
Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers



Abstract
1. Introduction
2. Epidemiological Studies of ITCs
3. Molecular Mechanism of ITCs in Cancer Prevention
3.1. Benzyl Isothiocyanate
3.1.1. Blood
3.1.2. Breast
3.1.3. Brain
3.1.4. Colon
3.1.5. Pancreatic
3.2. Phenethyl Isothiocyanate
3.2.1. Blood
3.2.2. Breast
3.2.3. Colon
3.2.4. Ovary
3.2.5. Prostate
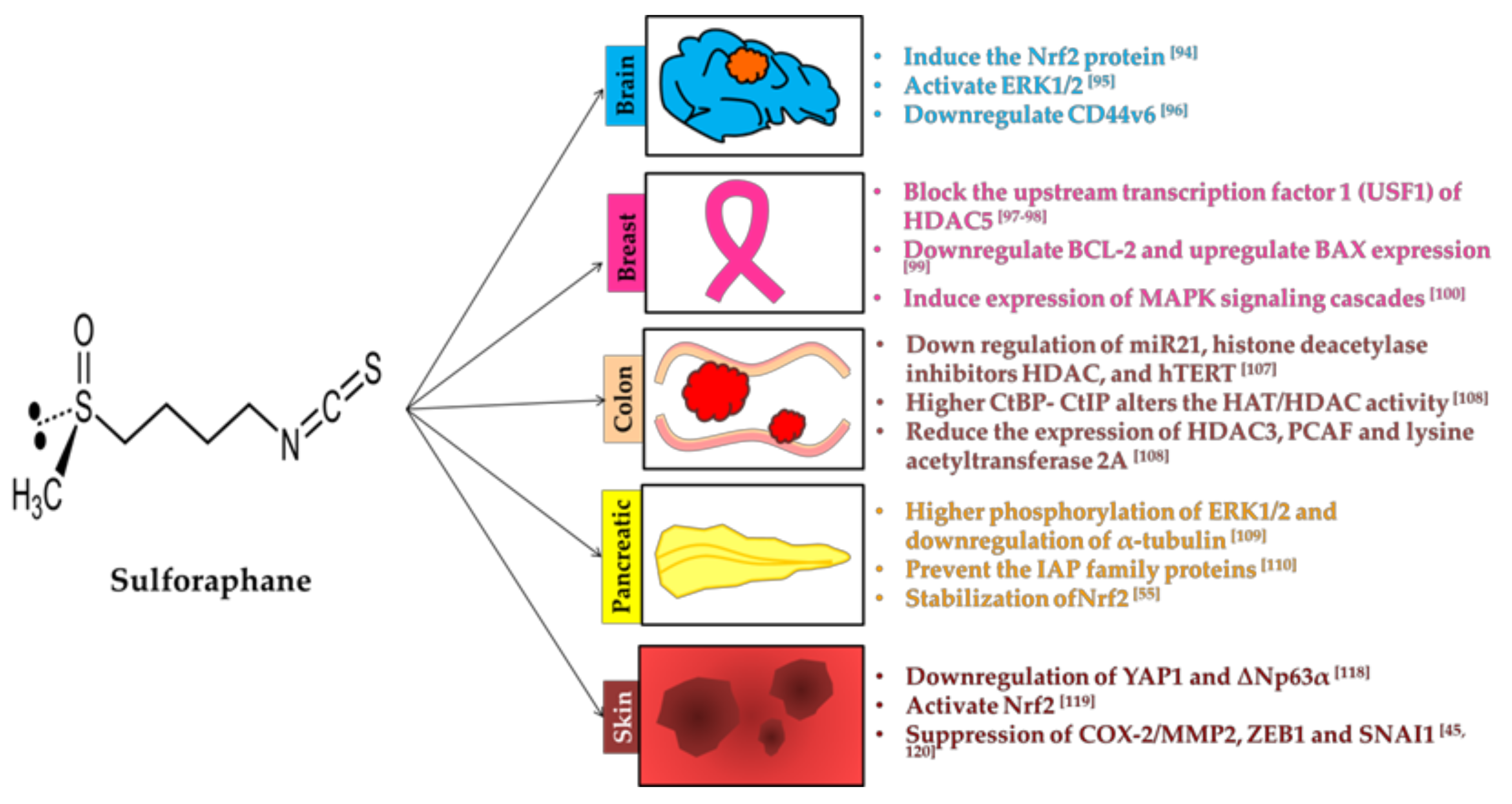
3.3. Sulforaphane
3.3.1. Brain
3.3.2. Breast
3.3.3. Colon
3.3.4. Prostate
3.3.5. Skin
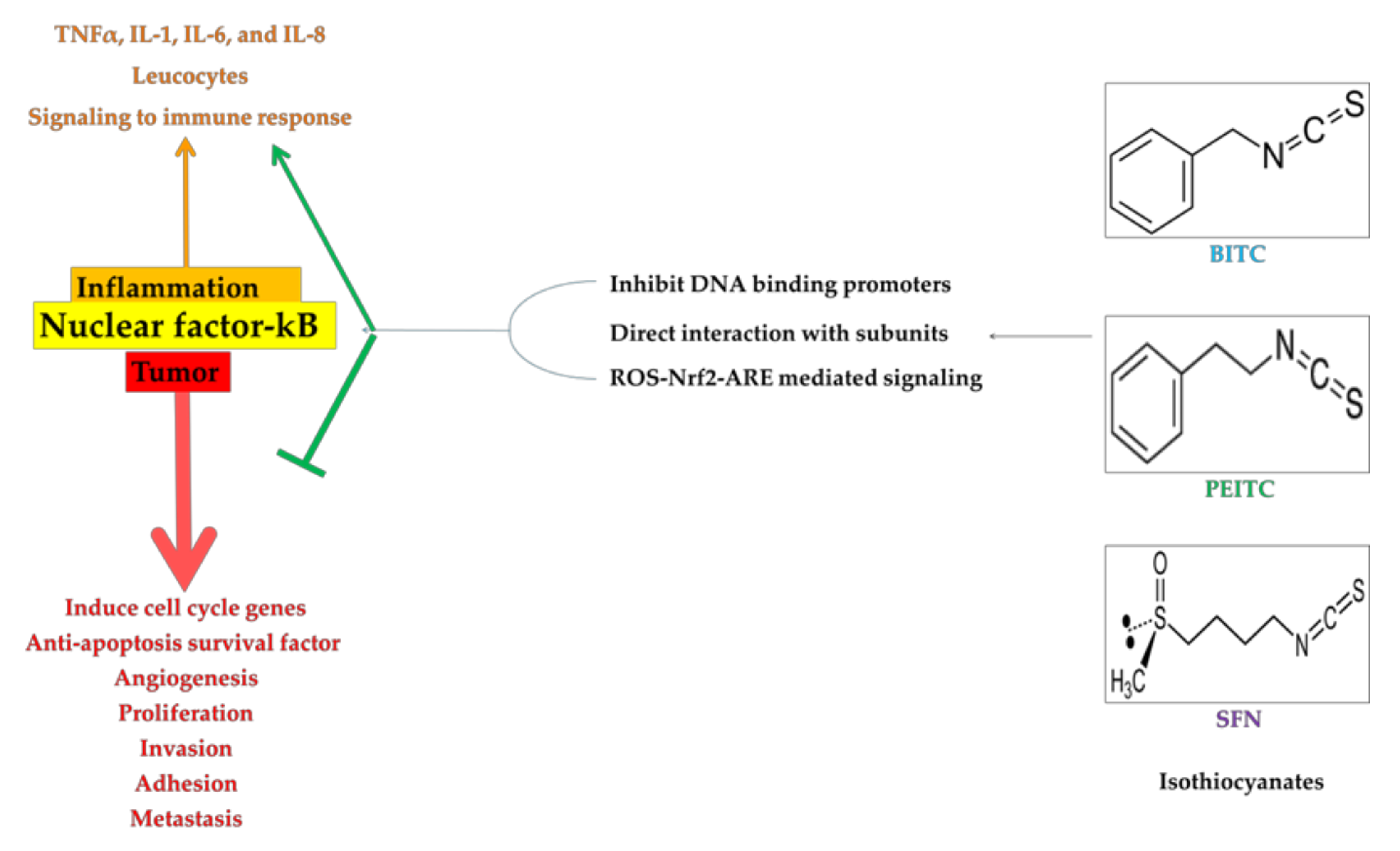
4. ROS-Nrf2-ARE Mediated Pathways in Downregulation of NF-κB
5. Direct and Indirect Inhibition of NF-κB by ITCs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Fleming, T. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [PubMed]
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. J. Paleontol. 2008, 25, 2097–2116. [Google Scholar]
- Edwards, I.R.; Aronson, J.K. Adverse drug reactions: Definitions, diagnosis, and management. Lancet 2000, 356, 1255–1259. [Google Scholar] [CrossRef]
- Block, G.; Patterson, B.; Subar, A. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutr. Cancer 1992, 18, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Tafrihi, M.; Nakhaei Sistani, R. E-Cadherin/beta-Catenin Complex: A Target for Anticancer and Antimetastasis Plants/Plant-derived Compounds. Nutr. Cancer 2017, 69, 702–722. [Google Scholar] [CrossRef] [PubMed]
- Popolo, A.; Pinto, A.; Daglia, M.; Nabavi, S.F.; Farooqi, A.A.; Rastrelli, L. Two likely targets for the anti-cancer effect of indole derivatives from cruciferous vegetables: PI3K/Akt/mTOR signalling pathway and the aryl hydrocarbon receptor. Semin. Cancer Biol. 2017, 46, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-S.; Jin, M.; Chun, J.-H.; Kim, S.-J.; Park, B.-S.; Shon, S.-H.; Kim, J.S. Functional analysis of three BrMYB28 transcription factors controlling the biosynthesis of glucosinolates in Brassica rapa. Plant Mol. Biol. 2016, 90, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-S.; Kim, J.S. Understanding of MYB transcription factors involved in glucosinolate biosynthesis in brassicaceae. Molecules 2017, 22, 1549. [Google Scholar]
- Seo, M.S.; Jin, M.; Sohn, S.H.; Kim, J.S. Expression profiles of Br MYB transcription factors related to glucosinolate biosynthesis and stress response in eight subspecies of Brassica rapa. FEBS Open Bio 2017, 7, 1646–1659. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, G.R.; Heaney, R.K.; Mullin, W.J.; VanEtten, C. Glucosinolates and their breakdown products in food and food plants. Crit. Rev. Food Sci. Nutr. 1983, 18, 123–201. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Inhibition of carcinogenesis by isothiocyanates. Drug Metab. Rev. 2000, 32, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, L.; Gonzalez, V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol. Cancer Ther. 2003, 2, 1045–1052. [Google Scholar] [PubMed]
- Lampe, J.W.; Peterson, S. Brassica, biotransformation and cancer risk: Genetic polymorphisms alter the preventive effects of cruciferous vegetables. J. Nutr. 2002, 132, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Mithen, R. Glucosinolates, isothiocyanates and human health. Phytochem. Rev. 2009, 8, 269–282. [Google Scholar] [CrossRef]
- Capasso, R.; Aviello, G.; Romano, B.; Borrelli, F.; De Petrocellis, L.; Di Marzo, V.; Izzo, A.A. Modulation of mouse gastrointestinal motility by allyl isothiocyanate, a constituent of cruciferous vegetables (Brassicaceae): Evidence for TRPA1-independent effects. Br. J. Pharmacol. 2012, 165, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Sturm, C.; Wagner, A.E. Brassica-Derived Plant Bioactives as Modulators of Chemopreventive and Inflammatory Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 1890. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Arunakaran, J.; Arunkumar, R.; Elumalai, P.; Senthilkumar, K. Impact of quercetin, diallyl disulfide and nimbolide on the regulation of nuclear factor kappa B expression in prostate and breast cancer cell lines. Nat. Prod. Chem. Res. 2013, 1, 1000115. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X.J. NF-κB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; William, C.S.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Richmond, A. NF-κB activation in melanoma. Pigment Cell Res. 2006, 19, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Monteillier, A.; Allard, P.-M.; Gindro, K.; Wolfender, J.-L.; Cuendet, M. Lung Cancer Chemopreventive Activity of Patulin Isolated from Penicillium vulpinum. Molecules 2018, 23, 636. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: Metabolism and excretion in humans. Cancer Epidemiol. Biomark. Prev. 2001, 10, 501–508. [Google Scholar]
