On the Power of Geometry over Tetrel Bonds


Abstract
1. Introduction
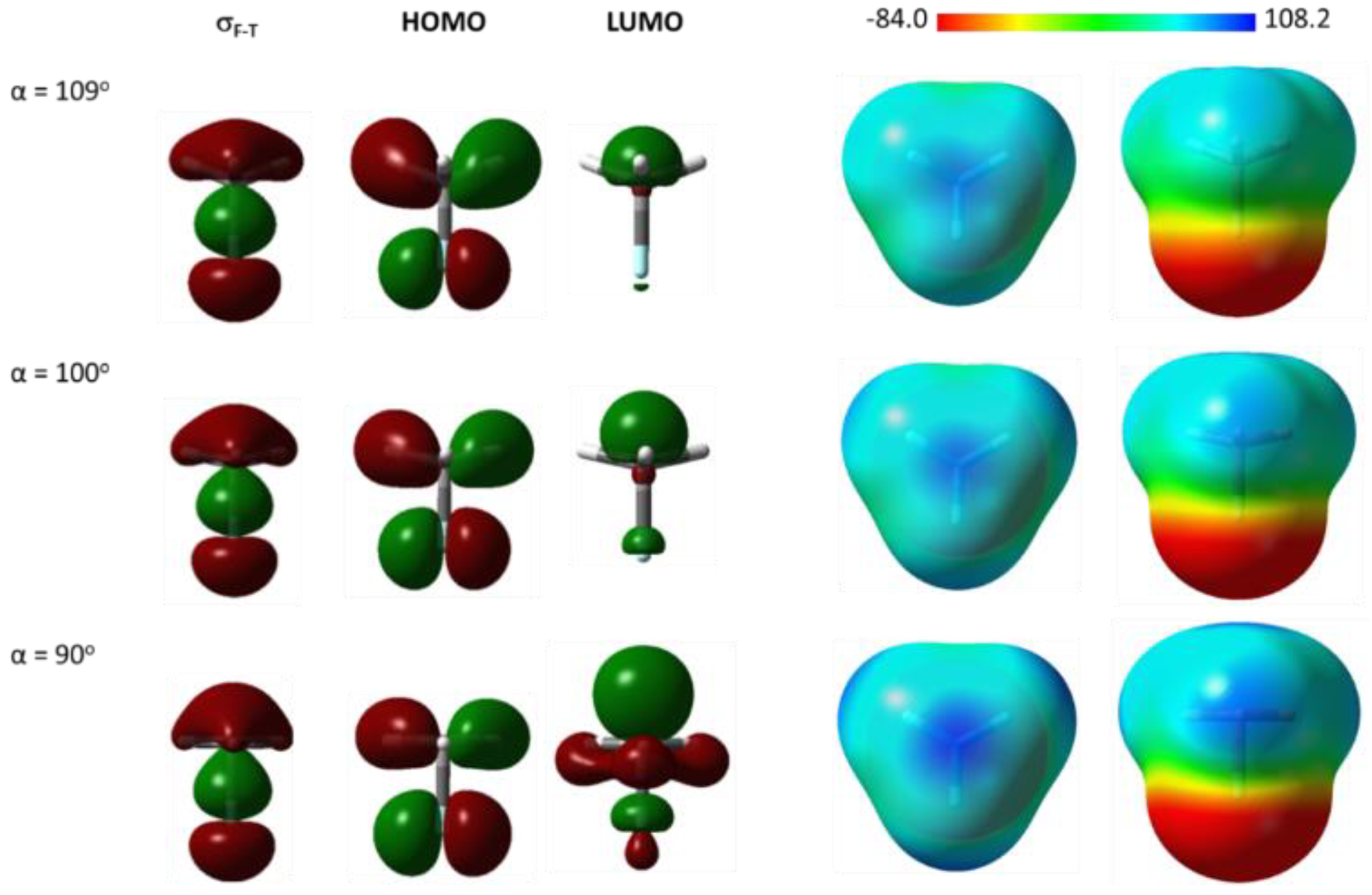
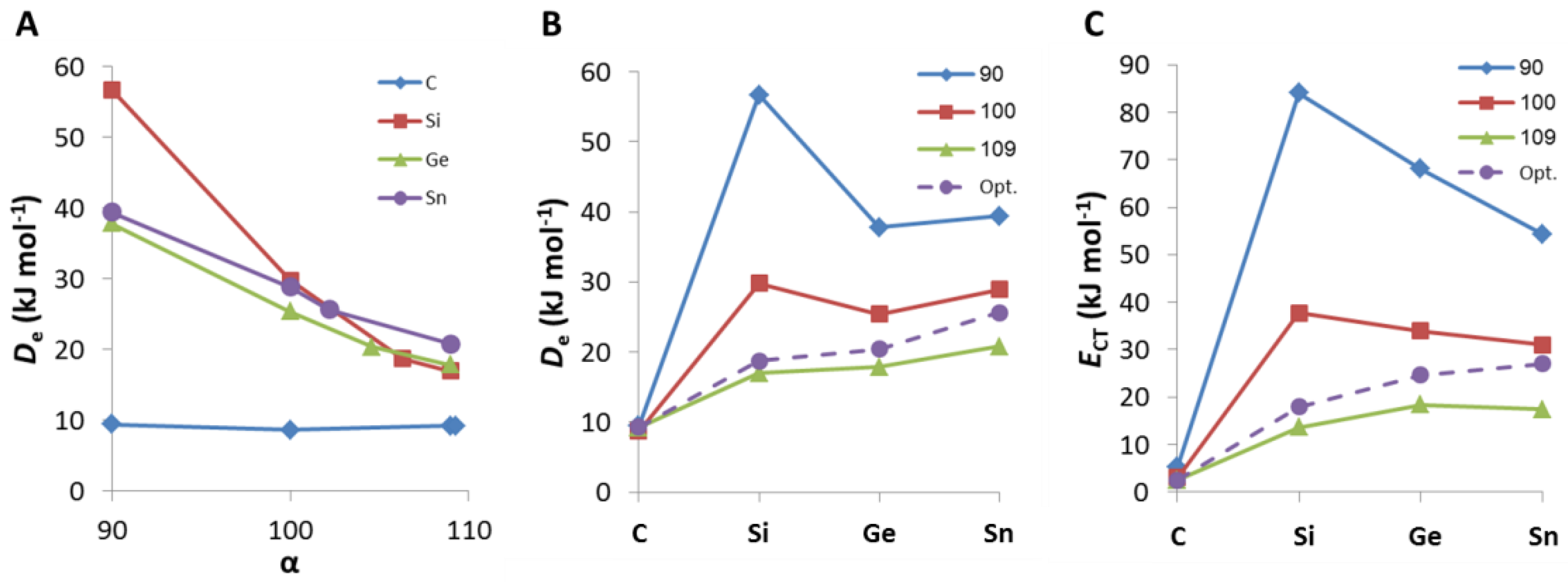
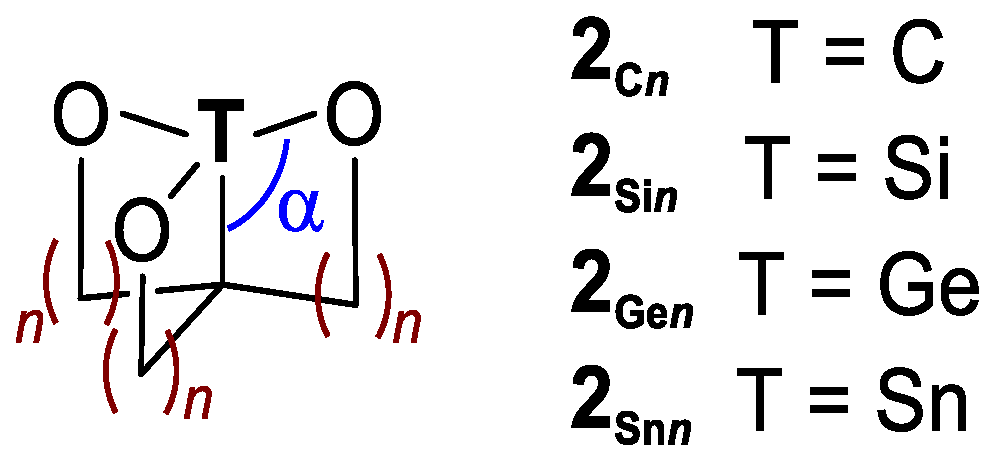
2. Results and Discussion
3. Conclusions
4. Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Angarov, V.; Kozuch, S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The origin of chalcogen-bonding interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. A new noncovalent force: Comparison of P···N interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The pnicogen bond in review: Structures, binding energies, bonding properties, and spin-spin coupling constants of complexes stabilized by pnicogen bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer International Publishing: New York, NY, USA, 2015. [Google Scholar]
- Adhikari, U.; Scheiner, S. Comparison of P⋯D (D = P,N) with other noncovalent bonds in molecular aggregates. J. Chem. Phys. 2011, 135, 184306. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel bonding interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Aerogen bonding interaction: A new supramolecular force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Anisimov, V.M.; Zolotoi, A.B.; Antipin, M.Y.; Lukin, P.M.; Nasakin, O.E.; Struchkov, Y.T. Hexacyanocyclopropane. Synthesis and structure. Mendeleev Commun. 1992, 2, 24–25. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Influence of ring size on the strength of carbon bonding complexes between anions and perfluorocycloalkanes. Phys. Chem. Chem. Phys. 2014, 16, 19192–19197. