Cube-Rhombellane Related Structures: A Drug Perspective


Abstract
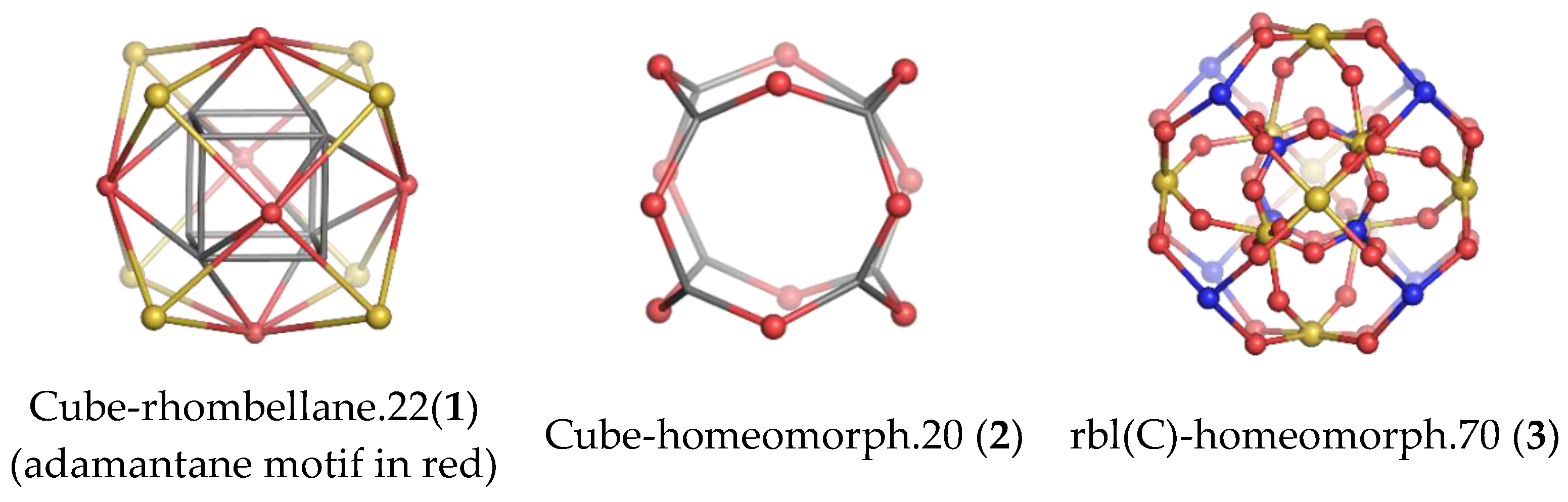
