
Microwave Irradiation Assists the Synthesis of a Novel Series of bis-Arm s-Triazine Oxy-Schiff Base and Oxybenzylidene Barbiturate Derivatives


Abstract
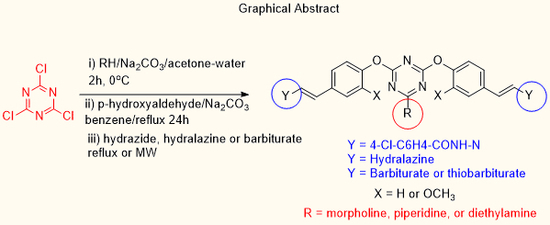
{kind=link}
{kind=link}
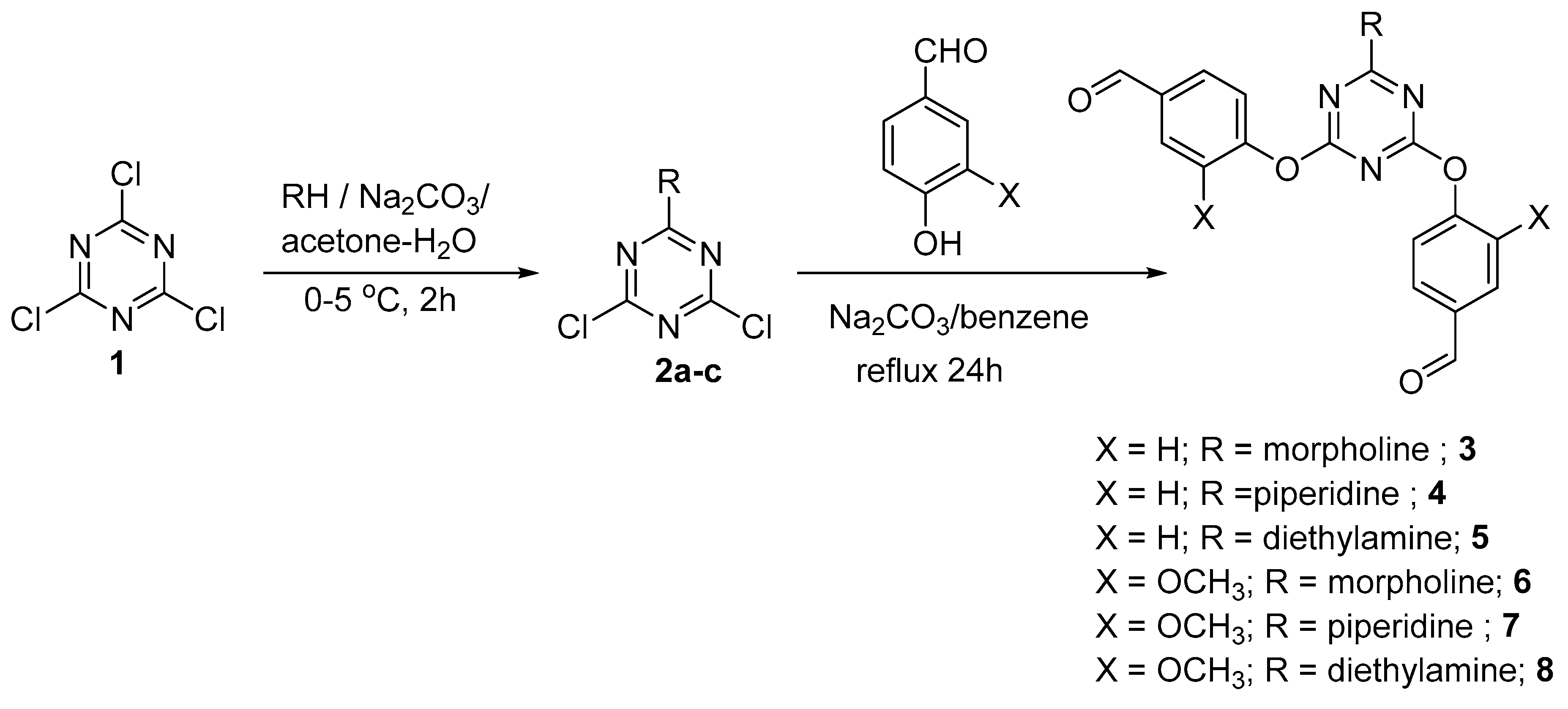{kind=link}
{kind=link}
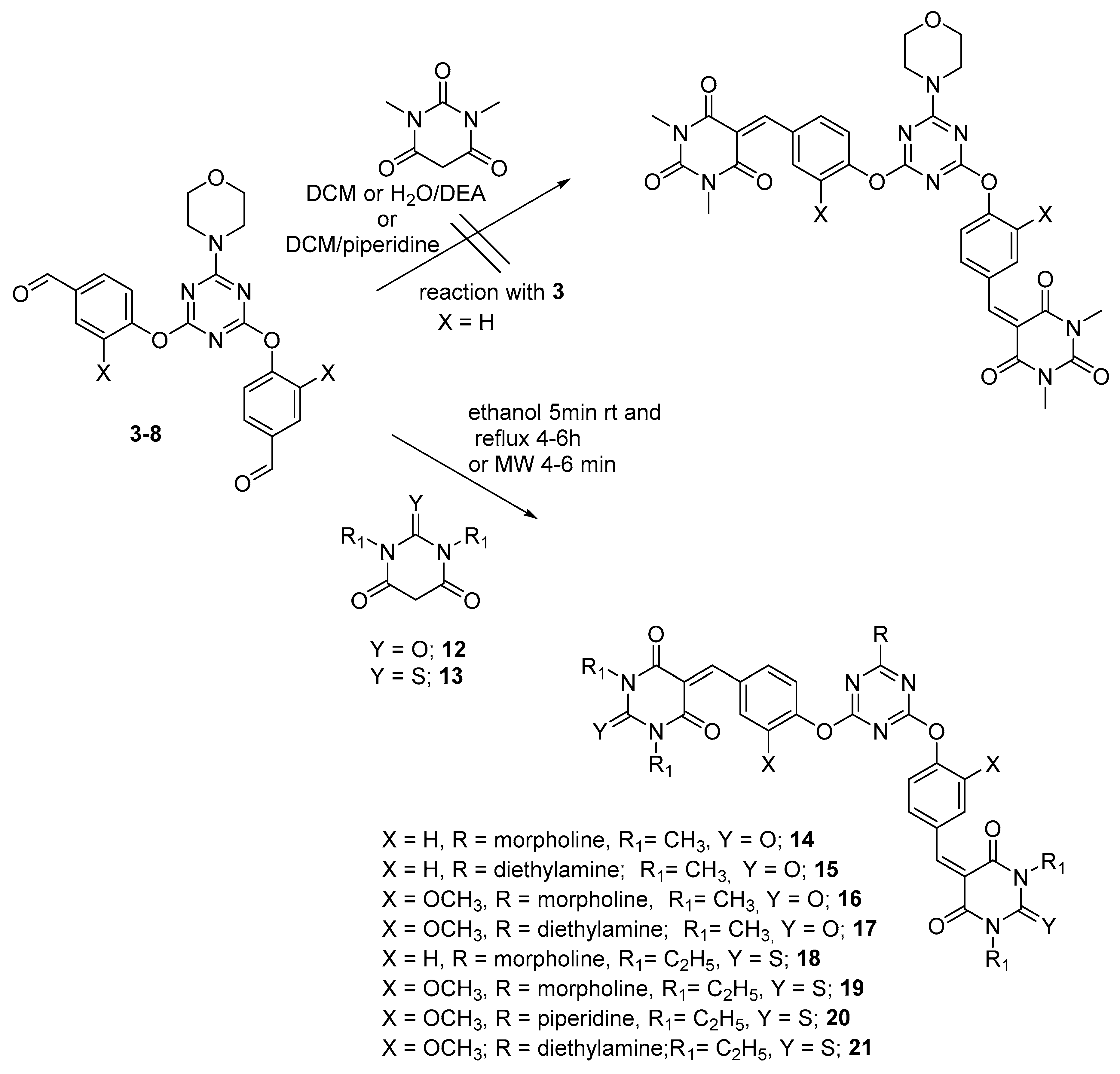{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Method for Synthesis of Dipodal 3–8
3.3. General Procedure for Preparation of Bis-Arm s-Triazine oxy-Schiff Base Derivatives 9a–c, 10a–c, and 11a–c
3.4. General Method for the Reaction of Dipodal 3–8 with Barbiturate Derivatives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Stadler, A.; Kappe, C.O. Microwave- Assisted Organic Synthesis; Lidström, P., Tierny, J.P., Eds.; Blackwell: Oxford, UK, 2005; Chapter 7; pp. 175–219. [Google Scholar]
- Caddick, S. Microwave assisted organic reactions. Tetrahedron 1995, 51, 10403–10432. [Google Scholar] [CrossRef]
- Hayes, B.L. Recent advances in microwave assisted synthesis. Aldrichim. Acta 2004, 27, 66–76. [Google Scholar]
- Moseley, J.D.; Kappe, C.O. A critical assessment of the greenness and energy efficiency of microwave-assisted organic synthesis. Green Chem. 2011, 13, 794–806. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinge, D. Controlled microwave heating in modern organic synthesis: Highlights from the 2004–2008. Mol. Divers. 2009, 13, 71–193. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Damm, M. Parallel microwave chemistry in silicon carbide microtiter platforms: A review. Mol. Divers. 2012, 16, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liu, Y.; Zhao, Y.; Wang, H.; Tan, L.; Fan, W.; Gong, P. Synthesis and biological evaluation of novel 6-hydrazinyl-2,4-bismorpholino pyrimidine and 1,3,5-triazine derivatives as potential antitumor agents. Arch. Pharm. 2012, 345, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Iino, Y.; Karakida, T.; Sugamata, N.; Andoh, T.; Takei, H.; Takahashi, M.; Yaguchi, S.; Matsuno, T.; Takehara, M.; Sakato, M.; et al. Antitumor effects of SEF19, a new nonsteroidal aromatase inhibitor, on 7,12-dimethyl benz[a]anthracene-induced mammary tumors in rats. Anticancer Res. 1998, 18, 171–176. [Google Scholar] [PubMed]
- Matsuno, T.; Kato, M.; Sasahara, H.; Watanbe, T.; Inaba, M.; Takahashi, M.; Yaguchi, S.-I.; Yoshioka, K.; Sakato, M.; Kawashima, S. Synthesis and antitumor activity of benzimidazolyl-1, 3, 5-triazine and benzimidazolyl pyrimidine derivatives. Chem. Pharm. Bull. 2000, 48, 1778–1781. [Google Scholar] [CrossRef] [PubMed]
