster, B.M.; Brandt, T.A.; Fannjiang, Y.; Ivanovska, I.; Basanez, G.; Kinnally, K.W.; Zimmerberg, J.; et al. Proapoptotic n-truncated Bcl-xl protein activates endogenous mitochondrial channels in living synaptic terminals. Proc. Natl. Acad. Sci. USA 2004, 101, 13590–13595. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Li, C.; Yang, J.; Petrenko, N.B.; Madesh, M.; Thompson, C.B.; Foskett, J.K. The endoplasmic reticulum gateway to apoptosis by Bcl-xl modulation of the insp3r. Nat. Cell Biol. 2005, 7, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis regulation by Bcl-xl modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.K.; DiLorenzo, J.C.; Sheu, K.-F.R. Pyruvate dehydrogenase complex is inhibited in calcium-loaded cerebrocortical mitochondria. Neurochem. Res. 1988, 13, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Satrustegui, J. Calcium signaling in brain mitochondria: Interplay of malate aspartate nadh shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J. Biol. Chem. 2009, 284, 7091–7099. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, M.J.; McHugh, N.J. Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Lett. 1996, 391, 323–329. [Google Scholar] [CrossRef]
- Territo, P.R.; Mootha, V.K.; French, S.A.; Balaban, R.S. Ca2+ activation of heart mitochondrial oxidative phosphorylation: Role of the F(0)/F(1)-ATPase. Am. J. Physiol. Cell Physiol. 2000, 278, C423–C435. [Google Scholar] [CrossRef] [PubMed]
- Boerries, M.; Most, P.; Gledhill, J.R.; Walker, J.E.; Katus, H.A.; Koch, W.J.; Aebi, U.; Schoenenberger, C.-A. Ca2+-dependent interaction of s100a1 with f1-atpase leads to an increased atp content in cardiomyocytes. Mol. Cell. Biol. 2007, 27, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Ehrlich, B.E. Atp modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron 1993, 10, 1175–1184. [Google Scholar] [CrossRef]
- Wagner, L.E.; Betzenhauser, M.J.; Yule, D.I. Atp binding to a unique site in the type-1 s2- inositol 1,4,5-trisphosphate receptor defines susceptibility to phosphorylation by protein kinase a. J. Biol. Chem. 2006, 281, 17410–17419. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Hu, X.; Eno, C.O.; Zhao, G.; Li, C.; White, C. An interaction between Bcl-xland the voltage-dependent anion channel (vdac) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 2013, 288, 19870–19881. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The bh4 domain of anti-apoptotic Bcl-xl, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (vdac1)-mediated transfer of pro-apoptotic ca2+signals to mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnoczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Holmuhamedov, E. Voltage-dependent anion channel (vdac) as mitochondrial governator—Thinking outside the box. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the antiapoptotic activity of Bcl-xl protein upon interaction with vdac1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef] [PubMed]
- Malia, T.J.; Wagner, G. Nmr structural investigation of the mitochondrial outer membrane protein vdac and its interaction with antiapoptotic Bcl-xl†. Biochemistry 2007, 46, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G. Bcl-xl promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001, 276, 19414–19419. [Google Scholar] [CrossRef] [PubMed]
- Vander-Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Nat. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of atg5/atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Diff. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Levine, B. The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene 2008, 27, S137–S148. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-xl-beclin 1 peptide complex: Beclin 1 is a novel bh3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Song, X.; Zhang, L.; Bartlett, D.L.; Lee, Y.J. Role of Bcl-xl/beclin-1 in interplay between apoptosis and autophagy in oxaliplatin and bortezomib-induced cell death. Biochem. Pharmacol. 2014, 88, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.