:1. Introduction
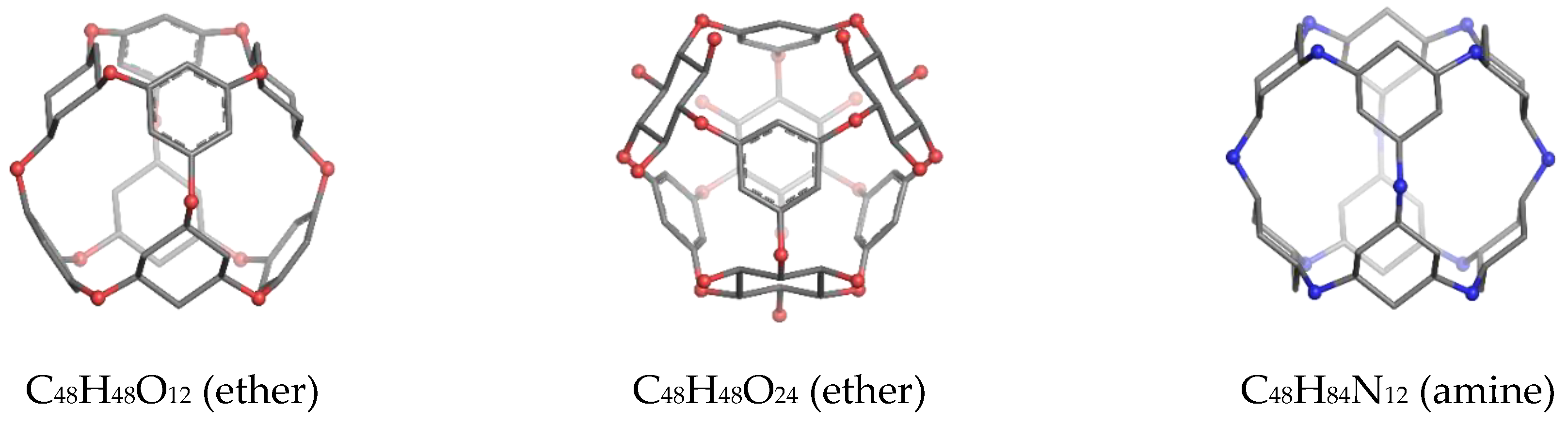
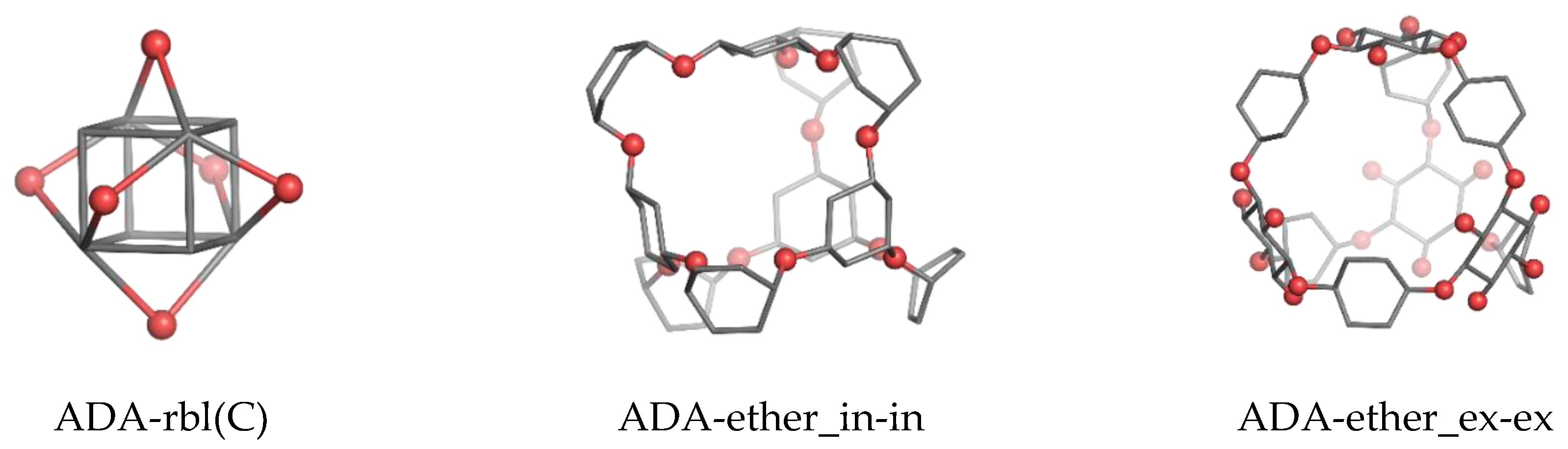