- Smolin, E.M.; Rapoport, L. Chemistry of Heterocyclic Compounds: s-Triazines and Derivatives, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; Volume 13. [Google Scholar]
- Giacomelli, G.; Porcheddu, A. Comprehensive Heterocyclic Chemistry III; Turnbull, K., Ed.; Elsevier Science & Technology: Oxford, UK, 2008; Volume 9, p. 197. [Google Scholar]
- Blotny, G. Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis. Tetrahedron 2006, 62, 9507–9522. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sączewski, F. Synthesis and antitumor activity of novel 2-amino-4-(3,5,5-trimethyl-2-pyrazolino)-1,3,5-triazine derivatives. Eur. J. Med. Chem. 2002, 37, 709–720. [Google Scholar] [CrossRef]
- Desai, N.C.; Makwana, A.H.; Senta, R.D. Synthesis, characterization and antimicrobial activity of some novel 4-(4-(arylamino)-6-(piperidin-1-yl)-1,3,5-triazine-2-ylamino)-N-(pyrimidin-2-yl)benzene sulfonamides. J. Saudi Chem. Soc. 2016, 20, 686–694. [Google Scholar] [CrossRef]
- Khan, F.G.; Yadav, M.V.; Sagar, A.D. Synthesis, characterization, and antimicrobial evaluation of novel trichalcones containing core s-triazine moiety. Med. Chem. Res. 2014, 23, 2633–2638. [Google Scholar] [CrossRef]
- Freeman, A.W.; Vreekamp, R.; Fréchet, J.M.J. Book of Abstracts. In Proceedings of the 214th ACS National Meeting, Las Vegas, NV, USA, 7–11 September 1997; PMSE-128; American Chemical Society: Washington, DC, USA, 1997. [Google Scholar]
- Afonso, C.A.M.; Lourenço, N.M.T.; Rosatella, A.A. Synthesis of 2,4,6-tri-substituted-1,3,5-triazines. Molecules 2002, 11, 81–102. [Google Scholar] [CrossRef]
- Cho, S.Y.; Chang, Y.; Kim, J.S.; Lee, S.C.; Kim, C. Dendrimers based on [1,3,5]-triazines. Macromol. Chem. Phys. 2001, 202, 263–269. [Google Scholar] [CrossRef]
- Kataoka, Y.; Kondo, T. Changing cellulose crystalline structure in forming wood cell walls. Macromolecules 1996, 29, 6353–6358. [Google Scholar] [CrossRef]
- Steffensen, M.B.; Hollink, E.; Kuschel, F.; Bauer, M.; Simanek, E.E. Dendrimers based on [1,3,5]-triazines. J. Polym. Sci. A Polym. Chem. 2006, 44, 3411–3433. [Google Scholar] [CrossRef] [PubMed]