-C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-xl and a bh3-like domain in beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase pink1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial parkin translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Muqit, M.M. PINK1 and parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The pink1/parkin-mediated mitophagy is compromised by pd-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. Pink1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xl and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Hickman, J.A.; Hardwick, J.M.; Kaczmarek, L.K.; Jonas, E.A. Bcl-xl inhibitor abt-737 reveals a dual role for Bcl-xl in synaptic transmission. J. Neurophysiol. 2008, 99, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Perkins, G.A.; Poblenz, A.T.; Harris, J.B.; Hung, M.; Ellisman, M.H.; Fox, D.A. Bcl-xl overexpression blocks bax-mediated mitochondrial contact site formation and apoptosis in rod photoreceptors of lead-exposed mice. Proc. Nat. Acad. Sci. USA 2003, 100, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Lovekamp-Swan, T.; Glendenning, M.L.; Schreihofer, D.A. A high soy diet enhances neurotropin receptor and Bcl-xl gene expression in the brains of ovariectomized female rats. Brain Res. 2007, 1159, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Lovekamp-Swan, T.; Glendenning, M.; Schreihofer, D.A. A high soy diet reduces programmed cell death and enhances Bcl-xl expression in experimental stroke. Neuroscience 2007, 148, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-F.; He, L.; Li, D.; Yuan, L.-H.; Yu, H.-L.; Ma, W.-W.; Yang, Y.; Xi, Y.-D.; Ding, J.; Xiao, Y.-X.; et al. Antagonizing effects of soybean isoflavones on β-amyloid peptides- induced oxidative damage in neuron mitochondria of rats. Basic Clin. Pharmacol. Toxicol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hata, R.; Zhu, P.; Sato, K.; Wen, T.-C.; Yang, L.; Fujita, H.; Mitsuda, N.; Tanaka, J.; Samukawa, K.; et al. Prevention of ischemic neuronal death by intravenous infusion of a ginseng saponin, ginsenoside rb1, that upregulates Bcl-xl expression. J. Cereb. Blood Flow Metab. 2006, 26, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Zhu, P.; Zhang, B.; Wen, T.-C.; Cao, F.; Ma, Y.-J.; Samukawa, K.; Mitsuda, N.; Tanaka, J.; Kuramoto, M.; et al. Intravenous infusion of dihydroginsenoside rb1 prevents compressive spinal cord injury and ischemic brain damage through upregulation of vegf and Bcl-xL. J. Neurotrauma 2007, 24, 1037–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Samukawa, K.; Fujita, H.; Kato, H.; Sakanaka, M. Oral administration of red ginseng extract promotes neurorestoration after compressive spinal cord injury in rats. Evid-Based Compl. Alt. Med. 2017, 2017, 1265464. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, S.O.; Kim, G.L.; Rhee, D.K. Estrogen receptor-beta of microglia underlies sexual differentiation of neuronal protection via ginsenosides in mice brain. CNS Neurosci. Ther. 2018, 24, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Hata, R.; Nakata, K.; Cao, F.; Samukawa, K.; Fujita, H.; Sakanaka, M. Intravenous infusion of ginsenoside Rb1 ameliorates compressive spinal cord injury through upregulation of Bcl-xL and VEGF. Int. J. Neurol. Neurother. 2015, 2, 1–6. [Google Scholar] [CrossRef]
- Lukiw, W.J. A role for docosahexaenoic acid-derived neuroprotectin d1 in neural cell survival and alzheimer disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, M.; Pyszko, J.; Strosznajder, J.B. Docosahexaenoic acid and tetracyclines as promising neuroprotective compounds with poly(adp-ribose) polymerase inhibitory activities for oxidative/genotoxic stress treatment. Neurochem. Int. 2013, 62, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Khare, P.; Rai, A.; Maurya, S.K.; Pathak, A.; Mohan, V.; Nagar, G.K.; Mudiam, M.K.R.; Godbole, M.M.; Bandyopadhyay, S. Anti-apoptotic role of omega-3-fatty acids in developing brain: Perinatal hypothyroid rat cerebellum as apoptotic model. Int. J. Dev. Neurosci. 2009, 27, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, D.; Xu, J.; Yanagita, T.; Xue, C.; Zhang, T.; Wang, W. DHA enriched phospholipids with different polar groups (PC and PS) had different improvements on MPTP-induced mice with Parkinson’s disease. J. Funct. Foods. 2018, 45, 414–426. [Google Scholar] [CrossRef]