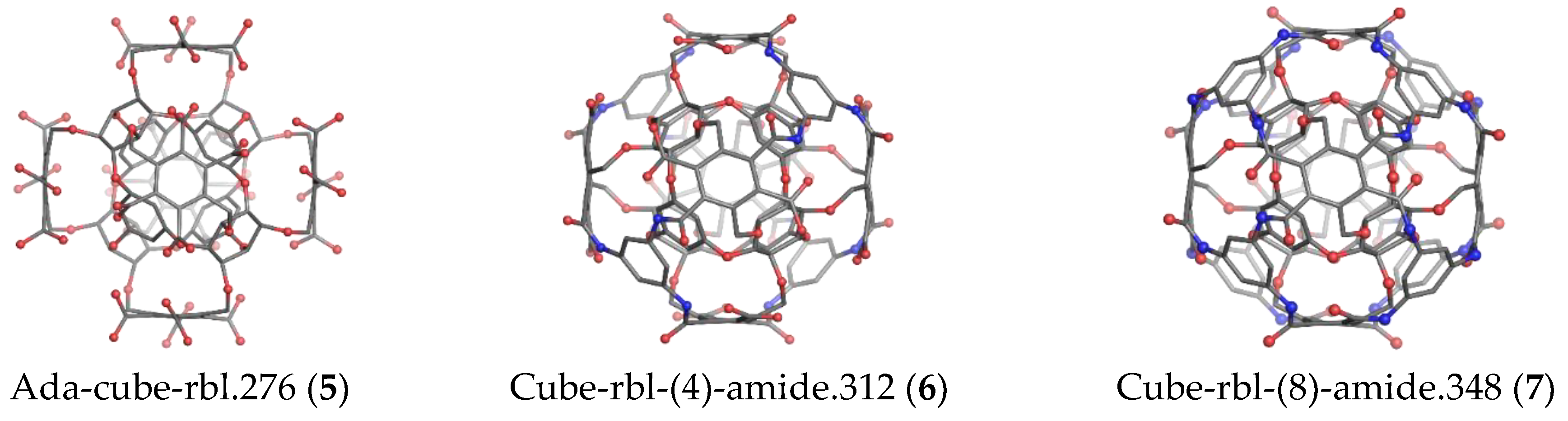
2. Results
2.1. DFT Results
2.2. Bioactivity Evaluation