- White, W.; Hudson, Z.M.; Feng, X.-D.; Han, S.; Lu, Z.-H.; Wang, S. Linear and star-shaped benzimidazolyl derivatives: Syntheses, photophysical properties and use as highly efficient electron transport materials in OLEDs. Dalton Trans. 2010, 39, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Machakanur, S.S.; Patil, B.R.; Badiger, D.S.; Bakale, R.P.; Gudasi, K.B.; Bligh, S.W.A. Synthesis, characterization and anticancer evaluation of novel tri-arm star shaped 1,3,5-triazine hydrazones. J. Mol. Str. 2012, 1011, 121–127. [Google Scholar] [CrossRef]
- Tahmassebi, D.C.; Sasaki, T.J. Synthesis of a new trialdehyde template for molecular imprinting. J. Org. Chem. 1994, 59, 679–681. [Google Scholar] [CrossRef]
- Koc, Z.E.; Ucan, H.I. Complexes of iron(III) salen and saloph Schiff bases with bridging 2,4,6-tris(2,5-dicarboxyphenylimino-4-formylphenoxy)-1,3,5-triazine and 2,4,6-tris(4-carboxyphenylimino-4′-formylphenoxy)-1,3,5-triazine. Trans. Metal. Chem. 2007, 32, 597–602. [Google Scholar] [CrossRef]
- Koc, Z.E.; Bingol, H.; Saf, A.O.; Torlak, E.; Coskun, A. Synthesis of novel tripodal-benzimidazole from 2, 4, 6-tris(p-formylphenoxy)-1,3,5-triazine: Structural, electrochemical and antimicrobial studies. J. Hazard. Mater. 2010, 183, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Celikbilek, S.; Koc, Z.E. Investigation of Dipodal oxy-Schiff base and its salen and salophen Fe(III)/Cr(III)/Mn(III) Schiff bases (N2O2) caped complexes and their magnetic and thermal behaviors. J. Mol. Struct. 2014, 1065–1066, 205–209. [Google Scholar] [CrossRef]
- Jarrahpour, A.; Khalili, D.; De Clercq, E.; Salmi, C.; Brunel, J.M. Synthesis, antibacterial, antifungal and antiviral activity evaluation of some new bis-Schiff bases of isatin and their derivatives. Molecules 2007, 12, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Al-Rashida, M.; Uroos, M.; Ali, A.S.; Khan, K.M. Schiff bases in medicinal chemistry: A patent review (2010-2015). Expert Opin. Ther. Patents 2017, 27, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Tiwari, A.K.; Singh, S.; Shukla, G.; Mishra, P.; Chandra, H.; Mishra, A.K. Synthesis, characterization and biological activity of Schiff base analogues of indole-3-carboxaldehyde. Eur. J. Med. Chem. 2008, 43, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Raghav, N. Biological activities of hydrazones: A review. Int. J. Pharm. Pharmaceut. Sci. 2011, 3, 26–32. [Google Scholar]
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.M.; El-Faham, A. Low temperature X-ray structure analyses combined with NBO studies of a new heteroleptic octa-coordinated Holmium(III) complex with N,N,N-tridentate hydrazono-phthalazine-type ligand. J. Mol. Struct. 2018, 1157, 222–229. [Google Scholar] [CrossRef]
