Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program





Abstract
1. Introduction
2. Results and Discussion
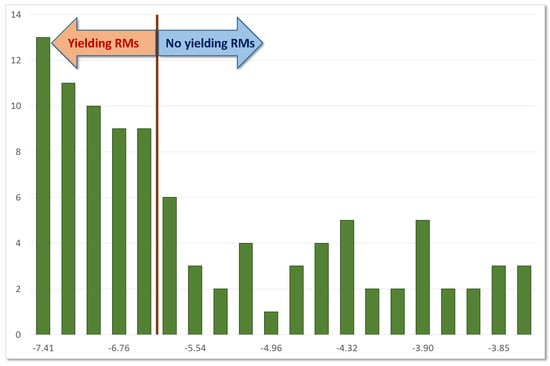
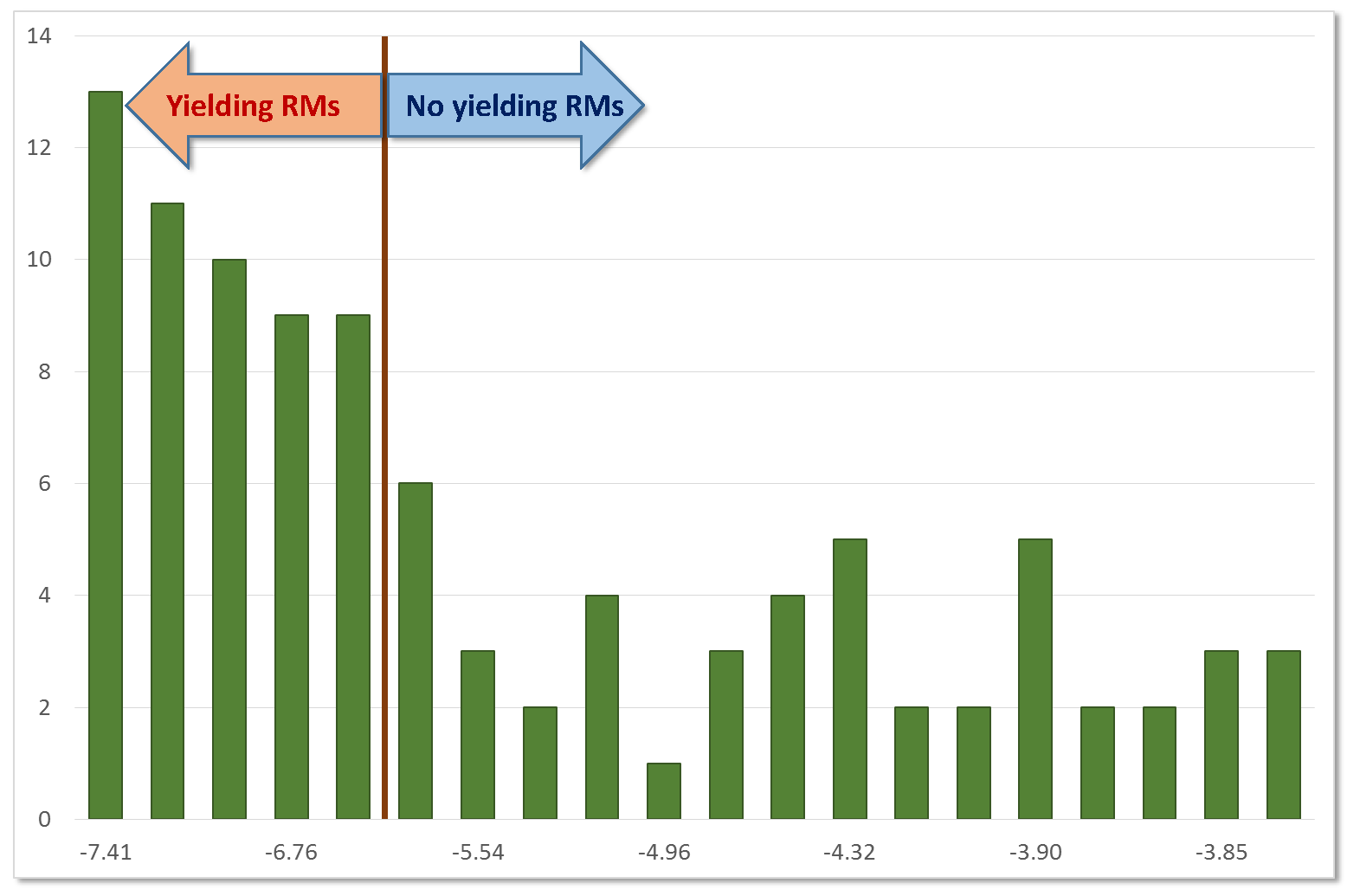
2.1. Calibration (Preliminary) Analyses
2.2. Predictive Models
2.2.1. Classifiers for Substrates Yielding RMs in the First and Any Generation
2.2.2. Classifiers for Specific Metabolic Reactions
2.2.3. Comparison of the Best Model with Weka Results
2.3. General Applicability in Machine Learning Analyses
3. Methods
3.1. Dataset Set-Up
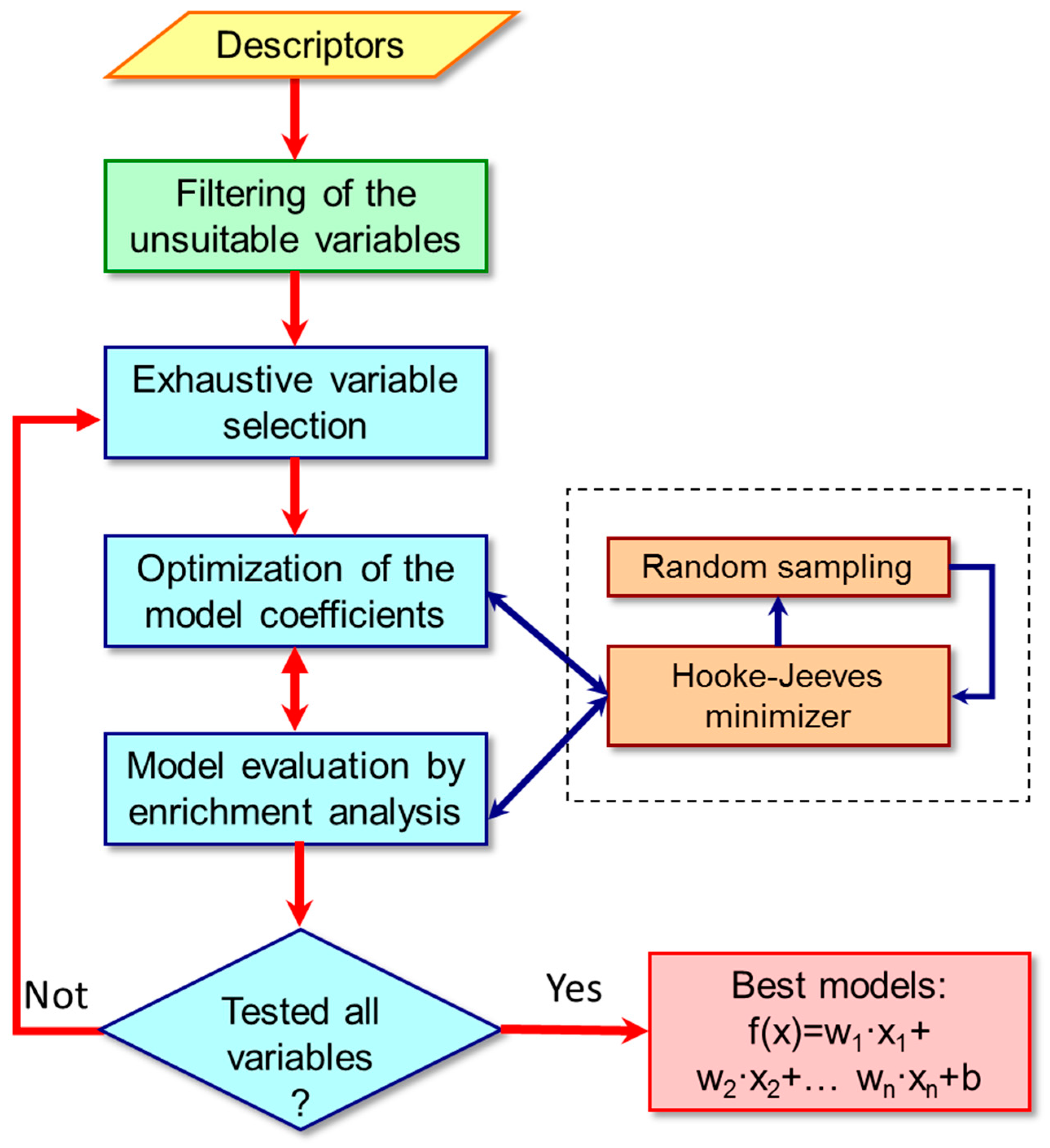
3.2. Classification Algorithm
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | asymmetry index |
| EFO | enrichment factor optimization |
| HOMO | highest occupied molecular orbital |
| LDA | linear discriminant analysis |
| LUMO | lowest unoccupied molecular orbital |
| MCC | Matthews correlation coefficients |
| RM | reactive metabolite |
| ROC | receiver operating characteristic |
References
- Cho, T.; Uetrecht, J. How reactive metabolites induce an immune response that sometimes leads to an idiosyncratic drug reaction. Chem. Res. Toxicol. 2017, 30, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Luch, A. On the impact of the molecule structure in chemical carcinogenesis. EXS 2009, 99, 151–179. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, D.K.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Ford, K.A. Role of electrostatic potential in the in silico prediction of molecular bioactivation and mutagenesis. Mol. Pharm. 2013, 10, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.J.; Sturino, C. Managing the liabilities arising from structural alerts: A safe philosophy for medicinal chemists. Curr. Med. Chem. 2011, 18, 3116–3135. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.S.; Miwa, G.T. Bioactivation of drugs: Risk and drug design. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Patlewicz, G.; Fitzpatrick, J.M. Current and future perspectives on the development, evaluation, and application of in silico approaches for predicting toxicity. Chem. Res. Toxicol. 