Investigation of New Orexin 2 Receptor Modulators Using In Silico and In Vitro Methods

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
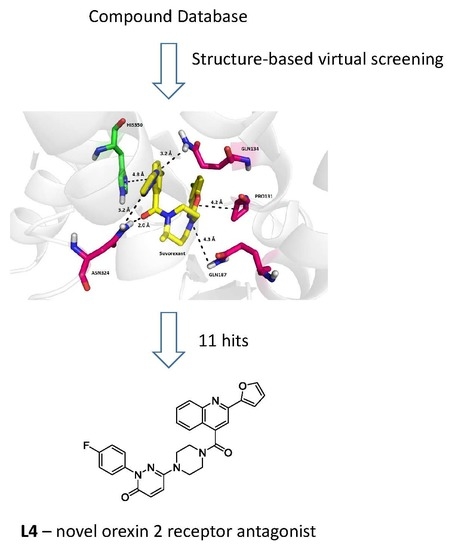
1. Introduction
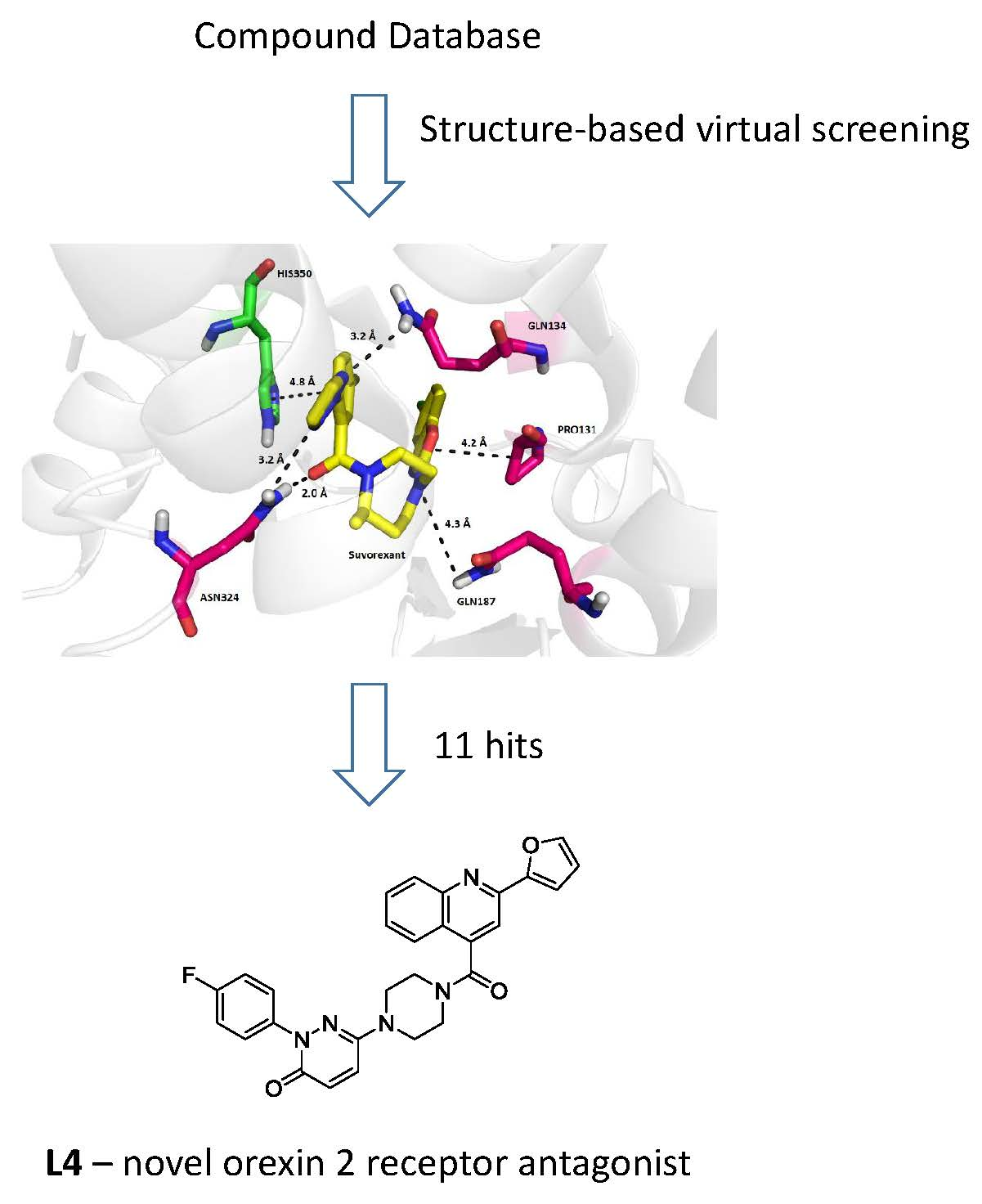
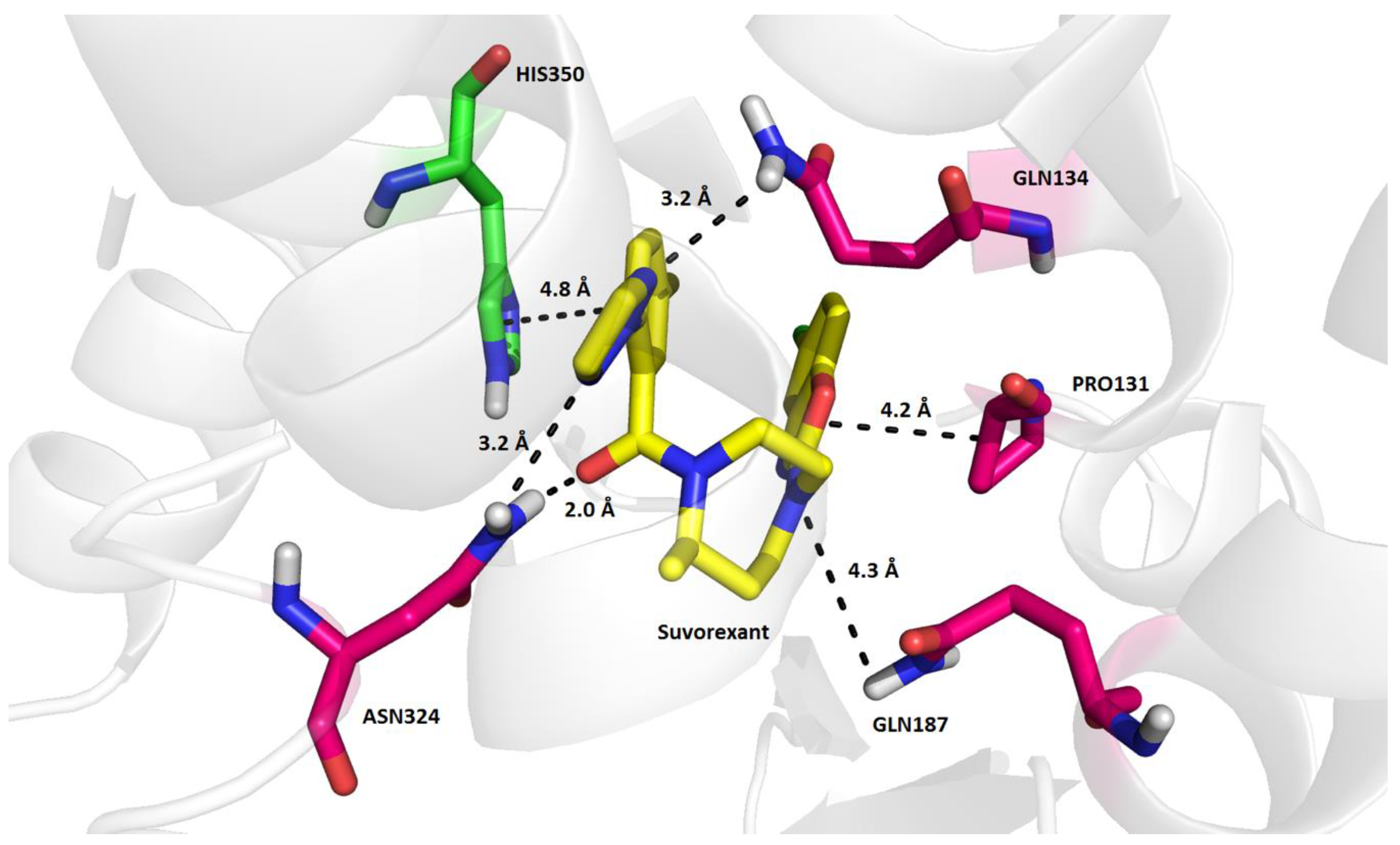
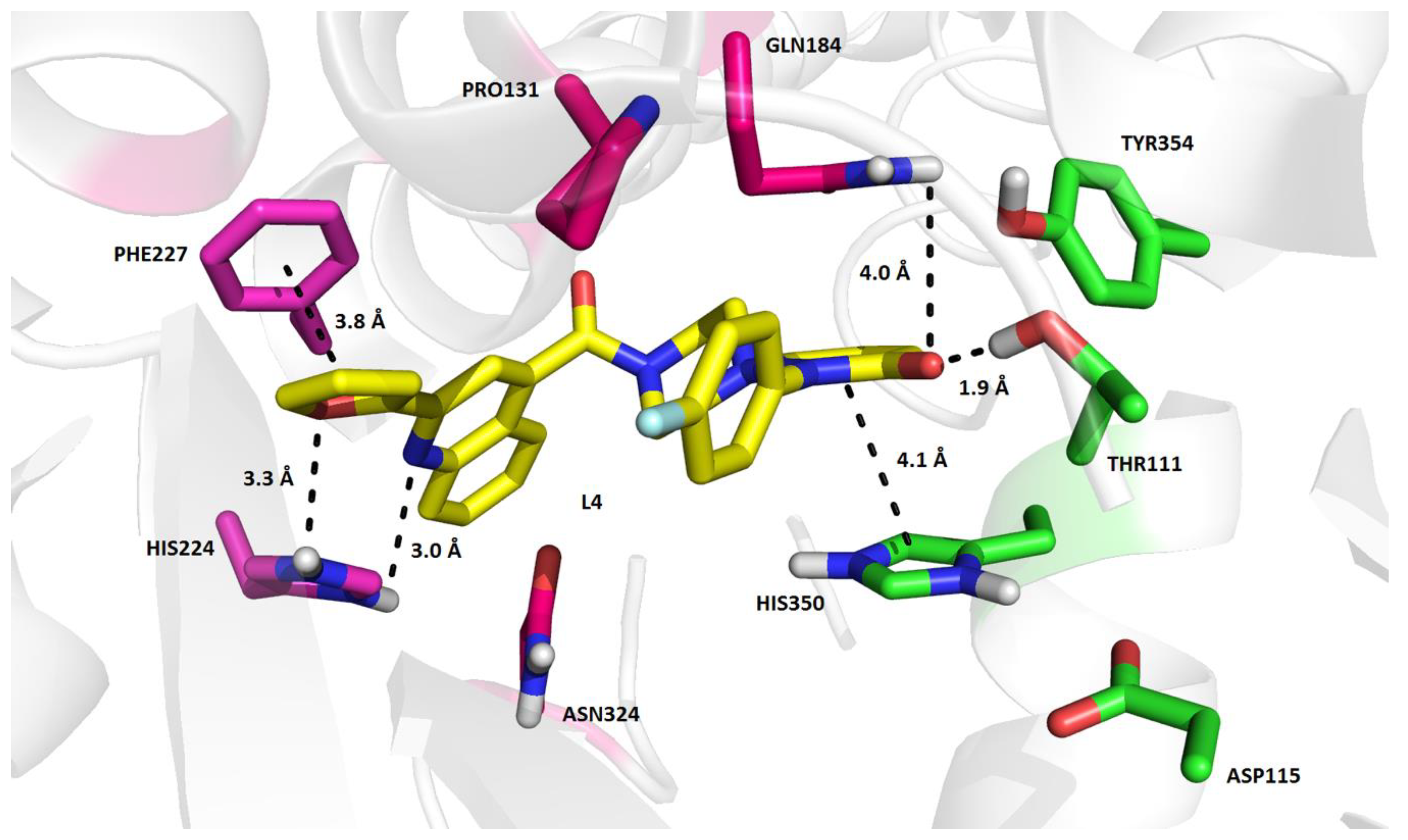
2. Results and Discussion
3. Materials and Methods
3.1. Structure-Based Virtual Screening
3.2. Chemicals
3.3. Cell Cultures
3.4. OX2R Calcium Assay
3.4.1. Assay Buffer Preparation
3.4.2. Calcium Assay Procedure
3.5. PAMPA Assay
3.6. MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeitzer, J.M.; Nishin, S.; Mignot, E. The neurobiology of hypocretins (orexins), narcolepsy and related therapeutic interventions. Trends Pharmacol. Sci. 2006, 27, 368–374. [Google Scholar] [CrossRef] [PubMed]
- De Lecea, L.; Kilduff, T.S.; Peyron, C.; Gao, X.-B.; Foye, P.E.; Danielson, P.E.; Fukuhara, C.; Battenberg, E.L.F.; Gautvik, V.T.; Bartlett, F.S.; et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. USA 1998, 95, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.C.; Richarson, J.A.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 1998, 92, 573–585. [Google Scholar] [CrossRef]
- Xu, T.-R.; Yang, Y.; Ward, R.; Gao, L.; Liu, Y. Orexin receptors: Multi-functional therapeutic targets for sleeping disorders, eating disorders, drug addiction, cancers and other physiological disorders. Cell. Signal. 2013, 25, 2413–2423. [Google Scholar] [CrossRef] [PubMed]
- Smart, D.; Jerman, J.C. The physiology and pharmacology of the orexins. Pharmacol. Ther. 2002, 94, 51–61. [Google Scholar] [CrossRef]
- Kilduff, T.S.; Peyron, C. The hypocretin/orexin ligand-receptor system: Implications for sleep and sleep disorders. Trends Neurosci. 2000, 23, 359–365. [Google Scholar] [CrossRef]