- Soliman, S.M.; Albering, J.H.; Farooq, M.; Wadaan, M.A.M.; El-Faham, A. Synthesis, structural and biological studies of two new Co(III) complexes with tridentate hydrazone ligand derived from the antihypertensive drug hydralazine. Inorg. Chim. Acta 2017, 466, 16–29. [Google Scholar] [CrossRef]
- Uhlmann, C.; Fröscher, W. Low risk of development of substance dependence for barbiturates and clobazam prescribed as antiepileptic drugs: Results from a questionnaire study. CNS Neurosci. Ther. 2009, 15, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Breyholz, H.J.; Wagner, S.; Faust, A.; Riemann, B.; Höltke, C.; Hermann, S.; Schober, O.; Schäfers, M.; Kopka, K. Radiofluorinated pyrimidine-2,4,6-triones as molecular probes for noninvasive MMP-targeted imaging. Chem. Med. Chem. 2010, 5, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, F.; Berger, S.T.A.; Remennikov, G.Y.; Polborn, K.; Mayr, H. Electrophilicity of 5-benzylidene-1,3-dimethylbarbituric and -thiobarbituric acids. J. Org. Chem. 2007, 72, 9170–9180. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Villar, J.D.; Oliveira, S.C.G. Synthesis and mechanism of formation of oxadeazaflavines by microwave thermal cyclization of ortho-halobenzylidene barbiturates. J. Braz. Chem. Soc. 2011, 20, 2101–2107. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, J.; Qin, C.; Thomson, L.M.; Parrish, A.R.; Safe, S.H.; Simanek, E.E. Triazine Dendrimers for Drug Delivery: Evaluation of solubilization properties, activity in cell culture, and in vivo toxicity of a candidate vehicle. Supramol. Chem. 2003, 15, 607–616. [Google Scholar] [CrossRef]
- Lim, J.; Simanek, E.E. Triazine dendrimers as drug delivery systems: From synthesis to therapy. Adv. Drug Deliv. Rev. 2012, 64, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ghabbour, H.; Khan, S.T.; de la Torre, B.G.; Albericio, F.; El-Faham, A. Novel pyrazolyl-s-triazine derivatives, molecular structure and antimicrobial activity. J. Mol. Struct. 2017, 1145, 244–253. [Google Scholar] [CrossRef]
- Padalkar, V.S.; Patil, V.S.; Nagaiyan, S. Synthesis of novel fluorescent 1,3,5-trisubstituted triazine derivatives and Photophysical property evaluation of fluorophores and its BSA. Chem. Cent. J. 2011, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Soliman, S.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.