- Rouzaud, G.; Young, S.A.; Duncan, A.J. Hydrolysis of glucosinolates to isothiocyanates after ingestion of raw or microwaved cabbage by human volunteers. Cancer Epidemiol. Biomark. Prev. 2004, 13, 125–131. [Google Scholar] [CrossRef]
- Conaway, C.C.; Getahun, S.M.; Liebes, L.L.; Pusateri, D.J.; Topham, D.K.; Botero-Omary, M.; Chung, F.-L. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr. Cancer 2000, 38, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, D.; Lehmann, C.; Florian, S.; Barknowitz, G.; Haack, M.; Mewis, I.; Wiesner, M.; Schreiner, M.; Glatt, H.; Brigelius-Flohé, R. Glucosinolates from pak choi and broccoli induce enzymes and inhibit inflammation and colon cancer differently. Food Funct. 2014, 5, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Cao, L.; Zhang, Q.; Song, Q.; Meng, Z.; Wu, X.; Xu, K. Inhibition of autophagy potentiates the anti-metastasis effect of phenethyl isothiocyanate through JAK2/STAT3 pathway in lung cancer cells. Mol. Carcinog. 2018, 57, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.L.; Tan, H.Q.; Chua, K.J.; Kang, A.; Lim, K.H.; Ling, K.L.; Yew, W.S.; Lee, Y.S.; Thiery, J.P.; Chang, M.W. Engineered commensal microbes for diet-mediated colorectal-cancer chemoprevention. Nat. Biomed. Eng. 2018, 2, 27–37. [Google Scholar] [CrossRef]
- Pereira, L.P.; Silva, P.; Duarte, M.; Rodrigues, L.; Duarte, C.M.; Albuquerque, C.; Serra, A.T. Targeting colorectal cancer proliferation, stemness and metastatic potential using brassicaceae extracts enriched in isothiocyanates: A 3D cell model-based study. Nutrients 2017, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.-Q.; Ho, S.C.; Mo, X.-F.; Lin, F.-Y.; Huang, W.-Q.; Luo, H.; Huang, J.; Zhang, C.-X. Glucosinolate and isothiocyanate intakes are inversely associated with breast cancer risk: A case–control study in China. Br. J. Nutr. 2018, 119, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.J.; Hwang, Y.-T.; Gomez, S.L.; Fung, T.T.; Yeh, S.-L.; Dash, C.; Allen, L.; Philips, S.; Hilakivi-Clarke, L.; Zheng, Y.-L.J. Correction to: Dietary intake of soy and cruciferous vegetables and treatment-related symptoms in Chinese-American and non-Hispanic White breast cancer survivors. Breast Cancer Res. Treat. 2018, 168, 481–482. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Clark, R.; Tu, P.; Bosworth, H.B.; Zullig, L.L. Breast cancer oral anti-cancer medication adherence: A systematic review of psychosocial motivators and barriers. Breast Cancer Res. Treat. 2017, 165, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zirpoli, G.R.; Guru, K.; Moysich, K.B.; Zhang, Y.; Ambrosone, C.B.; McCann, S.E. Consumption of raw cruciferous vegetables is inversely associated with bladder cancer risk. Cancer Epidemiol. Biomark. Prev. 2008, 17, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Abbaoui, B.; Lucas, C.R.; Riedl, K.M.; Clinton, S.K.; Mortazavi, A. Cruciferous Vegetables, Isothiocyanates, and Bladder Cancer Prevention. Mol. Nutr. Food Res. 2018, 62, 1800079. [Google Scholar] [CrossRef] [PubMed]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investig. New Drugs 2015, 33, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, A.; Rohrmann, S.; Timofeeva, M.; Risch, A.; Jansen, E.; Linseisen, J. Dietary glucosinolate intake, polymorphisms in selected biotransformation enzymes, and risk of prostate cancer. Cancer Epidemiol. Biomark. Prev. 2010, 19, 135–143. [Google Scholar] [CrossRef] [PubMed]