- Holmqvist, T.; Akerman, K.E.O.; Kukkonen, J.P. High specificity of human orexin receptors for orexins over neuropeptide Y and other neuropeptides. Neurosci. Lett. 2001, 305, 177–180. [Google Scholar] [CrossRef]
- Thannickal, T.C.; Moore, R.Y.; Nienhuis, R.; Ramanathan, L.; Gulyani, S.; Aldrich, M.; Cornford, M.; Siegel, J.M.; Nienhuis, R.; Ramanathan, L.; et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000, 27, 469–474. [Google Scholar] [CrossRef]
- Kumar, S.; Sagili, H. Etiopathogenesis and neurobiology of narcolepsy: A review. J. Clin. Diagn. Res. 2014, 8, 190–195. [Google Scholar] [PubMed]
- Scammell, T.E. Narcolepsy. N. Eng. J. Med. 2015, 373, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- Lammers, G.J. Drugs Used in Narcolepsy and Other Hypersomnias. Sleep Med. Clin. 2018, 13, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, P.; Malcolm, R. Mechanisms of modafinil: A review of current research. Neuropsychiatr. Dis. Treat. 2007, 3, 349–364. [Google Scholar] [PubMed]
- Busardo, F.P.; Kyriakou, C.; Napoletano, S.; Marinelli, E.; Zaami, S. Clinical applications of sodium oxybate (GHB): From narcolepsy to alcohol withdrawal syndrome. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4654–4663. [Google Scholar] [PubMed]
- Klimova, B.; Maresova, P.; Novotny, M.; Kuca, K. A Global View on Narcolepsy—A Review Study. Mini. Rev. Med. Chem. 2018, 18, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Toyama, S.; Shimoyama, N.; Tagaito, Y.; Nagase, H.; Saitoh, T.; Yanagisawa, M.; Shimoyama, M. Nonpeptide Orexin-2 Receptor Agonist Attenuates Morphine-induced Sedative Effects in Rats. Anesthesiology 2018, 128, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Turku, A.; Rinne, M.K.; af Gennäs, G.B.; Xhaard, H.; Lindholm, D.; Kukkonen, J.P. Orexin receptor agonist Yan 7874 is a weak agonist of orexin/hypocretin receptors and shows orexin receptor-independent cytotoxicity. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Janockova, J.; Misik, J.; Kubik, S.; Stuchlik, A.; Vales, K.; Korabecny, J.; Mezeiova, E.; Dolezal, R.; Soukup, O.; et al. Orexin Supplementation in Narcolepsy Treatment: A Review. Med. Res. Rev. 2018. accepted. [Google Scholar] [CrossRef]
- Van Riel, D.; Verdijk, R.; Kuiken, T. The olfactory nerve: A shortcut for influenza and other viral diseases into the central nervous system. J. Pathol. 2015, 235, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Tesoriero, C.; Codita, A.; Zhang, M.-D.; Cherninsky, A.; Karlsson, H.; Grassi-Zucconi, G.; Bertini, G.; Harkany, T.; Ljungberg, K.; Liljeström, P.; et al. H1N1 influenza virus induces narcolepsy-like sleep disruption and targets sleep–wake regulatory neurons in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E368–E377. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.A.; Winrow, C.J. Opportunities and perspectives for developing orexin receptor antagonists. Front. Neurosci. 2014, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.V.; Aspesi, A.V.; Evoy, K.E. Suvorexant: A dual orexin receptor antagonist for the treatment of sleep onset and sleep maintenance insomnia. Ann. Pharmacot. 2015, 49, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Karhu, L.; Turku, A.; Xhaard, H. Modeling of the OX1R-orexin-A complex suggests two alternative binding modes. BMC Struct. Biol. 2015, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Saitoh, T.; Kutsumura, N.; Irukayama-Tomobe, Y.; Ogawa, Y.; Kuroda, D.; Gouda, H.; Kumagai, H.; Fujii, H.; Yanagisawa, M.; et al. Design and Synthesis of Non-Peptide, Selective Orexin Receptor 2 Agonists. J. Med. Chem. 2015, 58, 7931–7937. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, A.; Barker, O.; Morris, G.B.; Law, R.J.; Slack, M.; Biggin, P.C. Toward an understanding of agonist binding to human Orexin-1 and Orexin-2 receptors with G-protein-coupled receptor modeling and site-directed mutagenesis. Biochemistry 2013, 52, 8246–8260. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Mobarec, J.C.; Kolb, P.; Rosenbaum, D.M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, A.; Bodkin, M.J.; Biggin, P.C. Discovery of the First Selective, Nonpeptidic Orexin 2 Receptor Agonists. J. Med. Chem. 2015, 58, 7928–7930. [Google Scholar] [CrossRef] [PubMed]
- Irukayama-Tomobe, Y.; Ogawa, Y.; Tominaga, H.; Ishikawa, Y.; Hosokawa, N.; Ambai, S.; Kawabe, Y.; Uchida, S.; Nakajima, R.; Saitoh, T.; et al. Nonpeptide orexin type-2 receptor agonist ameliorates narcolepsy-cataplexy symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 5731–5736. [Google Scholar] [CrossRef] [PubMed]
- Scammell, T.E.; Winrow, C.J. Orexin Receptors: Pharmacology and Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Roecker, A.J.; Coleman, P.J. Orexin receptor antagonists: Medicinal chemistry and therapeutic potential. Curr. Top. Med. Chem. 2008, 8, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.J.; Renger, J.J. Orexin receptor antagonists: A review of promising compounds patented since 2006. Expert Opin. Ther. Pat. 2010, 20, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Turku, A.; Borrel, A.; Leino, T.O.; Karhu, L.; Kukkonen, J.P.; Xhaard, H. Pharmacophore Model To Discover OX1 and OX2 Orexin Receptor Ligands. J. Med. Chem. 2016, 59, 8263–8275. [Google Scholar] [CrossRef] [PubMed]
- Lee-Iannotti, J.K.; Parish, J.M. Suvorexant: A promising, novel treatment for insomnia. Neuropsychiatr. Dis. Treat. 2016, 12, 491–495. [Google Scholar] [PubMed]