2016, 29, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Krasowski, M.D.; Ekins, S. Using cheminformatics to predict cross reactivity of “designer drugs” to their currently available immunoassays. J. Cheminform. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Aided. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Mazzolari, A.; Vistoli, G.; Testa, B. MetaQSAR: An integrated database engine to manage and analyze metabolic data. J. Med. Chem. 2018, 61, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.C.; Frank, E. Introducing machine learning concepts with WEKA. Methods Mol. Biol. 2016, 1418, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Villa, L.; Vistoli, G. Modeling of binding modes and inhibition mechanism of some natural ligands of farnesyl transferase using molecular docking. J. Med. Chem. 2002, 45, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Coulson, C.A.; Longuet-Higgins, H.C. The electronic structure of conjugated systems. II. Unsaturated hydrocarbons and their hetero-derivatives. Proc. Roy. Soc. Lond. A 1947, 192, 16–32. [Google Scholar] [CrossRef]
- Schüürmann, G. QSAR analysis of the acute fish toxicity of organic phosphorothionates using theoretically derived molecular descriptors. Environ. Toxicol. Chem. 1990, 9, 417–428. [Google Scholar] [CrossRef]
- Gareth, J.M.; Hastie, T.J. Functional linear discriminant analysis for irregularly sampled curves. J. R. Stat. Soc. B 2001, 63, 533–550. [Google Scholar] [CrossRef]
- Aha, D.; Kibler, D. Instance-based learning algorithms. Mach. Learn. 1991, 6, 37–66. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Quinlan, R. C4.5: Programs for Machine Learning; Morgan Kaufmann Publishers: San Mateo, CA, USA, 1993; ISBN 1-55860-238-0. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Liu, H.; Cocea, M. Semi-random partitioning of data into training and test sets in granular computing context. Granul. Comput. 2017, 2, 357–386. [Google Scholar] [CrossRef]
- Detrano, R.; Janosi, A.; Steinbrunn, W.; Pfisterer, M.; Schmid, J.J.; Sandhu, S.; Guppy, K.H.; Lee, S.; Froelicher, V. International application of a new probability algorithm for the diagnosis of coronary artery disease. Am. J. Cardiol. 1989, 64, 304–310. [Google Scholar] [CrossRef]