- Shi, Z.; Ren, H.; Luo, C.; Yao, X.; Li, P.; He, C.; Kang, J.-X.; Wan, J.-B.; Yuan, T.-F.; Su, H. Enriched endogenous omega-3 polyunsaturated fatty acids protect cortical neurons from experimental ischemic injury. Mol. Neurobiol. 2015, 53, 6482–6488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wu, P.; Zhang, J.H.; Li, Y.; Xu, S.; Wang, C.; Wang, L.; Zhang, G.; Dai, J.; Zhu, S.; et al. Docosahexaenoic acid alleviates oxidative stress-based apoptosis via improving mitochondrial dynamics in early brain injury after subarachnoid hemorrhage. Cell Mol. Neurobiol. 2018, 38, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ren, H.; Shi, Z.; Yao, X.; He, C.; Kang, J.-X.; Wan, J.-B.; Li, P.; Yuan, T.-F.; Su, H. Endogenous docosahexaenoic acid (dha) prevents aβ1–42 oligomer-induced neuronal injury. Mol. Neurobiol. 2015, 53, 3146–3153. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.; Pizzi, M. Targeted acetylation of nf-kappab/rela and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Surh, Y.J. Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic. Biol. Med. 2003, 34, 1100–1110. [Google Scholar] [CrossRef]
- Seo, J.-S.; Moon, M.-H.; Jeong, J.-K.; Seol, J.-W.; Lee, Y.-J.; Park, B.-H.; Park, S.-Y. Sirt1, a histone deacetylase, regulates prion protein-induced neuronal cell death. Neurobiol. Aging 2012, 33, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Peng, C.; Ding, J.Y.; Asmaro, K.; Sullivan, J.M.; Guthikonda, M.; Ding, Y. Acute administration of ethanol reduces apoptosis following ischemic stroke in rats. Neurosci. Res. 2013, 76, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Parmar, S.; Li, X.; Peng, C.; Ji, X.; Chakraborty, T.; Li, W.A.; Du, H.; Tan, X.; Ling, F.; et al. Reduced apoptosis by combining normobaric oxygenation with ethanol in transient ischemic stroke. Brain Res. 2013, 1531, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, Y.A.; Zakharova, I.O.; Avrova, N.F. The effects of alpha-tocoferol and H2O2 on the mitochondrial membrane potential and Bax/Bcl-xL ratio in PC12 cells. Neurochem. J. 2016, 10, 318–322. [Google Scholar] [CrossRef]
- Kerek, R.; Geoffroy, A.; Bison, A.; Martin, N.; Akchiche, N.; Pourié, G.; Helle, D.; Guéant, J.-L.; Bossenmeyer-Pourié, C.; Daval, J.-L. Early methyl donor deficiency may induce persistent brain defects by reducing stat3 signaling targeted by mir-124. Cell Death Dis. 2013, 4, e755. [Google Scholar] [CrossRef] [PubMed]
- Boue, S.M.; Wiese, T.E.; Nehls, S.; Burow, M.E.; Elliott, S.; Carter-Wientjes, C.H.; Shih, B.Y.; McLachlan, J.A. Evaluation of the estrogenic effects of legume extracts containing phytoestrogens. J. Agric. Food Chem. 2003, 51, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.-M.; Hoikkala, A.; Wähälä, K.; Adlercreutz, H. Metabolism of the soy isoflavones daidzein, genistein and glycitein in human subjects. J. Steroid Biochem. Mol. Biol. 2003, 87, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Fitzpatrick, L.A. Soy isoflavones: Are they useful in menopause? Mayo Clin. Proc. 2000, 75, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Morito, K.; Hirose, T.; Kinjo, J.; Hirakawa, T.; Okawa, M.; Nohara, T.; Ogawa, S.; Inoue, S.; Muramatsu, M.; Masamune, Y. Interaction of phytoestrogens with estrogen receptors α and β. Biol. Pharm. Bull. 2001, 24, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B. Soy isoflavone supplements antagonize reproductive behavior and estrogen receptor and dependent gene expression in the brain. Endocrinology 2001, 142, 2946–2952. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.C.; Campana, A.; Lange, P.S.; Lee, H.-H.; Banerjee, K.; Bryson, J.B.; Mahishi, L.; Alam, S.; Giger, R.J.; Barnes, S.; et al. A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a camp-independent pathway. J. Neurosci. 2010, 30, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Schreihofer, D.A. High-soy diet decreases infarct size after permanent middle cerebral artery occlusion in female rats. AJP Regul. Integr. Comp. Physiol. 2005, 289, R103–R108. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.X.; Chen, W.F.; Xie, J.X.; Wong, M.S. Neuroprotective effects of genistein on dopaminergic neurons in the mice model of Parkinson’s disease. Neurosci Res. 2008, 60, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wei, H.; Cai, M.; Lu, Y.; Hou, W.; Yang, Q.; Dong, H.; Xiong, L. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of erk activity in ovariectomized mice. Int. J. Boil. Sci. 2014, 10, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, X.Q.; Ding, C.; Du, X.L. Genistein attenuates isoflurane-induced neurotoxicity and improves impaired spatial learning and memory by regulating cAMP/CREB and BDNF-TrkB-PI3K/Akt signaling. Korean J. Physiol. Pharmacol. 2017, 21, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Grillot, D.A.; Gonzalez-Garcia, M.; Ekhterae, D.; Duan, L.; Inohara, N.; Ohta, S.; Seldin, M.F.; Nuñez, G. Genomic organization, promoter region analysis, and chromosome localization of the mouse Bcl-x gene. J. Immunol. 1997, 158, 4750–4757. [Google Scholar] [PubMed]
- Setchell, K.D.; Brown, N.M.; Zimmer-Nechemias, L.; Brashear, W.T.; Wolfe, B.E.; Kirschner, A.S.; Heubi, J.E. Evidence for lack of absorption of soy isoflavone glycosides in humans, supporting the crucial role of intestinal metabolism for bioavailability. Am. J. Clin. Nutr. 2002, 76, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Xphosphorylation of bax ser184 by akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar] [CrossRef] [PubMed]
- Koh, P.-O. Nicotinamide attenuates the ischemic brain injury-induced decrease of akt activation and bad phosphorylation. Neurosci. Lett. 2011, 498, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Demeter, M.R.; Ruan, H.; Comb, M.J. Bad ser-155 phosphorylation regulates bad/Bcl-xl interaction and cell survival. J. Biol. Chem. 2000, 275, 25865–25869. [Google Scholar] [CrossRef] [PubMed]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R.; Okonkwo, D.O.; Singleton, R.H.; Mutlu, L.K.; Helm, G.A.; Povlishock, J.T. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid β peptide in traumatic axonal injury. J. Neurotrauma 2002, 19, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.C.; Soriano, S.; Bredesen, D.E.; Koo, E.H. Caspase cleavage of the amyloid precursor protein modulates amyloid β-protein toxicity. J. Neurochem. 2003, 87, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Carthy, C.M.; Yanagawa, B.; Luo, H.; Granville, D.J.; Yang, D.; Cheung, P.; Cheung, C.; Esfandiarei, M.; Rudin, C.M.; Thompson, C.B.; et al. Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology 2003, 313, 147–157. [Google Scholar] [CrossRef]
- Hu, Y.; Benedict, M.A.; Wu, D.; Inohara, N.; Nunez, G. Bcl-xl interacts with apaf-1 and inhibits apaf-1-dependent caspase-9 activation. Proc. Nat. Acad. Sci. USA 1998, 95, 4386–4391. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.P. The chemical constituents of ginseng plants. Comp. Med. East West. 1977, 5, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Wargovich, M.J. Inflammation, cancer, and targets of ginseng. J Nutr. 2007, 37, 183S–185S. [Google Scholar] [CrossRef] [PubMed]
- Kitts, D.D.; Wijewickreme, A.N.; Hu, C. Antioxidant properties of a North American ginseng extract. Mol. Cell Biochem. 2000, 203, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-Y.; Choi, I.-S.; Shim, J.-Y.; Yun, E.-K.; Yun, Y.-S.; Jeong, G.; Song, J.-Y. The immunomodulator ginsan induces resistance to experimental sepsis by inhibiting toll-like receptor-mediated inflammatory signals. Eur. J. Immunol. 2006, 36, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Keum, Y.-S.; Han, S.S.; Chun, K.-S.; Park, K.-K.; Park, J.-H.; Lee, S.K.; Surh, Y.-J. Inhibitory effects of the ginsenoside rg3 on phorbol ester-induced cyclooxygenase-2 expression, nf-κb activation and tumor promotion. Mutat. Res. Mol. Mech. Mutagen. 2003, 523–524, 75–85. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, Y.; Wang, Y.; Ling, P.; Xie, X.; Jiang, C.; Zhang, Z.; Lian, X.-Y. Ginseng rb fraction protects glia, neurons and cognitive function in a rat model of neurodegeneration. PLoS ONE 2014, 9, e101077. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Sun, W.; Gong, W.; Ding, Y.; Zhuang, Y.; Hou, Q. Ginsenoside rg1 protects against transient focal cerebral ischemic injury and suppresses its systemic metabolic changes in cerabral injury rats. Acta Pharm. Sin. B 2015, 5, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, C.; Chen, S.; Li, Z.; Ma, L.; Jia, X.; Wang, K.; Bao, J.; Liang, Y.; Chen, M.W.; et al. Hormetic effect of panaxatriol saponins confers neuroprotection in PC12 cells and zebrafish through PI3K/AKT/mTOR and AMPK/SIRT1/FOXO3 pathways. Sci. Rep. 2017, 7, 41082. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, R.-Y.; Zhao, J.; Dong, Z.; Feng, D.-Y.; Wu, R.; Shi, M.; Zhao, G. Ginsenoside rd protects sh-sy5y cells against 1-methyl-4-phenylpyridinium induced injury. Int. J. Mol. Sci. 2015, 16, 14395–14408. [Google Scholar] [CrossRef] [PubMed]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Karl, E.; Zhang, Z.; Dong, Z.; Neiva, K.G.; Soengas, M.S.; Koch, A.E.; Polverini, P.J.; Núñez, G.; Nör, J.E. Unidirectional crosstalk between Bcl-xl and Bcl-2 enhances the angiogenic phenotype of endothelial cells. Cell Death Differ. 2007, 14, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, N.; Sathiyamoorthy, S.; Ramya, M.; Yang, D.-U.; Lee, H.N.; Yang, D.-C. Molecular docking studies of anti-apoptotic Bcl-2, Bcl-xl, and mcl-1 proteins with ginsenosides frompanax ginseng. J. Enzym. Inhib. Med. Chem. 2011, 27, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Dietary (n-3) fatty acids and brain development. J. Nutr. 2007, 137, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.S.; Jun, S.B.; Lovinger, D.; Kim, H.-Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Bazan, N.G. Docosahexaenoic acid and the aging brain. J. Nutr. 2008, 138, 2510–2514. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.K.; Carlson, S.E. Role of omega-3 fatty acids in brain development and function: Potential implications for the pathogenesis and prevention of psychopathology. Prostaglandins Leuk. Essent. Fat. Acids 2006, 75, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, H.S.; Agrawal, R.; Sharma, S.; Huo, Y.-X.; Ying, Z.; Gomez-Pinilla, F. Omega-3 fatty acid deficiency during brain maturation reduces neuronal and behavioral plasticity in adulthood. PLoS ONE 2011, 6, e28451. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-L.; Chen, S.-J.; Kao, C.-L.; Hung, S.-C.; Ding, D.-C.; Yu, C.-C.; Chen, Y.-J.; Ku, H.-H.; Lin, C.-P.; Lee, K.-H.; et al. Docosahexaenoic acid promotes dopaminergic differentiation in induced pluripotent stem cells and inhibits teratoma formation in rats with parkinson-like pathology. Cell Transpl. 2012, 21, 313–332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bazan, N.G. Lipid-mediated cell signaling protects against injury and neurodegeneration. J Nutr. 2010, 140, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.X.; Wang, J.; Wu, L.; Kang, Z.B. Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature 2004, 427, 504. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.X. Fat-1 transgenic mice: A new model for omega-3 research. Prostaglandins Leuk. Essent. Fat. Acids 2007, 77, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Upadhyay, G.; Kumar, S.; Kapoor, A.; Kumar, A.; Tiwari, M.; Godbole, M.M. Hypothyroidism alters the expression of Bcl-2 family genes to induce enhanced apoptosis in the developing cerebellum. J. Endocrinol. 2003, 176, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sinha, R.A.; Tiwari, M.; Pal, L.; Shrivastava, A.; Singh, R.; Kumar, K.; Kumar Gupta, S.; Godbole, M.M. Increased pro-nerve growth factor and p75 neurotrophin receptor levels in developing hypothyroid rat cerebral cortex are associated with enhanced apoptosis. Endocrinology 2006, 147, 4893–4903. [Google Scholar] [CrossRef] [PubMed]
- Antony, R.; Lukiw, W.J.; Bazan, N.G. Neuroprotectin d1 induces dephosphorylation of Bcl-xl in a pp2a-dependent manner during oxidative stress and promotes retinal pigment epithelial cell survival. J. Boil. Chem. 2010, 285, 18301–18308. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G. Neuroprotectin d1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and alzheimer’s disease. J. Lipid Res. 2008, 50, S400–S405. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Tarcsafalvi, A.; Seng, N.; Hodeify, R.; Price, P. Cdk2 phosphorylation of Bcl-xl after stress converts it to a pro-apoptotic protein mimicking bax/bak. Cell Death Discov. 2016, 2, 15066. [Google Scholar] [CrossRef] [PubMed]
- Cittelly, D.M.; Nesic-Taylor, O.; Perez-Polo, J.R. Phosphorylation of Bcl-xL after spinal cord injury. J. Neurosci. Res. 2007, 85, 1894–1911. [Google Scholar] [CrossRef] [PubMed]