- McManus, H.; Moysich, K.B.; Tang, L.; Joseph, J.; McCann, S.E. Usual Cruciferous Vegetable Consumption and Ovarian Cancer: A Case-Control Study. Nutr. Cancer Lett. 2018, 70, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Bassan, P.; Bhushan, S.; Kaur, T.; Arora, R.; Arora, S.; Vig, A.P. Extraction, profiling and bioactivity analysis of volatile glucosinolates present in oil extract of Brassica juncea var. raya. Physiol. Mol. Biol. Plants 2018, 24, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.M.; Bracha, S.; Wong, C.P.; Beaver, L.M.; Stevens, J.F.; Ho, E. Sulforaphane absorption and histone deacetylase activity following single dosing of broccoli sprout supplement in normal dogs. Vet. Med. Sci. 2018, 4, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.L.; Liu, X.; Charron, C.S.; Novotny, J.A.; Jeffery, E.H.; Seifried, H.E.; Ross, S.A.; Miller, M.J.; Swanson, K.S.; Holscher, H.D. Broccoli consumption affects the human gastrointestinal microbiota. J. Nutr. Biochem. 2018, 63, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, R.; Schreiner, M.; Krumbein, A.; Ciska, E.; Holst, B.; Rowland, I.; De Schrijver, R.; Hansen, M.; Gerhäuser, C.; Mithen, R.J.; et al. Glucosinolates in Brassica vegetables: The influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 2009, 53, S219. [Google Scholar] [CrossRef] [PubMed]
- Herz, C.; Márton, M.-R.; Tran, H.T.T.; Gründemann, C.; Schell, J.; Lamy, E. Benzyl isothiocyanate but not benzyl nitrile from Brassicales plants dually blocks the COX and LOX pathway in primary human immune cells. J. Funct. Foods 2016, 23, 135–143. [Google Scholar] [CrossRef]
- Lee, W.; Nam, J.H.; Cho, H.J.; Lee, J.Y.; Cho, W.K.; Kim, U.; We, Y.M.; Ma, J.Y.; Hoe, H.S. Epimedium koreanum Nakai inhibits PMA-induced cancer cell migration and invasion by modulating NF-kappaB/MMP-9 signaling in monomorphic malignant human glioma cells. Oncol. Rep. 2017, 38, 3619–3631. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Abe, N.; Yoshimoto, M.; Zhu, B.; Murata, Y.; Nakamura, Y. Benzyl isothiocyanate inhibits IL-13 expression in human basophilic KU812 cells. Biosci. Biotechnol. Biochem. 2015, 79, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Larkin, S.; Williams, T. Cyclooxygenase-2: Regulation and relevance in inflammation. Biochem. Pharmacol. 1995, 50, 1535–1542. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical–biological functions and (Q) SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Cho, H.-J.; Jeong, Y.-J.; Shin, J.-M.; Park, K.-K.; Park, Y.-Y.; Bae, Y.-S.; Chung, I.-K.; Kim, M.; Kim, C.-H.; et al. Isothiocyanates inhibit the invasion and migration of C6 glioma cells by blocking FAK/JNK-mediated MMP-9 expression. Oncol. Rep. 2015, 34, 2901–2908. [Google Scholar] [CrossRef] [PubMed]
- Pore, S.K.; Hahm, E.-R.; Latoche, J.D.; Anderson, C.J.; Shuai, Y.; Singh, S.V. Prevention of breast cancer-induced osteolytic bone resorption by benzyl isothiocyanate. Carcinogenesis 2017, 39, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Nagalingam, A.; Kuppusamy, P.; Muniraj, N.; Langford, P.; Győrffy, B.; Saxena, N.K.; Sharma, D. Benzyl Isothiocyanate potentiates p53 signaling and antitumor effects against breast cancer through activation of p53-LKB1 and p73-LKB1 axes. Sci. Rep. 2017, 7, 40070. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Myatt, S.S.; Lam, E.W.-F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Bommareddy, A.; Kim, S.-H.; Sehrawat, A.; Hahm, E.-R.; Singh, S.V. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS ONE 2012, 7, e32597. [Google Scholar] [CrossRef] [PubMed]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Vega, M.I.; León-Contreras, J.C.; Hernández-Pando, R.; Medina-Campos, O.N.; Rodríguez, E.; Tapia, E.; Pedraza-Chaverri, J.J. Sulforaphane induces differential modulation of mitochondrial biogenesis and dynamics in normal cells and tumor cells. Food Chem. Toxicol. 2017, 100, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, L.; Zhang, G.-D.; Wang, H.-O.; Liu, M.-Y.; Jiang, Y.; Qi, L.-S.; Li, Q.; Yang, P. Potential mechanisms of benzyl isothiocyanate suppression of invasion and angiogenesis by the U87MG human glioma cell line. Asian Pac. J. Cancer Prev. 2014, 19, 8225–8228. [Google Scholar] [CrossRef]