- Gorman, R.P.; Sejnowski, T.J. Analysis of hidden units in a layered network trained to classify sonar targets. Neural Netw. 1988, 1, 75–89. [Google Scholar] [CrossRef]
- Pouriyeh, S.; Vahid, S.; Sannino, G.; De Pietro, G.; Arabnia, H.; Gutierrez, J. A comprehensive investigation and comparison of machine learning techniques in the domain of heart disease. In Proceedings of the 22nd IEEE Symposium on Computers and Communications (ISCC 2017), Heraklion, Greece, 3–6 July 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 204–207. [Google Scholar]
- Karayilan, T.; Kiliç, O. Prediction of heart disease using neural network. In Proceedings of the 2nd International Conference on Computer Science and Engineering (UBMK 2017), Antalya, Turkey, 5–8 October 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 719–723. [Google Scholar]
- Stewart, J.J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Mazzolari, A.; Ricci, C.; Vistoli, G. Enhancing the reliability of GPCR models by accounting for flexibility of their pro-containing helices: The case of the human mAChR1 receptor. Mol. Inform. 2015, 34, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Doane, D.P.; Seward, L.E. Measuring skewness: A forgotten statistic? J. Stat. Educ. 2011, 19, 1–18. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
| Cluster Size | Sampling Cycles | Variables | EF Cut-Off Top 5% | Ionization State | Mean Top 1% | Mean Top 10% | Best Top 10% |
|---|---|---|---|---|---|---|---|
| 10 | 12 | 3 | 2.0 | N | 82.2 | 25.95 | 32 |
| 20 | 12 | 3 | 2.0 | N | 68.9 | 28.2 | 34 |
| 40 | 12 | 3 | 2.0 | N | 59.4 | 32.7 | 42 |
| 60 | 12 | 3 | 2.0 | N | 58.3 | 34.5 | 45 |
| 80 | 12 | 3 | 2.0 | N | 55.7 | 36.1 | 46 |
| 100 | 12 | 3 | 2.0 (12) | N | 47.8 | 38.2 | 46 |
| 100 | 6 | 3 | 2.0 | N | 48.8 | 38.2 | 47 |
| 100 | 24 | 3 | 2.0 | N | 47.2 | 37.9 | 46 |
| 100 | 12 | 3 | 2.5 (18) | N | 44.4 | 38.1 | 45 |
| 100 | 12 | 3 | 1.5 (6) | N | 48.8 | 36.4 | 45 |
| 100 | 12 | 3 | 1.0 (2) | N | 49.4 | 32.5 | 43 |
| 100 | 12 | 3 | 0.0 (0) | N | 51.1 | 32.7 | 43 |
| 100 | 12 | 1 | 2.0 | N | 37.4 | 25.1 | 30 |
| 100 | 12 | 2 | 2.0 | N | 49.4 | 32.2 | 40 |