-K.; Wadood, A.; Warad, I.; Sloop, J.C. New diethyl ammonium salt of thiobarbituric acid derivative: Synthesis, molecular structure investigations and docking studies. Molecules 2015, 20, 20642–20658. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Majid, A.M.; Lotfy, G.; Arshad, F.; Yousuf, S.; Choudhary, M.I.; Ashraf, S.; Ul-Haq, Z. Synthesis and dynamics studies of barbituric acid derivatives as urease inhibitors. Chem. Cent. J. 2015, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jursic, B.S. A simple method for knoevenagel condensation of α,β-conjugated and aromatic aldehydes with barbituric acid. J. Heterocycl. Chem. 2001, 38, 655–657. [Google Scholar] [CrossRef]
- Kim, E.G. Method for Preparing Fluorescent Materials Containing Triazine Groups. Repub. Korea Patent KR 796988, B1 20080122, 2008. [Google Scholar]
- Wang, S.-J.; Lu, Z.-R.; Dong, X.; Chen, B.; Hua, C.-W.; Gou, X.-F.; Zhao, J.-L. Synthesis and recognition of metal ions of Schiff base macrocyclic compounds of 1,3,5-triazine. Chin. J. Org. Chem. 2015, 37, 739–741, 750. [Google Scholar]
- Li, X.; Hua, C.; Gou, X.; Zhao, J.; Chen, B. Synthesis and characterization of novel Schiff base macrocyclic compounds of 1,3,5-triazine. Chin. J. Org. Chem. 2012, 32, 939–942. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlous, K.A.; Almarhoon, Z.; Badjah-Hadj-Ahmed, A.-Y.; AL Othman, Z.A.; El-Faham, A. Microwave Irradiation Assists the Synthesis of a Novel Series of bis-Arm s-Triazine Oxy-Schiff Base and Oxybenzylidene Barbiturate Derivatives. Molecules 2018, 23, 2976. https://doi.org/10.3390/molecules23112976
Dahlous KA, Almarhoon Z, Badjah-Hadj-Ahmed A-Y, AL Othman ZA, El-Faham A. Microwave Irradiation Assists the Synthesis of a Novel Series of bis-Arm s-Triazine Oxy-Schiff Base and Oxybenzylidene Barbiturate Derivatives. Molecules. 2018; 23(11):2976. https://doi.org/10.3390/molecules23112976
Chicago/Turabian StyleDahlous, Kholood A., Zainab Almarhoon, Ahmed-Yacine Badjah-Hadj-Ahmed, Zeid A. AL Othman, and Ayman El-Faham. 2018. "Microwave Irradiation Assists the Synthesis of a Novel Series of bis-Arm s-Triazine Oxy-Schiff Base and Oxybenzylidene Barbiturate Derivatives" Molecules 23, no. 11: 2976. https://doi.org/10.3390/molecules23112976
APA StyleDahlous, K. A., Almarhoon, Z., Badjah-Hadj-Ahmed, A.-Y., AL Othman, Z. A., & El-Faham, A. (2018). Microwave Irradiation Assists the Synthesis of a Novel Series of bis-Arm s-Triazine Oxy-Schiff Base and Oxybenzylidene Barbiturate Derivatives. Molecules, 23(11), 2976. https://doi.org/10.3390/molecules23112976