- Veas-Perez de Tudela, M.; Delgado-Esteban, M.; Maestre, C.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Vecino, R.; Bolaños, J.P.; Almeida, A. Regulation of Bcl-xL-ATP synthase interaction by mitochondrial cyclin b1-cyclin-dependent kinase-1 determines neuronal survival. J. Neurosci. 2015, 35, 9287–9301. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Goodwin, M.; Vu, T.; Brantley-Finley, C.; Gaarde, W.A.; Chambers, T.C. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-xl is mediated by jnk and occurs in parallel with inactivation of the raf-1/mek/erk cascade. J. Biol. Chem. 2000, 275, 29980–29985. [Google Scholar] [CrossRef] [PubMed]
- Calderon, F.; Kim, H.Y. Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 2004, 90, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Tosun, İ.; İnkaya, A.N. Resveratrol as a health and disease benefit agent. Food Rev. Int. 2010, 26, 85–101. [Google Scholar] [CrossRef]
- Khan, M.A.; Chen, H.; Wan, X.; Tania, M.; Xu, A.; Chen, F.; Zhang, D. Regulatory effects of resveratrol on antioxidant enzymes: A mechanism of growth inhibition and apoptosis induction in cancer cells. Mol. Cells 2013, 35, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Schmatz, R.; Perreira, L.B.; Stefanello, N.; Mazzanti, C.; Spanevello, R.; Gutierres, J.; Bagatini, M.; Martins, C.C.; Abdalla, F.H.; da Silva Serres, J.D.; et al. Effects of resveratrol on biomarkers of oxidative stress and on the activity of delta aminolevulinic acid dehydratase in liver and kidney of streptozotocin-induced diabetic rats. Biochimie 2012, 94, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Kuršvietienė, L.; Stanevičienė, I.; Mongirdienė, A.; Bernatonienė, J. Multiplicity of effects and health benefits of resveratrol. Medicina 2016, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.M.; Hsieh, T.C. Resveratrol: A cardioprotective substance. Ann. N. Y. Acad Sci. 2011, 1215, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.G.; D’Orazio, J.A.; Pearson, K.J. Resveratrol and cancer: Focus on in vivo evidence. Endocr. Relat. Cancer 2014, 21, R209–R225. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Das, D.K. Anti-inflammatory responses of resveratrol. Inflamm. Allergy Drug Targets 2007, 6, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.A.; Lee, K.-E.; Kim, H.-S.; Park, E.-M. Acute Resveratrol treatment modulates multiple signaling pathways in the ischemic brain. Neurochem. Res. 2012, 37, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pang, L.; Fang, F.; Zhang, G.; Zhang, J.; Xie, M.; Wang, L. Resveratrol attenuates brain damage in a rat model of focal cerebral ischemia via up-regulation of hippocampal Bcl-2. Brain Res. 2012, 1450, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Koronowski, K.B.; Dave, K.R.; Saul, I.; Camarena, V.; Thompson, J.W.; Neumann, J.T.; Young, J.I.; Perez-Pinzon, M.A. Resveratrol preconditioning induces a novel extended window of ischemic tolerance in the mouse brain. Stroke 2015, 46, 2293–2298. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gong, Q.; Dong, H.; Shi, J. Resveratrol, a neuroprotective supplement for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Edelstein, L.C.; Gelinas, C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Sarnico, I.; Ingrassia, R.; Boroni, F.; Branca, C.; Benarese, M.; Faraco, G.; Blasi, F.; Chiarugi, A.; Spano, P.; et al. The acetylation of rela in lys310 dictates the nf-κb-dependent response in post-ischemic injury. Cell Death Dis. 2010, 1, e96. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Franzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Gao, H.; Zhang, W.; Zhang, W.; Wang, Y. Resveratrol alleviates nerve injury after cerebral ischemia and reperfusion in mice by inhibiting inflammation and apoptosis. Int. J. Clin. Exp. Med. 2015, 8, 3219–3226. [Google Scholar] [PubMed]