| 100 | 12 | 4 | 2.0 | N | 49.9 | 39.7 | 45 |
| 100 | 12 | 5 | 2.0 | N | 51.1 | 41.6 | 47 |
| 100 | 12 | 6 | 2.0 | N | 55.5 | 43.6 | 48 |
| 100 | 12 | 8 | 2.0 | N | 56.7 | 44.3 | 48 |
| 100 | 12 | 10 | 2.0 | N | 57.2 | 45.5 | 48 |
| 100 | 12 | 2 | 2.0 | I | 44.4 | 34.3 | 42 |
| 100 | 12 | 3 | 2.0 | I | 47.2 | 39.4 | 46 |
| 100 | 12 | 4 | 2.0 | I | 49.4 | 40.6 | 47 |
| 100 | 12 | 5 | 2.0 | I | 50.2 | 42.2 | 47 |
| 100 | 12 | 6 | 2.0 | I | 54.4 | 44.0 | 48 |
| Mod. | Gen./React. | State | Cluster Size | Equation | Statistics |
|---|---|---|---|---|---|
| 1 | First | N | 70 | 1.00 HBT + 2.47 Lipole − 0.0001 Electronic_Energy + 0.13 Dipole + + 2.55 Dn_Total − 2.73 De_Total | Precision = 0.42 Accuracy = 0.85 MCC = 0.33 |
| 2 | First | I | 70 | 1.00 Rotors − 1.55 HBA + 5.09 Lipole − 0.0018 Electronic_Energy + +5.40 Dn_Total − 5.22 PiS_Total | Precision = 0.35 Accuracy = 0.81 MCC = 0.24 |
| 3 | All | N | 140 | 1.00 HBA + 1.09 Lipole − 0.0089 Heat_Formation + 0.070 Filled_Levels + − 2.03 De_Total + 4.47 PiS_Total | Precision = 0.42 Accuracy = 0.77 MCC = 0.28 |
| 4 | All | I | 140 | 1.00 Lipole − 0.033 PSA − 0.0059 ASA − 0.0004 Electronic_Energy + − 0.23 De_Total + 2.04 PiS_Total | Precision = 0.46 Accuracy = 0.74 MCC = 0.29 |
| 5 | Csp2/Csp ox | N | 30 | −1.00 Angles + 19.13 Rotors − 0.43 HBA + 15.47 HBT − 9.89 Impropers + + 21.32 Lipole | Precision = 0.67 Accuracy = 0.83 MCC = 0.55 |
| 6 | Quinone ox | N | 20 | 1.00 Angles + 1.07 Rotors + 68.34 Radius_Gyration − 8.38 HBA + − 30.28 HBD − 1.09 ASA | Precision = 0.63 Accuracy = 0.87 MCC = 0.54 |
| 7 | NH/NOH ox | N | 20 | −1.00 HBD + 0.041 Impropers − 0.15 Dipole − 0.0007 E_HOMO + + 0.68 Mulliken_Electronegativity + − 0.46 Schuurmann_alpha | Precision = 0.63 Accuracy = 0.87 MCC = 0.54 |
| 8 | Csp2/Csp Ox | I | 30 | −1.00 Rotors − 10.12 HBA + 1.47 HBD + − 1.36 Impropers + 1.04 PSA + + 0.40 E_LUMO | Precision = 0.61 Accuracy = 0.78 MCC = 0.46 |
| 9 | Quinone ox | I | 20 | 1.00 Angles + 1.53 Rotors + + 14.46 Radius_Gyration − 0.42 HBA + − 14.65 HBD − 0.46 ASA | Precision = 0.67 Accuracy = 0.83 MCC = 0.55 |
| 10 | NH/NOH ox | I | 20 | −1.00 HBD + 0.0083 Impropers + − 0.20 Lipole + 0.035 LogPMLP + + 0.015 Dipole + 0.14 Ionization_Potential | Precision = 0.70 Accuracy = 0.87 MCC = 0.61 |