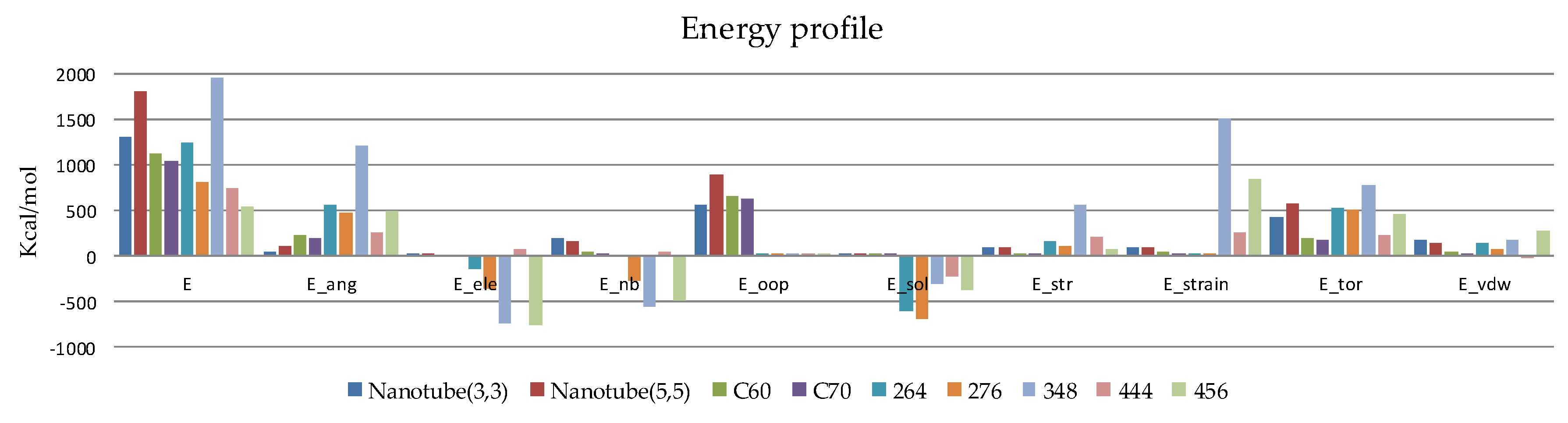
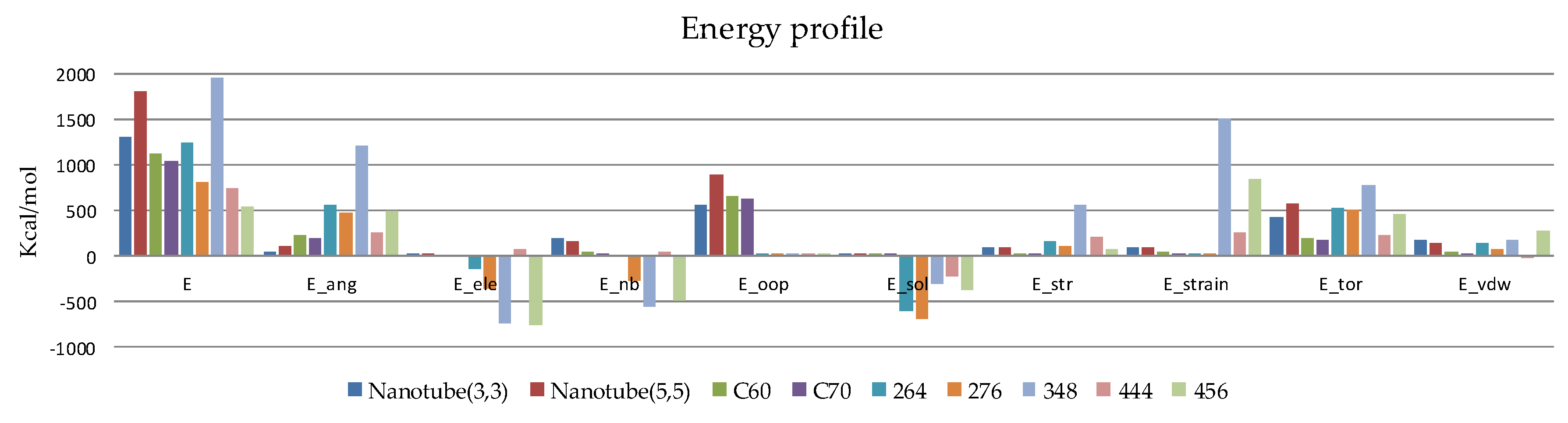
2.2.1. MM Computations