- Kizmazoglu, C.; Aydin, H.E.; Sevin, I.E.; Kalemci, O.; Yüceer, N.; Atasoy, M.A. Neuroprotective effect of resveratrol on acute brain ischemia reperfusion injury by measuring annexin v, p53, Bcl-2 levels in rats. J. Korean Neurosurg. Soc. 2015, 58, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Y.; Zhang, H.; Yan, H.; Wu, X.; Zhang, C. Administration of the resveratrol analogues isorhapontigenin and heyneanol-a protects mice hematopoietic cells against irradiation injuries. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, H.; Jia, G.; Li, L.; Chen, H.; Bi, J.; Wang, C.L. The Synergistic Neuroprotective Effects of Combined Rosuvastatin and Resveratrol Pretreatment against Cerebral Ischemia/Reperfusion Injury. J. Stroke Cerebrovasc. Dis. 2018, 27, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Kumar, V.; Kashyap, M.P.; Khanna, V.K.; Randhawa, G.S.; Pant, A.B. Ischemic insult induced apoptotic changes in pc12 cells: Protection by trans resveratrol. Eur. J. Pharmacol. 2011, 666, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.S.; Mohr, C.; Rossi, D.J. Opposite actions of alcohol on tonic gabaa receptor currents mediated by nnos and pkc activity. Nat. Neurosci. 2013, 16, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Fitzgerald, P.J.; MacPherson, K.P.; DeBrouse, L.; Colacicco, G.; Flynn, S.M.; Masneuf, S.; Pleil, K.E.; Li, C.; Marcinkiewcz, C.A.; et al. Chronic alcohol remodels prefrontal neurons and disrupts nmdar-mediated fear extinction encoding. Nat. Neurosci. 2012, 15, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Taffe, M.A.; Kotzebue, R.W.; Crean, R.D.; Crawford, E.F.; Edwards, S.; Mandyam, C.D. Long-lasting reduction in hippocampal neurogenesis by alcohol consumption in adolescent nonhuman primates. Proc. Nat. Acad. Sci. USA 2010, 107, 11104–11109. [Google Scholar] [CrossRef] [PubMed]
- Stampfer, M.J.; Kang, J.H.; Chen, J.; Cherry, R.; Grodstein, F. Effects of moderate alcohol consumption on cognitive function in women. N. Engl. J. Med. 2005, 352, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Elkind, M.; Boden-Albala, B.; Lin, I.F.; Kargman, D.E.; Hauser, W.A.; Shea, S.; Paik, M.C. The protective effect of moderate alcohol consumption on ischemic stroke. JAMA 1999, 281, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ruitenberg, A.; van Swieten, J.C.; Witteman, J.C.; Mehta, K.M.; van Duijn, C.M.; Hofman, A.; Breteler, M.M.B. Alcohol consumption and risk of dementia: The rotterdam study. Lancet 2002, 359, 281–286. [Google Scholar] [CrossRef]
- Ronksley, P.E.; Brien, S.E.; Turner, B.J.; Mukamal, K.J.; Ghali, W.A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ 2011, 342, d671. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Fu, P.; Ji, X.; Peng, C.; Fredrickson, V.; Sy, C.; Meng, R.; Ling, F.; Du, H.; Tan, X.; et al. Synergetic neuroprotection of normobaric oxygenation and ethanol in ischemic stroke through improved oxidative mechanism. Stroke 2013, 44, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Kochanski, R.; Peng, C.; Higashida, T.; Geng, X.; Hüttemann, M.; Guthikonda, M.; Ding, Y. Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: An inhibitory effect on hyperglycolysis and nadph oxidase activation. J. Neurochem. 2013, 126, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Li, J.; Cao, X.; Chen, Y.; Ye, C.; Chen, K. Protective effect of n-butyl alcohol extracts from Rhizoma Pinelliae Pedatisectae against cerebral ischemia-reperfusion injury in rats. J. Ethnopharmacol. 2016, 188, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-S.; Zhao, J.; Zheng, W.-P.; Zhao, Y. Neuroprotective effect of 4-hydroxybenzyl alcohol against transient focal cerebral ischemia via anti-apoptosis in rats. Brain Res. 2010, 1308, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Billen, L.P.; Kokoski, C.L.; Lovell, J.F.; Leber, B.; Andrews, D.W. Bcl-xl inhibits membrane permeabilization by competing with bax. PLoS Biol. 2008, 6, e147. [Google Scholar] [CrossRef] [PubMed]
- Siler-Marsiglio, K.I.; Madorsky, I.; Pan, Q.; Paiva, M.; Neeley, A.W.; Shaw, G.; Heaton, M.B. Effects of acute ethanol exposure on regulatory mechanisms of Bcl-2-associated apoptosis promoter, bad, in neonatal rat cerebellum: Differential effects during vulnerable and resistant developmental periods. Alcohol. Clin. Exp. Res. 2006, 30, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Jeong, E.Y.; Kim, Y.S.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S. C-jun N-terminal kinase regulates the interaction between 14-3-3 and bad in ethanol-induced cell death. J. Neurosci. Res. 2008, 86, 3221–3229. [Google Scholar] [CrossRef] [PubMed]
- Bazovkina, D.V.; Tsybko, A.S.; Filimonova, E.A.; Ilchibaeva, T.V.; Naumenko, V.S. Influence of chronic alcohol treatment on the expression of the bdnf, bax, Bcl-xl, and casp3 genes in the mouse brain: Role of the c1473g polymorphism in the gene encoding tryptophan hydroxylase 2. Mol. Biol. 2016, 50, 262–269. [Google Scholar] [CrossRef]