- Tang, N.-Y.; Chueh, F.-S.; Yu, C.-C.; Liao, C.-L.; Lin, J.-J.; Hsia, T.-C.; Wu, K.-C.; Liu, H.-C.; Lu, K.-W.; Chung, J.-G. Benzyl isothiocyanate alters the gene expression with cell cycle regulation and cell death in human brain glioblastoma GBMá8401 cells. Oncol. Rep. 2016, 35, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.S.; Lin, J.J.; Lin, C.C.; Lien, J.C.; Peng, S.F.; Fan, M.J.; Hsu, F.T.; Chung, J.G. Benzyl isothiocyanate inhibits human brain glioblastoma multiforme GBM 8401 cell xenograft tumor in nude mice in vivo. Environ. Toxicol. 2018, 33, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Miyagawa, M.; Liu, X.; Zhu, B.; Munemasa, S.; Nakamura, T.; Murata, Y.; Nakamura, Y. Methyl-β-cyclodextrin potentiates the BITC-induced anti-cancer effect through modulation of the Akt phosphorylation in human colorectal cancer cells. Biosci. Biotechnol. Biochem. 2018, 82, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, W.; Dong, X.; Chen, Y.; Lu, Y.; Lin, B.; Guo, J.; Li, M. Benzyl-isothiocyanate induces apoptosis and inhibits migration and invasion of hepatocellular carcinoma cells in vitro. J. Cancer 2017, 8, 240–248. [Google Scholar] [CrossRef] [PubMed]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updates 2008, 11, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-C.; Huang, A.-C.; Hsu, S.-C.; Kuo, C.-L.; Yang, J.-S.; Wu, S.-H.; Chung, J.-G. Benzyl isothiocyanate (BITC) inhibits migration and invasion of human colon cancer HT29 cells by inhibiting matrix metalloproteinase-2/-9 and urokinase plasminogen (uPA) through PKC and MAPK signaling pathway. J. Agric. Food Chem. 2010, 58, 2935–2942. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.R.; Pramanik, K.C.; Srivastava, S.K. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clin. Cancer Res. 2011, 17, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Traganos, F.; Darzynkiewicz, Z. Kinetics of histone H2AX phosphorylation and Chk2 activation in A549 cells treated with topotecan and mitoxantrone in relation to the cell cycle phase. Cytometry 2008, 73A, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Wicker, C.A.; Sahu, R.P.; Kulkarni-Datar, K.; Srivastava, S.K.; Brown, T.L.J. BITC sensitizes pancreatic adenocarcinomas to TRAIL-induced apoptosis. Cancer Growth Metastasis 2009, 2. [Google Scholar] [CrossRef]
- Kasiappan, R.; Jutooru, I.; Karki, K.; Hedrick, E.; Safe, S. Benzyl isothiocyanate (BITC) induces reactive oxygen species-dependent repression of STAT3 protein by down-regulation of specificity proteins in pancreatic cancer. J. Biol. Chem. 2016, 291, 27122–27133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, S.; Wang, J.; Fang, Q.; Chai, Q. Phenethyl isothiocyanate inhibits growth of human chronic myeloid leukemia K562 cells via reactive oxygen species generation and caspases. Mol. Med. Rep. 2014, 10, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P. FasL binds preassembled Fas. Science 2000, 288, 2328–2329. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood 2008, 112, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Trachootham, D.; Lu, W.; Carew, J.; Giles, F.J.; Keating, M.; Arlinghaus, R.B.; Huang, P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 2008, 22, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Srivastava, S.K. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med. 2012, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.J.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 2007, 131, 18–43. [Google Scholar] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly (ADP-ribose) polymerase (PARP) cleavage in apoptosis Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Huang, A.C.; Yang, J.S.; Liao, C.L.; Lu, H.F.; Chou, S.T.; Ma, C.Y.; Hsia, T.C.; Ko, Y.C.; Chung, J.G. Benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells. J. Orthop. Res. 2011, 29, 1199–1209. [Google Scholar] [PubMed]
