
Sample Volume Reduction Using the Schwarzschild Objective for a Circular Dichroism Spectrophotometer and an Application to the Structural Analysis of Lysine-36 Trimethylated Histone H3 Protein


Abstract
1. Introduction
2. Results and Discussion
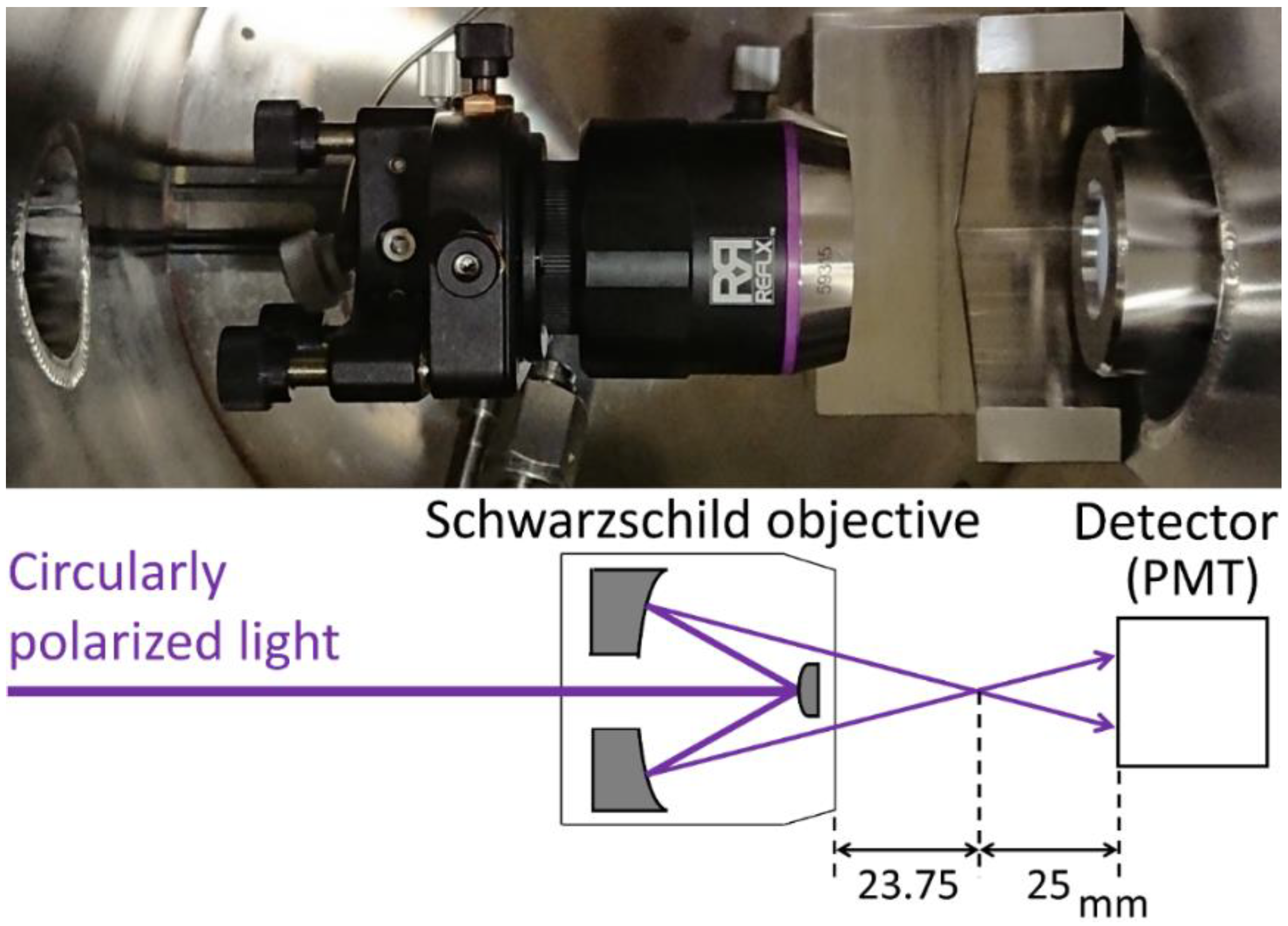
2.1. Beam Focusing by the SO
2.2. Determination of the Sample Position
2.3. Small-Capacity Sample Cell
2.4. Performance of CD Measurement Using the SO
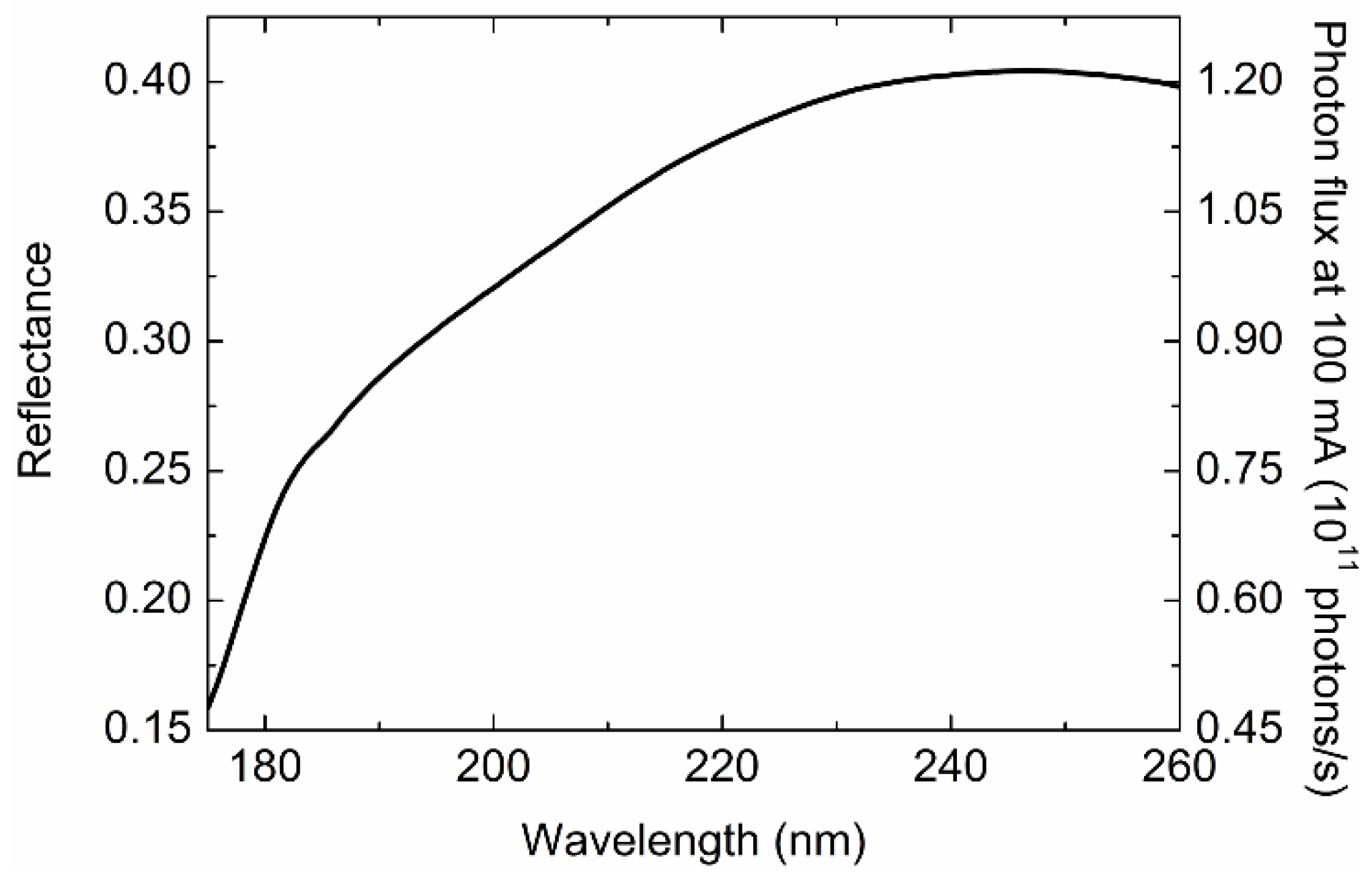
2.4.1. Radiation Damage to Protein Sample
2.4.2. Distortion of CD Spectrum by the SO
2.5. Structural Analysis of H3K36me3
2.5.1. CD Spectrum of H3K36me3
2.5.2. Secondary Structural Contents of H3K36me3
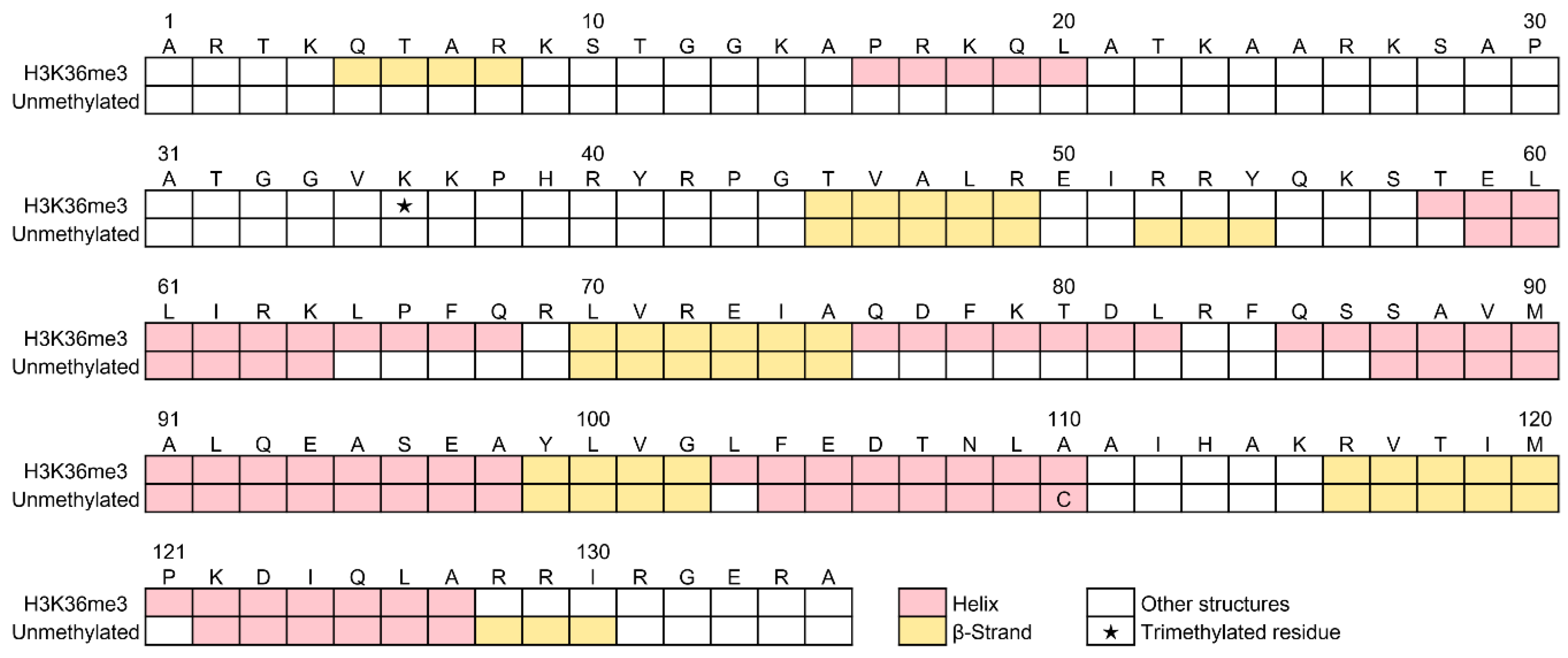
2.5.3. Predicted Positions of α-Helices and β-Strands
3. Materials and Methods
3.1. Sample Preparation
3.2. CD Measurements
3.3. Analysis of Secondary Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, Y.; Bigelow, L.; Borovilos, M.; Dementieva, I.; Duggan, E.; Hatzos, C.; Joachimiak, G.; Li, H.; Mulligan, R.; Quartey, P.; et al. High-throughput protein purification for X-ray crystallography and NMR. Adv. Protein Chem. Struct. Biol. 2008, 75, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, P.S.; Araujo, A.P.U.; Lopes, J.L.S. Going deep into protein secondary structure with synchrotron radiation circular dichroism spectroscopy. Biophys. Rev. 2017, 9, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.S.; Hoffmann, A.; Baumgärtel, P.; Seckler, R.; Reichardt, G.; Horsley, D.A.; Schuler, B.; Bakajin, O. Microfluidic mixers for the investigation of rapid protein folding kinetics using synchrotron radiation circular dichroism spectroscopy. Anal. Chem. 2008, 80, 9534–9541. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yuri, M.; Onuki, H.; Ishizaka, S. Microscopic imaging of circular dichroism using a polarizing undulator. Jpn. J. Appl. Phys. 2000, 39, 310–315. [Google Scholar] [CrossRef]
- Sawada, M.; Namatame, H.; Taniguchi, M. Optical design of a compact and practical UV beamline at HiSOR-BL 12. J. Phys. Conf. Ser. 2013, 425, 162010. [Google Scholar] [CrossRef]