- Lee, J.-H.; Tajuddin, N.F.; Druse, M.J. Effects of ethanol and ipsapirone on the expression of genes encoding anti-apoptotic proteins and an antioxidant enzyme in ethanol-treated neurons. Brain Res. 2009, 1249, 54–60. [Google Scholar] [CrossRef] [PubMed]


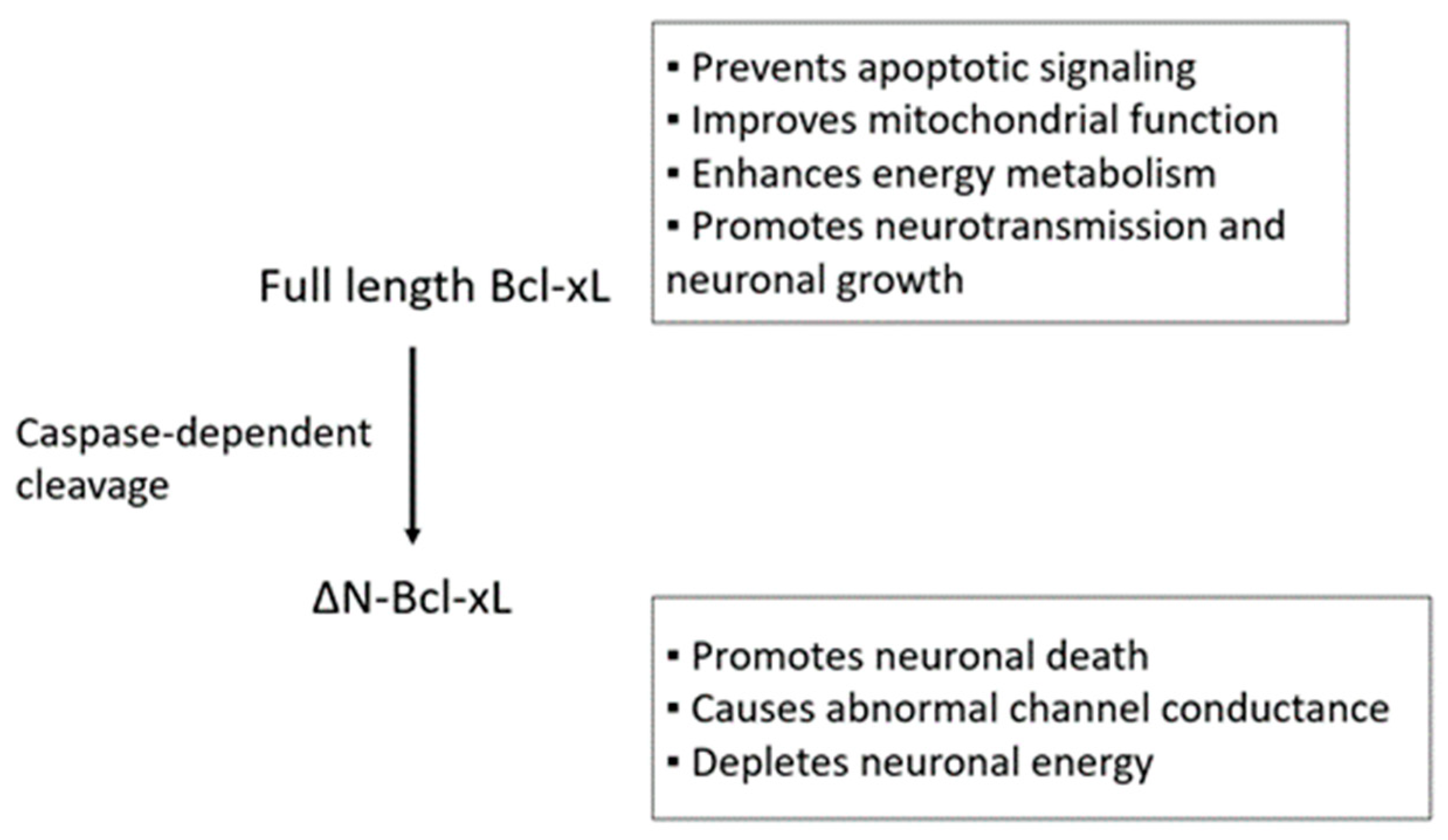{kind=link}
| CNS-Associated Disorders | References |
|---|---|
| Ischemic stroke | [18,30,31,32,33,34,35] |
| Spinal cord injury | [36,37,38,39,40] |
| Alzheimer | [41,42,43,44] |
| Parkinson | [45,46,47,48,49,50] |
| Anxiety and Depression | [51,52,53] |
| Traumatic Brain injury | [54,55,56] |
| Spinal Muscular Dystrophy | [57,58,59,60,61] |
| Schizophrenia | [62,63,64] |
| Nutrients and Foods | Roles | References |
|---|---|---|
| High soy diet | Upregulation of Bcl-xL mRNA and protein in rats | [106] |
| High soy diet | Upregulation of Bcl-xL mRNA and protein in rats MCAO model | [107] |
| Orally gavaged soy isoflavones | Upregulation of Bcl-xL protein against Aβ-induced neurotoxicity in rats | [108] |
| Intravenous infusion of ginsenoside | STAT5-dependent upregulation of Bcl-xL protein in rodent ischemia models (rats and gerbils) and primary cortical neurons | [109] |
| Intravenous infusion of dihydroginsenoside Rb1 | Upregulation of Bcl-xL mRNA in spinal cord injury and cerebral ischemia models | [110] |
| Oral administration of red ginseng extract | Upregulation of Bcl-xL protein in rat spinal cord injury model | [111] |
| Oral administration and intravenous infusion of ginsenoside | Upregulation of Bcl-xL protein in mice during pneumococcal infection | [112] |
| Intravenous infusion of ginsenoside | Upregulation of Bcl-xL protein in rat spinal cord injury model | [113] |
| DHA or neuroprotectin D1 treatment in vitro | Upregulation of Bcl-xL gene in HN cell line | [114] |
| DHA treatment in vitro | Upregulation of Bcl-xL mRNA in HT22 cell line against MNNG-induced toxicity | [115] |
| Oral supplementation of DHA and EPA | Upregulation of Bcl-xL protein in hypothyroid rats | [116] |
| Oral supplementation of DHA | Upregulation of Bcl-xL mRNA in mice brains after challenge with dopaminergic toxin | [117] |
| Application with Fat-1 mouse model | Cortical neurons isolated from Fat-1 mice, genetically modified model with high endogenous omega-3 fatty acid levels, retained Bcl-xL protein against OGD | [118] |
| DHA treatment in vitro | Upregulation of Bcl-xL protein in primary cortical neurons against oxyhemoglobin treatment | [119] |
| Primary cortical neurons isolated from Fat-1 mice | Upregulation of Bcl-xL protein in primary cortical neurons against Aβ 1-42 treatment | [120] |
| Intraperitoneal delivery of resveratrol | Upregulation of acetylation of H3 histones on Bcl-xL promoter in mice | [121] |
| Resveratrol treatment in vitro | Upregulation of Bcl-xL protein in PC12 cells | [122] |
| Resveratrol treatment in vitro | Upregulation of Bcl-xL protein in SH-SY5Y cells | [123] |
| Intraperitoneal injection of ethanol | Upregulation of Bcl-xL protein in rat MCAO model | [124] |
| Intraperitoneal injection of ethanol | Upregulation of Bcl-xL protein in rat MCAO model | [125] |
| α-Tocopherol treatment in vitro | Downregulation of Bax/Bcl-xL protein ratio in PC 12 cells | [126] |
| Methyl donor deficient diet (no folate or vitamin B12) | Downregulation of Bcl-xL protein via miR-124-mediated Stat3 inhibition | [127] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-A.; Broman, K.; Stumpf, A.; Kazyak, S.; Jonas, E.A. Nutritional Regulators of Bcl-xL in the Brain. Molecules 2018, 23, 3019. https://doi.org/10.3390/molecules23113019
Park H-A, Broman K, Stumpf A, Kazyak S, Jonas EA. Nutritional Regulators of Bcl-xL in the Brain. Molecules. 2018; 23(11):3019. https://doi.org/10.3390/molecules23113019
Chicago/Turabian StylePark, Han-A, Katheryn Broman, Allison Stumpf, Sara Kazyak, and Elizabeth A. Jonas. 2018. "Nutritional Regulators of Bcl-xL in the Brain" Molecules 23, no. 11: 3019. https://doi.org/10.3390/molecules23113019
APA StylePark, H.-A., Broman, K., Stumpf, A., Kazyak, S., & Jonas, E. A. (2018). Nutritional Regulators of Bcl-xL in the Brain. Molecules, 23(11), 3019. https://doi.org/10.3390/molecules23113019