- Winrow, C.J.; Gotter, A.L.; Cox, C.D.; Doran, S.M.; Tannenbaum, P.L.; Breslin, M.J.; Garson, S.L.; Fox, S.V.; Harrell, C.M.; Stevens, J.; et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J. Neurogenet. 2011, 25, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D., Jr.; Merz, K.M.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. ACS Cent. Sci. 2017, 3, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, R.; Nepovimova, E.; Melikova, M.; Kuca, K. Structure-Based Virtual Screening for Novel Modulators of Human Orexin 2 Receptor with Cloud Systems and Supercomputers. In ACIIDS 2017: Advanced Topics in Intelligent Information and Database Systems; Król, D., Nguyen, N.T., Shirai, K., Eds.; Springer: Cham, Switzerland, 2017; pp. 161–171. [Google Scholar]
Sample Availability: Samples of the compounds are available upon request. |



{kind=link}
{kind=link}
{kind=link}
| Codename | IUPAC Name | Chemical Structure | Mw [g mol−1] | logP 1 |
|---|---|---|---|---|
| L1 | 1-(1-{5H,6H-benzo[h]quinazolin-2-yl}-5-cyclopropyl-1H-pyrazole-4-carbonyl)-4-benzylpiperazine |  | 490.60 | 4.90 |
| L2 | 4-{2-[4-(naphthalene-2-sulfonyl)piperazin-1-yl]acetyl}-1,2,3,4-tetrahydroquinoxalin-2-one |  | 464.54 | 1.58 |
| L3 | 4-[4-(2,3-dihydro-1,4-benzodioxine-2-carbonyl)piperazine-1-carbonyl]-2-(furan-2-yl)quinoline |  | 469.49 | 3.00 |
| L4 | 2-(4-fluorophenyl)-6-{4-[2-(furan-2-yl)quinoline-4-carbonyl]piperazin-1-yl}-2,3-dihydropyridazin-3-one |  | 495.50 | 3.60 |
| L5 | (12E)-22-hydroxy-20-(2-hydroxyquinolin-3-yl)-4-methyl-3,17-dioxatricyclo[12.8.0.016,21]docosa-1(14),12,15,21-tetraene-2,8,18-trione |  | 515.55 | 6.46 |
| L6 | 3,5-dimethyl-13-(4-oxo-4H-chromen-3-yl)-8-phenyl-12-oxa-3,5,9-triazatricyclo[7.4.0.02,7]trideca-1,7-diene-4,6-dione |  | 455.46 | 2.69 |