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Implications of monomer deformation for tetrel and pnicogen bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. Halogen bonds: Benchmarks and theoretical analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 (Revision A.03); Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Kirshenboim, O.; Kozuch, S. How to twist, split and warp a σ-hole with hypervalent halogens. J. Phys. Chem. A 2016, 120, 9431–9445. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.D. 469. The electronic orbitals, shapes, and spectra of polyatomic molecules. Part IV. Tetratomic hydride molecules, AH3. J. Chem. Soc. (Resumed) 1953, 2296–2301. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kawaguchi, K.; Kawashima, T. Water-coordinated neutral silane complex: A frozen intermediate of hydrolysis of alkoxysilanes. J. Am. Chem. Soc. 2004, 126, 16318–16319. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO; Version 3.1; TCI: Madison, WI, USA, 1998. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
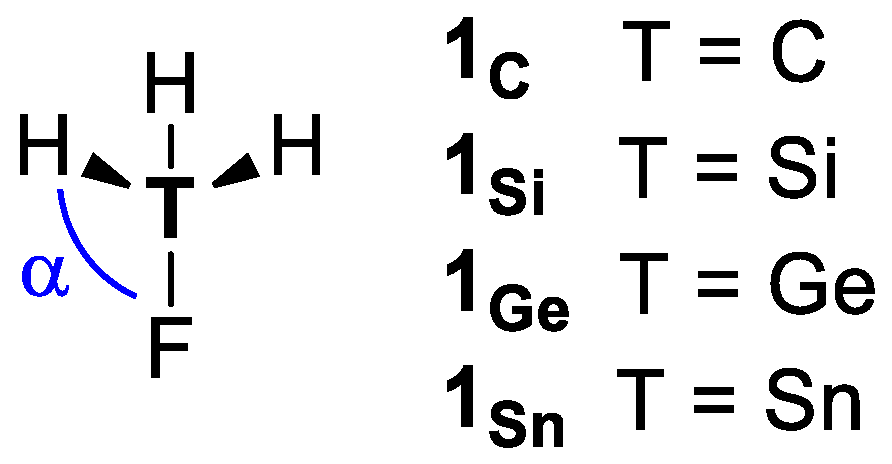{kind=link}
{kind=link}
| T | α | σF-T | LUMO | dF-T | %s b | %p b | Vs,max |
|---|---|---|---|---|---|---|---|
| C | Opt. (109.1°) a | −1413.9 | 154.7 | 1.376 | 21.52 | 78.28 | 81.2 |
| 109° | −1413.2 | 154.7 | 1.376 | 21.48 | 78.32 | 81.2 | |
| 100° | −1328.6 | 147.6 | 1.426 | 16.09 | 83.71 | 84.1 | |
| 90° | −1232.6 | 84.6 | 1.511 | 8.53 | 91.25 | 95.2 | |
| Si | Opt. (108.3°) a | −1328.8 | 27.2 | 1.598 | 21.40 | 76.03 | 142.5 |