- Hussain, R.; Jávorfi, T.; Siligardi, G. Circular dichroism beamline B34 at the Diamond Light Source. J. Synchrotron Rad. 2012, 19, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Bürck, J.; Roth, S.; Windisch, D.; Wadhwani, P.; Moss, D.; Ulrich, A.S. UV-CD12: Synchrotron radiation circular dichroism beamline at ANKA. J. Synchrotron Rad. 2015, 22, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Wien, F.; Miles, A.J.; Lees, J.G.; Hoffmann, S.V.; Wallace, B.A. VUV irradiation effects on proteins in high-flux synchrotron radiation circular dichroism spectroscopy. J. Synchrotron Rad. 2005, 12, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.J.; Janes, R.W.; Brown, A.; Clarke, D.T.; Sutherland, J.C.; Tao, Y.; Wallace, B.A.; Hoffmann, S.V. Light flux density threshold at which protein denaturation is induced by synchrotron radiation circular dichroism beamlines. J. Synchrotron Rad. 2008, 15, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.T.; Jones, G. CD12: A new high-flux beamline for ultraviolet and vacuum-ultraviolet circular dichroism on the SRS, Daresbury. J. Synchrotron Rad. 2004, 11, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Wien, F.; Wallace, B.A. Calcium fluoride micro cells for synchrotron radiation circular dichroism spectroscopy. Appl. Spectrosc. 2005, 59, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Sakai, K.; Matsushita, Y.; Fukuyama, T.; Gekko, K. Optical cell with a temperature-control unit for a vacuum-ultraviolet circular dichroism spectrophotometer. Anal. Sci. 2003, 19, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Warner, L.R.; Brown, R.J.; Yingst, S.M.C.; Oxford, J.T. Isoform-specific heparin sulfate binding within the amino-terminal noncollagenous domain of collagen α1 (XI). J. Biol. Chem. 2006, 281, 39507–39516. [Google Scholar] [CrossRef] [PubMed]
- Šťovíčková, L.; Tatarkovič, M.; Logerová, H.; Vavřinec, J.; Setnička, V. Identification of spectral biomarkers for type 1 diabetes mellitus using the combination of chiroptical and vibrational spectroscopy. Analyst 2015, 140, 2266–2272. [Google Scholar] [CrossRef] [PubMed]
- Habartová, L.; Bunganič, B.; Tatarkovič, M.; Zavoral, M.; Vondroušová, J.; Syslová, K.; Setnička, V. Chiroptical spectroscopy and metabolomics for blood-based sensing of pancreatic cancer. Chirality 2018, 30, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Manzo, G.; Scorciapino, M.A.; Wadhwani, P.; Bürck, J.; Montaldo, N.P.; Pintus, M.; Sanna, R.; Casu, M.; Giuliani, A.; Pirri, G.; et al. Enhanced amphiphilic profile of a short β-stranded petide improves its antimicrobial activity. PLoS ONE 2015, 10, e0116379. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Fujii, K.; Wien, F.; Houée-Lévin, C.; Lacombe, S.; Salado-Leza, D.; Porcel, E.; Masoud, R.; Yamamoto, S.; Réfrégiers, M.; et al. Structure change from β-strand and turn to α-helix in histone H2A-H2B induced by DNA damage response. Biophys. J. 2016, 111, 69–78. [Google Scholar] [CrossRef] [PubMed]
- van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Pandita, T.K.; Richardson, C. Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic. Acids Res. 2009, 37, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.R.; Ramnarain, D.; Horikoshi, N.; Iyengar, P.; Pandita, R.L.; Shay, J.W.; Pandita, T.K. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiat. Res. 2013, 179, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat. Res. 2013, 750, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-L.; Shen, C.; Zhu, W.-G. Histone modifications in DNA damage response. Sci. China Life Sci. 2016, 59, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. Rev. 2017. [Google Scholar] [CrossRef]
- Downs, J.A.; Nussenzweig, M.C.; Nussenzweig, A. Chromatin dynamics and the preservation of genetic information. Nature 2007, 447, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenet. Chromatin 2012, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E. Reshaping chromatin after DNA damage: The choreography of histone proteins. J. Mol. Biol. 2015, 427, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.J.; Fasman, G.D. Altered conformational effects of naturally acetylated histone f2a1 (IV) in f2a1-deoxyribonucleic acid complexes. Circular dichroism studies. J. Biol. Chem. 1974, 249, 2911–2914. [Google Scholar] [PubMed]
- Prevelige, P.E., Jr.; Fasman, G.D. Structural studies of acetylated and control inner core histones. Biochemistry 1987, 26, 2944–2955. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Moore, S.C.; Laszckzak, M.; Ausió, J. Acetylation increases the α-helical content of the histone tails of the nucleosome. J. Biol. Chem. 2000, 275, 35013–35020. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Arya, G. Structure and binding of the H4 histone tail and the effects of lysine 16 acethylation. Phys. Chem. Chem. Phys. 2011, 13, 2911–2921. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Yamamoto, S.; Fujii, K.; Yokoya, A. Secondary structure alterations of histone H2A and H2B in X-irradiated human cancer cells: Altered histones persist in cells for at least 24 hours. Radiat. Res. 2015, 184, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, J.; Sakuraba, S.; Kono, H. H3 histone tail conformation within the nucleosome and the impact of K14 acetylation studied using enhanced sampling simulation. PLoS Comput. Biol. 2016, 12, e1004788. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Fujii, K.; Yamamoto, S.; Matsuo, K.; Namatame, H.; Taniguchi, M.; Yokoya, A. DNA damage response induces structural alterations in histone H3-H4. J. Radiat. Res. 2017, 58, 59–65. [Google Scholar] [CrossRef] [PubMed]
- du Preez, L.L.; Patterton, H.-G. The effect of epigenetic modifications on the secondary structures and possible binding positions of the N-terminal tail of histone H3 in the nucleosome: A computational study. J. Mol. Model. 2017, 23, 137. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Matsuo, K.; Fujii, K.; Yokoya, A.; Taniguchi, M.; Namatame, H. Circular dichroism spectroscopic study on structural alterations of histones induced by post-translational modifications in DNA damage responses: Lysine-9 methylation of H3. J. Radiat. Res. 2018, 59, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Matsuo, K.; Namatame, H. Structural analysis of lysine-4 methylated histone H3 proteins using synchrotron radiation circular dichroism spectroscopy. Chirality 2018, 30, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, N.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Rothbart, S.B.; Lu, R.; Xu, B.; Chen, W.-Y.; Tripathy, A.; Rockowitz, S.; Zheng, D.; Patel, D.J.; Allis, C.D.; et al. An H3K36 methylation-engaging tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zeng, H.; Lam, R.; Tempel, W.; Amaya, M.F.; Xu, C.; Dombrovski, L.; Qiu, W.; Wang, Y.; Min, J. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 2011, 6, e18919. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Avvakumov, N.; Watanabe, R.; Abraham, C.G.; Lalonde, M.