| L7 | (6E)-5-imino-6-({1-[(2-methylphenyl)methyl]-1H-indol-3-yl}methylidene)-2-[2-oxo-2-(pyrrolidin-1-yl)ethyl]-5H,6H,7H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-7-one |  | 510.61 | 4.47 |
| L8 | N-[(1R,9S)-6-oxo-11-(pyridine-2-carbonyl)-7,11-diazatricyclo[7.3.1.02,7]trideca-2,4-dien-5-yl]benzamide |  | 414.46 | 1.14 |
| L9 | 3-{2-[2-(3,4-dihydro-2H-1,5-benzodioxepin-7-yl)pyrrolidin-1-yl]-2-oxoethyl}-5-(4-fluorophenyl)-2,3-dihydro-1,3,4-oxadiazol-2-one |  | 439.44 | 2.91 |
| L10 | N-[3-(2,3-dihydro-1-benzofuran-5-yl)phenyl]-2-oxo-1,2,3,4-tetrahydroquinoline-4-carboxamide |  | 384.435 | 3.70 |
| L11 | N-[2-(2,5-difluorophenyl)-1H-1,3-benzodiazol-5-yl]-1-ethyl-1H-1,2,3-benzotriazole-5-carboxamide |  | 418.40 | 4.47 |
| Suvorexant | 5-chloro-2-{3-methyl-4-[5-methyl-2-(2H-1,2,3-triazol-2-yl)benzoyl]piperazin-1-yl}-1,3-benzoxazole |  | 450.92 | 3.99 |
| Ligand | Binding Energy Estimate [kcal/mol] | |
|---|---|---|
| iDock | AutoDock Vina | |
| L1 | −11.8 | −13.7 |
| L2 | −11.5 | −14.8 |
| L3 | −12.0 | −13.6 |
| L4 | −11.9 | −15.0 |
| L5 | −12.4 | −15.4 |
| L6 | −12.1 | −13.5 |
| L7 | −11.8 | −14.6 |
| L8 | −11.0 | −12.3 |
| L9 | −11.2 | −13.2 |
| L10 | −12.2 | −13.7 |
| L11 | −12.1 | −13.8 |
| Suvorexant | −11.1 | −12.5 |
| Ligand | IC50 µM ± SEM |
|---|---|
| L3 | 8.9 ± 0.43 |
| L4 | 2.2 ± 0.47 |
| L6 | 12.2 ± 3.1 |
| L7 | 25 ± 2.6 |
| L10 | 27.8 ± 0.5 |
| Suvorexant | 0.071 ± 0.013 |
| Ligand | Pe ± SEM (×10−6 cm s−1) 1 | CNS Predicted Availability 2 | Cytotoxicity CHO-K1 IC50 µM ± SEM |
|---|---|---|---|
| L1 | 10.2 ± 0.9 | CNS + | 78.8 ± 6.88 |
| L2 | 31.6 ± 3.4 | CNS + | 165 ± 5.36 |
| L3 | 17.9 ± 2.2 | CNS + | 279 ± 36.6 |
| L4 | 18.3 ± 0.1 | CNS + | 99.8 ± 10.7 |
| L5 | 12.0 ± 2.5 | CNS + | 59.0 ± 6.41 |
| L6 | 17.6 ± 6.4 | CNS + | >100 |
| L7 | 3.37 ± 2.1 | CNS +/− | 61.8 ± 3.74 |