| 11 | Heart data | N/A | 75 | −1.00 Pain + + 0.063 maximum_heart_rate_achieved + − 0.47 exercise_induced_angina + − 2.07 oldpeak + − 2.68 number_of_major_vessels + − 1.54 thal | Precision = 0.86 Accuracy = 0.87 MCC = 0.71 |
| Algorithm | MCC | Algorithm | MCC |
|---|---|---|---|
| Mod. 1 | 0.33 (21) | IterativeClass | 0.24 |
| BayesNet | 0.12 | RandomSubspace | 0.16 |
| FLDA | 0.25 | DecisionTable | 0.13 |
| LDA | 0.17 | JRip | 0.14 |
| Logistic | 0.11 | PART | 0.19 |
| Multilayer | 0.30 (7) | DecisionStump | 0.13 |
| IBk | 0.33 (14) | J48 | 0.33 (17) |
| Kstar | 0.27 | LMT | 0.17 |
| LWL | 0.13 | RandomForest | 0.37 (9) |
| AdaBoostM1 | 0.13 | RandomTree | 0.16 |
| Bagging | 0.21 | REPTree | 0.14 |
| Regression | 0.27 | LogitBoost | 0.18 |
| FilteredClass | 0.16 | Randomcommitee | 0.32 (13) |
| A1DE | 0.14 | NNge | 0.30 (7) |
| CHIRP | 0.23 | ExtraTree | 0.25 |
| Dataset | Attributes | Instances | Accuracy | |||||
|---|---|---|---|---|---|---|---|---|
| C4.5 | NB | K-NN | EFO (0.0) | EFO (1.0) | EFO (1.5) | |||
| Sonar | 60 | 208 | 0.68 | 0.71 | 0.84 | - | 0.76 | 0.69 |
| Heart | 13 | 270 | 0.74 | 0.86 | 0.59 | 0.87 | 0.73 | 0.73 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzolari, A.; Vistoli, G.; Testa, B.; Pedretti, A. Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program. Molecules 2018, 23, 2955. https://doi.org/10.3390/molecules23112955
Mazzolari A, Vistoli G, Testa B, Pedretti A. Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program. Molecules. 2018; 23(11):2955. https://doi.org/10.3390/molecules23112955
Chicago/Turabian StyleMazzolari, Angelica, Giulio Vistoli, Bernard Testa, and Alessandro Pedretti. 2018. "Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program" Molecules 23, no. 11: 2955. https://doi.org/10.3390/molecules23112955
APA StyleMazzolari, A., Vistoli, G., Testa, B., & Pedretti, A. (2018). Prediction of the Formation of Reactive Metabolites by A Novel Classifier Approach Based on Enrichment Factor Optimization (EFO) as Implemented in the VEGA Program. Molecules, 23(11), 2955. https://doi.org/10.3390/molecules23112955