-E.; Hong, Z.; Allen, C.; Roy, S.; Nuñez, J.K.; Nickoloff, J.; et al. Molecular basis for H3K36me3 recognition by the tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012, 19, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hilringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Battaglioli, E.; Mattevi, A. Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 2005, 280, 41360–41365. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Blumenthal, R.M. Coodinated chromatin control: Structural and functional linkage of DNA and histone methylation. Biochemistry 2010, 49, 2999–3008. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yang, H.; Dong, Z.; Fang, J.; Wang, P.; Zhu, T.; Gong, W.; Fang, R.; Shi, Y.G.; Li, Z.; et al. Structural insight into substrate recognition by histone demethylase LSD2/KDM1b. Cell Res. 2013, 23, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Burg, J.M.; Gonzalez, J.J.; Maksimchuk, K.R.; McCafferty, D.G. Lysine-specific demethylase 1A (KDM1A/LSD1): Product recognition and kinetic analysis of full-length histones. Biochemistry 2016, 55, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Chu, F.; Racki, L.R.; de la Cruz, C.C.; Burlingame, A.L.; Panning, B.; Narlikar, G.J.; Shokat, K.M. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 2007, 128, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Venyaminov, S.Y.; Woody, R.W. Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 1999, 8, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR, methods with and expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Yonehara, R.; Gekko, K. Secondary-structure analysis of proteins by vacuum-ultraviolet circular dichroism spectroscopy. J. Biochem. 2004, 135, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Yonehara, R.; Gekko, K. Improved estimation of the secondary structures of proteins by vacuum-ultraviolet circular dichroism spectroscopy. J. Biochem. 2005, 138, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.J.; Wien, F.; Lees, J.G.; Rodger, A.; Janes, R.W.; Wallace, B.A. Calibration and standardisation of synchrotron radiation circular dichroism and conventional circular dichroism spectrophotometers. Spectroscopy 2003, 17, 653–661. [Google Scholar] [CrossRef]