| L8 | 4.0 ± 0.3 | CNS +/− | ~700 |
| L9 | 32.3 ± 1.5 | CNS + | 143.6 ± 11.1 |
| L10 | 18.9 ± 2.4 | CNS + | 53.9 ± 5.03 |
| L11 | ND | ND | 28.3 ± 4.07 |
| Tacrine | 6.0 ± 0.6 | CNS + | ND |
| Donepezil | 21.9 ± 2.1 | CNS + | ND |
| Rivastigmine | 20.0 ± 2.1 | CNS + | ND |
| Ibuprofen | 18.0 ± 4.3 | CNS + | ND |
| Chlorothiazide | 1.1 ± 0.5 | CNS − | ND |
| Furosemide | 0.2 ± 0.07 | CNS − | ND |
| Ranitidine | 0.04 ± 0.02 | CNS − | ND |
| Sulfasalazine | 0.09 ± 0.05 | CNS − | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janockova, J.; Dolezal, R.; Nepovimova, E.; Kobrlova, T.; Benkova, M.; Kuca, K.; Konecny, J.; Mezeiova, E.; Melikova, M.; Hepnarova, V.; et al. Investigation of New Orexin 2 Receptor Modulators Using In Silico and In Vitro Methods. Molecules 2018, 23, 2926. https://doi.org/10.3390/molecules23112926
Janockova J, Dolezal R, Nepovimova E, Kobrlova T, Benkova M, Kuca K, Konecny J, Mezeiova E, Melikova M, Hepnarova V, et al. Investigation of New Orexin 2 Receptor Modulators Using In Silico and In Vitro Methods. Molecules. 2018; 23(11):2926. https://doi.org/10.3390/molecules23112926
Chicago/Turabian StyleJanockova, Jana, Rafael Dolezal, Eugenie Nepovimova, Tereza Kobrlova, Marketa Benkova, Kamil Kuca, Jan Konecny, Eva Mezeiova, Michaela Melikova, Vendula Hepnarova, and et al. 2018. "Investigation of New Orexin 2 Receptor Modulators Using In Silico and In Vitro Methods" Molecules 23, no. 11: 2926. https://doi.org/10.3390/molecules23112926
APA StyleJanockova, J., Dolezal, R., Nepovimova, E., Kobrlova, T., Benkova, M., Kuca, K., Konecny, J., Mezeiova, E., Melikova, M., Hepnarova, V., Ring, A., Soukup, O., & Korabecny, J. (2018). Investigation of New Orexin 2 Receptor Modulators Using In Silico and In Vitro Methods. Molecules, 23(11), 2926. https://doi.org/10.3390/molecules23112926