- Cang, S.; Ma, Y.; Chiao, J.-W.; Liu, D. Phenethyl isothiocyanate and paclitaxel synergistically enhanced apoptosis and alpha-tubulin hyperacetylation in breast cancer cells. J. Exp. Hematol. Oncol. 2014, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Tsou, M.F.; Tien, N.; Lu, C.C.; Chiang, J.H.; Yang, J.S.; Lin, J.P.; Fan, M.J.; Lu, J.J.; Yeh, S.P.; Chung, J.G. Phenethyl isothiocyanate promotes immune responses in normal BALB/c mice, inhibits murine leukemia WEHI-3 cells, and stimulates immunomodulations in vivo. Environ. Toxicol. 2013, 28, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.-C.; Hsu, S.-C.; Kuo, C.-L.; Ip, S.-W.; Yang, J.-S.; Hsu, Y.-M.; Huang, H.-Y.; Wu, S.-H.; Chung, J.-G. Phenethyl isothiocyanate inhibited tumor migration and invasion via suppressing multiple signal transduction pathways in human colon cancer HT29 cells. J. Agric. Food Chem. 2010, 58, 11148–11155. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Kim, B.R.; Chen, C.; Hebbar, V.; Kong, A.-N.T. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis 2003, 24, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dey, M. Dietary Phenethyl Isothiocyanate Protects Mice from Colitis Associated Colon Cancer. Int. J. Mol. Sci. 2017, 18, 1908. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, S.; Kandala, P.K.; Gupta, P.; Srivastava, S.K. Inhibition of EGFR-AKT axis results in the suppression of ovarian tumors in vitro and in preclinical mouse model. PLoS ONE 2012, 7, e43577. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.Y.; Yang, Y.L.; Yan, H.; Huang, Q.; Liu, K.J.; Zhang, S. Phenethyl isothiocyanate suppresses the metastasis of ovarian cancer associated with the inhibition of CRM1-mediated nuclear export and mTOR-STAT3 pathway. Cancer Biol. Ther. 2017, 18, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Miskimins, W.K. Metformin and phenethyl isothiocyanate combined treatment in vitro is cytotoxic to ovarian cancer cultures. J. Ovarian Res. 2012, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Ikejiri, F.; Honma, Y.; Kasukabe, T.; Urano, T.; Suzumiya, J. TH588, an MTH1 inhibitor, enhances phenethyl isothiocyanateinduced growth inhibition in pancreatic cancer cells. Oncol. Lett. 2018, 15, 3240–3244. [Google Scholar] [PubMed]
- Kasukabe, T.; Honma, Y.; Okabe-Kado, J.; Higuchi, Y.; Kato, N.; Kumakura, S. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol. Rep. 2016, 36, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Xiao, D. Phenethyl isothiocyanate sensitizes androgen-independent human prostate cancer cells to Docetaxel-induced apoptosis in vitro and in vivo. Pharm. Res. 2010, 27, 722–731. [Google Scholar]
- Xiao, D.; Singh, S.V. p66Shc Is Indispensable for Phenethyl Isothiocyanate–Induced Apoptosis in Human Prostate Cancer Cells. Cancer Res. 2010, 70, 3150–3158. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; McGough, R.; Aswad, B.; Block, J.A.; Terek, R. Hypoxia induces HIF-1α and VEGF expression in chondrosarcoma cells and chondrocytes. J. Orthop. Res. 2004, 22, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Hahm, E.-R.; Rigatti, L.H.; Normolle, D.P.; Yuan, J.-M.; Singh, S.V. Inhibition of Glycolysis in Prostate Cancer Chemoprevention by Phenethyl Isothiocyanate. Cancer Prev. Res. 2018, 11, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Singh, S.V. Fatty acid synthesis intermediates represent novel noninvasive biomarkers of prostate cancer chemoprevention by phenethyl isothiocyanate. Cancer Prev. Res. 2017, 10, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shu, L.; Kim, H.; Khor, T.O.; Wu, R.; Li, W.; Kong, A.N.T.J. Phenethyl isothiocyanate (PEITC) suppresses prostate cancer cell invasion epigenetically through regulating microRNA-194. Mol. Nutr. Food Res. 2016, 60, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.-Y.; Huang, Y.-T.; Yu, C.-S.; Ko, Y.-C.; Wu, S.-H.; Ji, B.-C.; Yang, J.-S.; Yang, J.-L.; Hsia, T.-C.; Chen, Y.-Y.; et al. Phenethyl isothiocyanate (PEITC) promotes G2/M phase arrest via p53 expression and induces apoptosis through caspase-and mitochondria-dependent signaling pathways in human prostate cancer DU 145 cells. Anticancer Res. 2011, 31, 1691–1702. [Google Scholar] [PubMed]