- Provencher, S.W.; Glöckner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- van Stokkum, I.H.M.; Spoelder, H.J.W.; Bloemendal, M.; van Grondelle, R.; Groen, F.C.A. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990, 191, 110–118. [Google Scholar] [CrossRef]
- Miles, A.J.; Wallace, B.A. Synchrotron radiation circular dichroism spectroscopy of proteins and applications in structural and functional genomics. Chem. Soc. Rev. 2006, 35, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.G.; Miles, A.J.; Wien, F.; Wallace, B.A. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 2006, 22, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Watanabe, H.; Gekko, K. Improved sequence-based prediction of protein secondary structures by combining vacuum-ultraviolet circular dichroism spectroscopy with neutral network. Proteins 2008, 73, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure Content (%) | H3K36me3 | Unmethylated H3 1 |
|---|---|---|
| Helix | 35.6 ± 1.3 | 25.0 ± 1.2 |
| β-strand | 18.7 ± 2.5 | 21.3 ± 1.5 |
| Turn | 22.3 ± 0.9 | 21.1 ± 1.0 |
| Unordered | 23.3 ± 2.8 | 32.7 ± 1.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izumi, Y.; Matsuo, K. Sample Volume Reduction Using the Schwarzschild Objective for a Circular Dichroism Spectrophotometer and an Application to the Structural Analysis of Lysine-36 Trimethylated Histone H3 Protein. Molecules 2018, 23, 2865. https://doi.org/10.3390/molecules23112865
Izumi Y, Matsuo K. Sample Volume Reduction Using the Schwarzschild Objective for a Circular Dichroism Spectrophotometer and an Application to the Structural Analysis of Lysine-36 Trimethylated Histone H3 Protein. Molecules. 2018; 23(11):2865. https://doi.org/10.3390/molecules23112865
Chicago/Turabian StyleIzumi, Yudai, and Koichi Matsuo. 2018. "Sample Volume Reduction Using the Schwarzschild Objective for a Circular Dichroism Spectrophotometer and an Application to the Structural Analysis of Lysine-36 Trimethylated Histone H3 Protein" Molecules 23, no. 11: 2865. https://doi.org/10.3390/molecules23112865
APA StyleIzumi, Y., & Matsuo, K. (2018). Sample Volume Reduction Using the Schwarzschild Objective for a Circular Dichroism Spectrophotometer and an Application to the Structural Analysis of Lysine-36 Trimethylated Histone H3 Protein. Molecules, 23(11), 2865. https://doi.org/10.3390/molecules23112865