| 109° | −1332.2 | 28.7 | 1.596 | 21.62 | 75.81 | 137.3 | |
| 100° | −1282.5 | −22.8 | 1.617 | 18.39 | 78.95 | 200.6 | |
| 90° | −1212.8 | −123.7 | 1.655 | 13.54 | 83.79 | 256.5 | |
| Ge | Opt. (106.2°) a | −1250.8 | 19.4 | 1.737 | 19.96 | 79.06 | 164.5 |
| 109° | −1267.7 | 21.4 | 1.731 | 21.20 | 77.83 | 148.8 | |
| 100° | −1212.4 | −18.6 | 1.751 | 17.05 | 81.98 | 197.0 | |
| 90° | −1145.6 | −120.0 | 1.788 | 11.39 | 87.66 | 238.0 | |
| Sn | Opt. (104.4°) a | −1158.6 | −32.6 | 1.927 | 18.66 | 80.49 | 196.6 |
| 109° | −1182.5 | −46.3 | 1.920 | 20.75 | 78.41 | 170.9 | |
| 100° | −1134.9 | −58.2 | 1.935 | 16.56 | 82.57 | 218.6 | |
| 90° | −1079.2 | −158.9 | 1.962 | 11.20 | 87.94 | 255.7 |
| T | α | dT-F | dT···N | % Cov. Rad. b | De c | E2n→σ*d |
|---|---|---|---|---|---|---|
| C | Opt. (109.3°) a | 1.380 | 3.154 | 208 | 9.3 | 2.5 |
| 109° | 1.381 | 3.155 | 208 | 9.2 | 2.5 | |
| 100° | 1.432 | 3.116 | 205 | 8.7 | 3.0 | |
| 90° | 1.521 | 3.013 | 198 | 9.5 | 5.3 | |
| Si | Opt. (106.3°) a | 1.608 | 2.847 | 153 | 18.7 | 18.0 |
| 109° | 1.602 | 2.944 | 158 | 17.0 | 13.6 | |
| 100° | 1.625 | 2.576 | 138 | 29.8 | 37.7 | |
| 90° | 1.670 | 2.162 | 116 | 56.7 | 84.1 e | |
| Ge | Opt. (104.6°) a | 1.749 | 2.931 | 149 | 20.4 | 24.6 |
| 109° | 1.738 | 3.043 | 154 | 17.8 | 18.4 | |
| 100° | 1.763 | 2.804 | 142 | 25.4 | 33.9 | |
| 90° | 1.808 | 2.532 | 128 | 37.8 | 68.1 | |
| Sn | Opt. (102.2°) a | 1.945 | 2.934 | 136 | 25.7 | 27.0 |
| 109° | 1.930 | 3.086 | 143 | 20.8 | 17.4 | |
| 100° | 1.950 | 2.887 | 134 | 28.9 | 31.0 | |
| 90° | 1.982 | 2.703 | 125 | 39.4 | 54.3 |