- Holloway, P.M.; Gillespie, S.; Becker, F.; Vital, S.A.; Nguyen, V.; Alexander, J.S.; Evans, P.C.; Gavins, F.N. Sulforaphane induces neurovascular protection against a systemic inflammatory challenge via both Nrf2-dependent and independent pathways. Vasc. Pharmacol. 2016, 85, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mattar, P.; Dixit, R.; Lawn, S.O.; Wilkinson, G.; Kinch, C.; Eisenstat, D.; Kurrasch, D.M.; Chan, J.A.; Schuurmans, C. RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J. Neurosci. 2014, 34, 2169–2190. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Castellana, D.; Klingbeil, P.; Hernández, I.C.; Vitacolonna, M.; Orlicky, D.J.; Roffler, S.R.; Brodt, P.; Zöller, M. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 2009, 11, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Royston, I.; Majda, J.; Baird, S.; Meserve, B.; Griffiths, J. Human T cell antigens defined by monoclonal antibodies: The 65,000-dalton antigen of T cells (T65) is also found on chronic lymphocytic leukemia cells bearing surface immunoglobulin. J. Immunol. 1980, 125, 725–731. [Google Scholar] [PubMed]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5–LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018, 143, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Royston, K.J.; Udayakumar, N.; Lewis, K.; Tollefsbol, T.O. A novel combination of withaferin A and sulforaphane inhibits epigenetic machinery, cellular viability and induces apoptosis of breast cancer cells. Int. J. Mol. Sci. 2017, 18, 1092. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, A.; Wiczk, A.; Kaczyńska, A.; Antosiewicz, J.; Herman-Antosiewicz, A. Sulforaphane inhibits growth of phenotypically different breast cancer cells. Eur. J. Nutr. 2013, 52, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Xu, C.; Chen, C.; Hebbar, V.; Kong, A.-N.T.J.C.C. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chem. Pharmacol. 2006, 57, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Flores-Rozas, H.; Kelman, Z.; Dean, F.B.; Pan, Z.-Q.; Harper, J.W.; Elledge, S.J.; O’Donnell, M.; Hurwitz, J. Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase delta holoenzyme. Proc. Natl. Acad. Sci. USA 1994, 91, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.-W.; Dashwood, W.-M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Parnaud, G.; Li, P.; Cassar, G.; Rouimi, P.; Tulliez, J.; Combaret, L.; Gamet-Payrastre, L.J. Mechanism of sulforaphane-induced cell cycle arrest and apoptosis in human colon cancer cells. Nutr. Cancer 2004, 48, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Jakubíková, J.; Sedlák, J.; Mithen, R.; Bao, Y. Role of PI3K/Akt and MEK/ERK signaling pathways in sulforaphane-and erucin-induced phase II enzymes and MRP2 transcription, G2/M arrest and cell death in Caco-2 cells. Biochem. Pharmacol. 2005, 69, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Pappa, G.; Strathmann, J.; Löwinger, M.; Bartsch, H.; Gerhäuser, C. Quantitative combination effects between sulforaphane and 3, 3′-diindolylmethane on proliferation of human colon cancer cells in vitro. Carcinogenesis 2007, 28, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.L.; Kala, R.; Tollefsbol, T. Mechanisms for the inhibition of colon cancer cells by sulforaphane through epigenetic modulation of microRNA-21 and human telomerase reverse transcriptase (hTERT) down-regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Okonkwo, A.; Mitra, J.; Johnson, G.S.; Li, L.; Dashwood, W.M.; Hegde, M.; Yue, C.; Dashwood, R.H.; Rajendran, P. Heterocyclic Analogs of Sulforaphane Trigger DNA Damage and Impede DNA Repair in Colon Cancer Cells: Interplay of HATs and HDACs. Mol. Nutr. Food Res. 2018, 62, e1800228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yang, G.; Tian, H.; Hu, Y.; Wu, S.; Geng, Y.; Lin, K.; Wu, W. Sulforaphane metabolites cause apoptosis via microtubule disruption in cancer. Endocr. Relat. Cancer 2018, 25, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Rausch, V.; Liu, L.; Kallifatidis, G.; Baumann, B.; Mattern, J.; Gladkich, J.; Wirth, T.; Schemmer, P.; Büchler, M.W.; Zöller, M. Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res. 2010, 70, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lew, K.L.; Xiao, H.; Herman-Antosiewicz, A.; Xiao, D.; Brown, C.K.; Singh, S.V. D, L-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis 2007, 28, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-H.; Kang, K.-S.; Kwak, M.-K. Effect of redox modulating NRF2 activators on chronic kidney disease. Molecules 2014, 19, 12727–12759. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Dashwood, R.H. Histone deacetylases as targets for dietary cancer preventive agents: Lessons learned with butyrate, diallyl disulfide, and sulforaphane. Curr. Drug Targets 2006, 7, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.-Y.; Khor, T.O.; Shu, L.; Kong, A.-N.T. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Chapple, S.J.; Siow, R.C.; Mann, G.E. Crosstalk between Nrf2 and the proteasome: Therapeutic potential of Nrf2 inducers in vascular disease and aging. Int. J. Biochem. Cell Biol. 2012, 44, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.; Wickramasekara, S.; Palomera-Sanchez, Z.; Black, C.; Maier, C.; Williams, D.; Dashwood, R.; Ho, E. SUV39H1/H3K9me3 attenuates sulforaphane-induced apoptotic signaling in PC3 prostate cancer cells. Oncogenesis 2014, 3, e131. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Higashi, S.; Kimura, H.; Yamamoto, H.; Mori, M.; Matsuura, S.; Matsuura, N. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013, 104, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.L.; Ciavattone, N.; Grun, D.; Adhikary, G.; Eckert, R.L. Sulforaphane reduces YAP/∆ Np63α signaling to reduce cancer stem cell survival and tumor formation. Oncotarget 2017, 8, 73407–73418. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Zhang, L.; Bao, Y.; Li, B.; He, C.; Gao, M.; Feng, X.; Xu, W.; Zhang, X.; Wang, S. Epithelial-mesenchymal transition, a novel target of sulforaphane via COX-2/MMP2, 9/Snail, ZEB1 and miR-200c/ZEB1 pathways in human bladder cancer cells. J. Nutr. Biochem. 2013, 24, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cha, Y.-N.; Surh, Y.-J. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 690, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Lin, A.H.; Liu, C.T.; Tsai, C.W.; Chang, I.S.; Chen, H.W.; Lii, C.K. Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFκB activation. Mol. Nutr. Food Res. 2013, 57, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.L.; Shi, M.; Tang, H.; Han, W.; Spivack, S.D. Candidate dietary phytochemicals modulate expression of phase II enzymes GSTP1 and NQO1 in human lung cells. J. Nutr. 2010, 140, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Ernst, I.M.; Wagner, A.E.; Schuemann, C.; Storm, N.; Hoppner, W.; Doring, F.; Stocker, A.; Rimbach, G. Allyl-, butyl- and phenylethyl-isothiocyanate activate Nrf2 in cultured fibroblasts. Pharmacol. Res. 2011, 63, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Qin, X.; Zhang, G.; Lu, X.; Zhu, Y.; Zhang, H.; Dai, B.; Shi, G.; Ye, D.J.T.B. Oxidized low-density lipoprotein is associated with advanced-stage prostate cancer. Tumor Biol. 2015, 36, 3573–3582. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-C.; Liao, Y.-C.; Wang, J.-Y.; Lin, Y.-C.; Chen, C.-H.; Juo, S.-H.H. Oxidized low-density lipoprotein is a common risk factor for cardiovascular diseases and gastroenterological cancers via epigenomical regulation of microRNA-210. Oncotarget 2015, 6, 24105–24118. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Dikovskaya, D.; Gaurilcikaite, E.; Knatko, E.V.; Healy, Z.R.; Mohan, H.; Koh, G.; Laurell, A.; Ball, G.; Olagnier, D.; et al. Transcription factors NRF2 and HSF1 have opposing functions in autophagy. Sci. Rep. 2017, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- Olagnier, D.; Lababidi, R.R.; Hadj, S.B.; Sze, A.; Liu, Y.; Naidu, S.D.; Ferrari, M.; Jiang, Y.; Chiang, C.; Beljanski, V.; et al. Activation of Nrf2 Signaling Augments Vesicular Stomatitis Virus Oncolysis via Autophagy-Driven Suppression of Antiviral Immunity. Mol. Ther. 2017, 25, 1900–1916. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.; Liu, Z.; Yao, J.; Patel, N.; Chen, J.; Wu, Y.; Ahn, E.E.; Fodstad, O.; Tan, M. Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). J. Biol. Chem. 2013, 288, 9165–9176. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic DNA-Mediated, STING-Dependent Pro-Inflammatory Gene Induction Necessitates canonical NF-κB activation Through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Xiao, Z.; Hood, B.L.; Dakshanamurthy, S.; Wang, X.; Govind, S.; Conrads, T.P.; Veenstra, T.D.; Chung, F.L. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J. Biol. Chem. 2008, 283, 22136–22146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Molecular mechanism of rapid cellular accumulation of anticarcinogenic isothiocyanates. Carcinogenesis 2001, 22, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.; Sahu, R.P.; Kandala, P.K.; Srivastava, S.K. Benzyl isothiocyanate–mediated inhibition of histone deacetylase leads to NF-κB turnoff in human pancreatic carcinoma cells. Mol. Cancer Ther. 2010, 1535–7163. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Singh, S.V. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis 2004, 25, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kawakami, M.; Yoshihiro, A.; Miyoshi, N.; Ohigashi, H.; Kawai, K.; Osawa, T.; Uchida, K. Involvement of the mitochondrial death pathway in chemopreventive benzyl isothiocyanate-induced apoptosis. J. Biol. Chem. 2002, 277, 8492–8499. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-N.; Kim, D.-H.; Kim, E.-H.; Lee, M.-H.; Kundu, J.K.; Na, H.-K.; Cha, Y.-N.; Surh, Y.-J. Sulforaphane inhibits phorbol ester-stimulated IKK-NF-κB signaling and COX-2 expression in human mammary epithelial cells by targeting NF-κB activating kinase and ERK. Cancer Lett. 2014, 351, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Nallasamy, P.; Si, H.; Babu, P.V.A.; Pan, D.; Fu, Y.; Brooke, E.A.; Shah, H.; Zhen, W.; Zhu, H.; Liu, D. Sulforaphane reduces vascular inflammation in mice and prevents TNF-α-induced monocyte adhesion to primary endothelial cells through interfering with the NF-κB pathway. J. Nutr. Biochem. 2014, 25, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.T.; ME, L.L. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 2016, 49, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Wu, K.; Wang, W.; Wang, S.; Lin, N.; Zhao, R.; Cassidy, A.; Bao, Y. Sulforaphane down-regulates COX-2 expression by activating p38 and inhibiting NF-κB-DNA-binding activity in human bladder T24 cells. Int. J. Oncol. 2009, 34, 1129–1134. [Google Scholar] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative stress in cancer and fibrosis: Opportunity for therapeutic intervention with antioxidant compounds, enzymes, and nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.L.; Sonis, S.T. Mechanisms of cellular fibrosis associated with cancer regimen-related toxicities. Front. Pharmacol. 2014, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Milczarek, M.; Misiewicz-Krzeminska, I.; Lubelska, K.; Wiktorska, K. Combination treatment with 5-fluorouracil and isothiocyanates shows an antagonistic effect in Chinese hamster fibroblast cells line-V79. Acta Pol. Pharm. 2011, 68, 331–342. [Google Scholar] [PubMed]
- Mohammed, E.D.; El-Naga, R.N.; Lotfy, R.A.; Al-Gendy, A.A.; El-Demerdash, E. Anti-fibrotic potential of a Matthiola arabica isothiocyanates rich fraction: Impact on oxidative stress, inflammatory and fibrosis markers. Die Pharm. 2017, 72, 614–624. [Google Scholar]
- Kim, J.; Bang, H.; Ahn, M.; Choi, Y.; Kim, G.O.; Shin, T. Allyl isothiocyanate reduces liver fibrosis by regulating Kupffer cell activation in rats. J. Vet. Med. Sci. 2018, 80, 893–897. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isothiocyanate | Benzyl Isothiocyanate (BITC) | Phenethyl Isothiocyanate (PEITC) | Sulforaphane (SFN) |
|---|---|---|---|
| Dietary Source | Cabbage, garden cress, Indian cress | Watercress/turnip | Broccoli, Brussels sprouts, cabbage |
| Precursor |  Glucotropaeolin |  Gluconasturtiin |  Glucoraphanin |
| Structure |  |  |  |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soundararajan, P.; Kim, J.S. Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers. Molecules 2018, 23, 2983. https://doi.org/10.3390/molecules23112983
Soundararajan P, Kim JS. Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers. Molecules. 2018; 23(11):2983. https://doi.org/10.3390/molecules23112983
Chicago/Turabian StyleSoundararajan, Prabhakaran, and Jung Sun Kim. 2018. "Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers" Molecules 23, no. 11: 2983. https://doi.org/10.3390/molecules23112983
APA StyleSoundararajan, P., & Kim, J. S. (2018). Anti-Carcinogenic Glucosinolates in Cruciferous Vegetables and Their Antagonistic Effects on Prevention of Cancers. Molecules, 23(11), 2983. https://doi.org/10.3390/molecules23112983