| T | n | dT-C Monomer | dT-C Complex | α Monomer | α Complex | dT···N | % Cov. Rad. c | De | Vs,max d |
|---|---|---|---|---|---|---|---|---|---|
| C | 1 | 1.481 | 1.486 | 97.6 | 97.1 | 2.953 | 194.3 | 0.9 | 42.1 |
| 2 | 1.561 | - | 107.8 | - | - | - | NB | −107.2 | |
| 3 | 1.542 | - | 111.4 | - | - | - | NB | −153.5 | |
| Si | 1 | 1.884 | 1.954 | 88.7 | 83.8 | 1.931 | 103.8 | 111.3 | 260.3 |
| 2 | 1.830 | 1.879 | 101.0 | 95.6 | 2.084 | 112.0 | 42.3 | 147.6 | |
| 3 a | 1.854 | 1.860 | 108.5 | 107.7 | 3.549 | 190.8 | −3.2 | −28.7 | |
| Ge | 1 | 1.974 | 2.043 | 83.2 | 79.8 | 2.047 | 103.9 | 112.6 | 339.2 |
| 2 | 1.899 | 1.935 | 98.9 | 95.4 | 2.220 | 112.7 | 47.9 | 186.6 | |
| 3 | 1.934 | 1.944 | 109.4 | 107.8 | 3.198 | 162.3 | −1.2 | 9.8 | |
| Sn b | 2 | 2.077 | 2.104 | 93.0 | 91.0 | 2.318 | 107.3 | 73.9 | 264.5 |
| 3 | 2.118 | 2.147 | 107.9 | 104.1 | 2.470 | 114.4 | 30.3 | 140.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solel, E.; Kozuch, S. On the Power of Geometry over Tetrel Bonds. Molecules 2018, 23, 2742. https://doi.org/10.3390/molecules23112742
Solel E, Kozuch S. On the Power of Geometry over Tetrel Bonds. Molecules. 2018; 23(11):2742. https://doi.org/10.3390/molecules23112742
Chicago/Turabian StyleSolel, Ephrath, and Sebastian Kozuch. 2018. "On the Power of Geometry over Tetrel Bonds" Molecules 23, no. 11: 2742. https://doi.org/10.3390/molecules23112742
APA StyleSolel, E., & Kozuch, S. (2018). On the Power of Geometry over Tetrel Bonds. Molecules, 23(11), 2742. https://doi.org/10.3390/molecules23112742