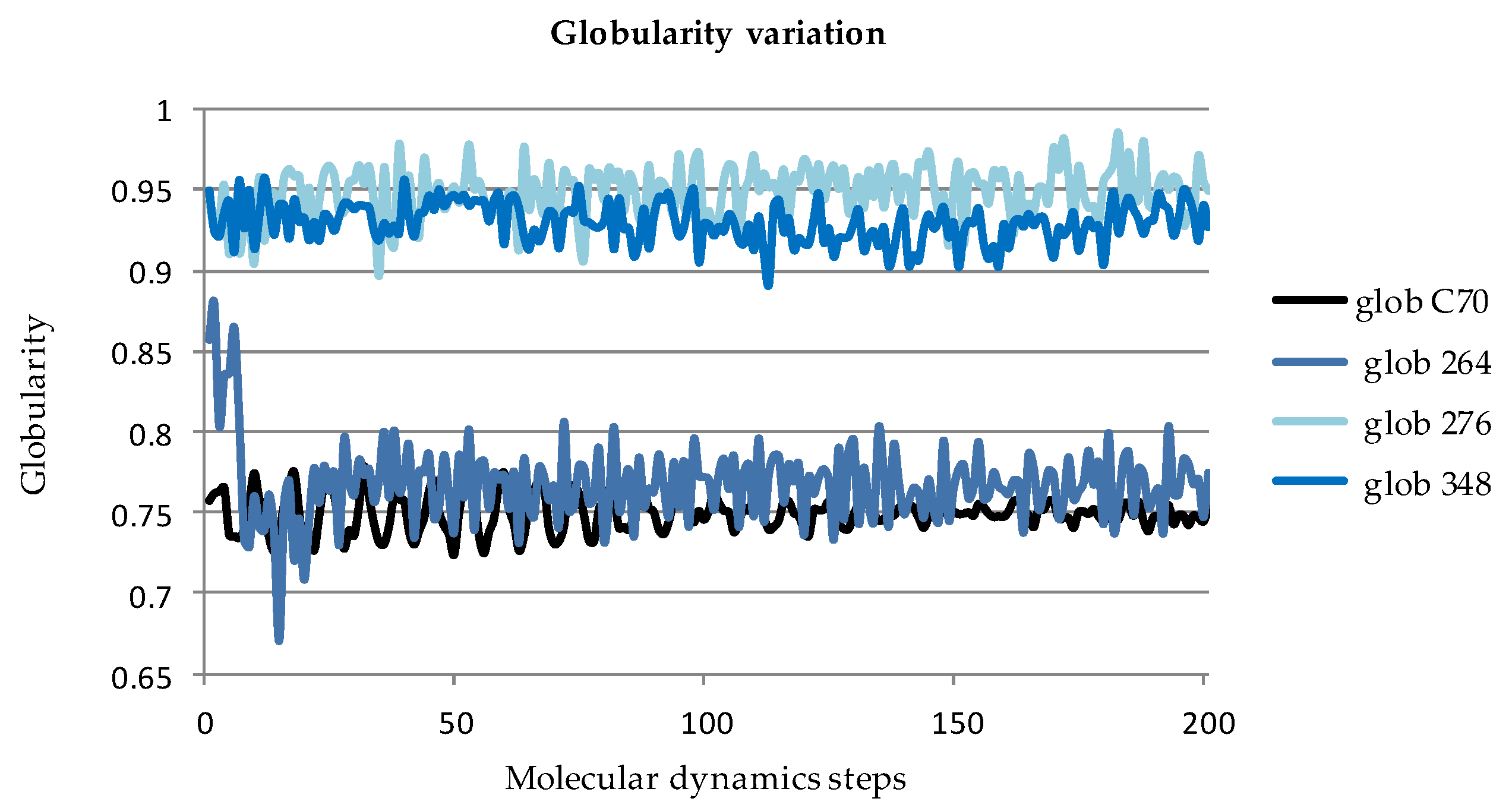
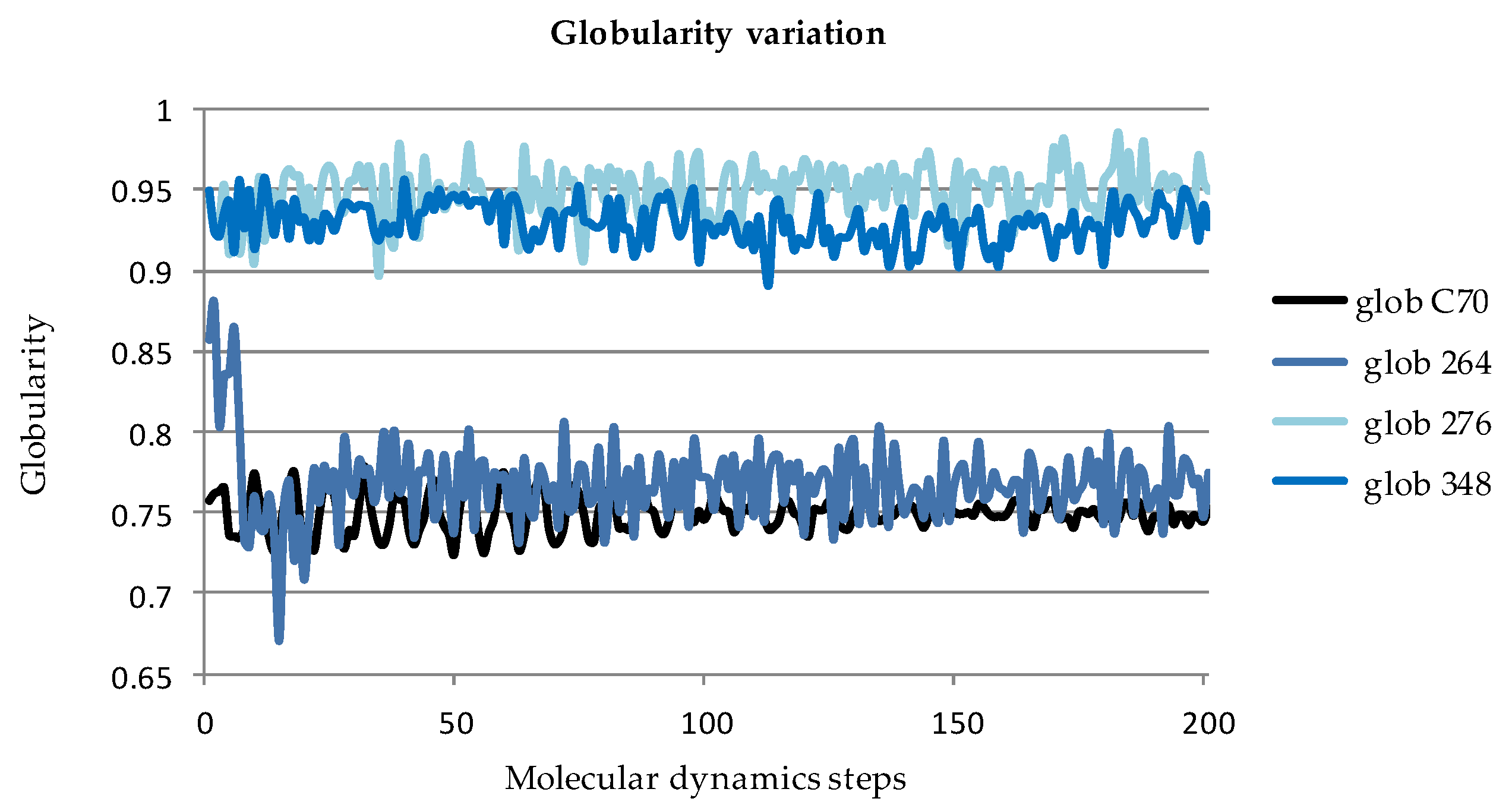
2.2.2. Globularity
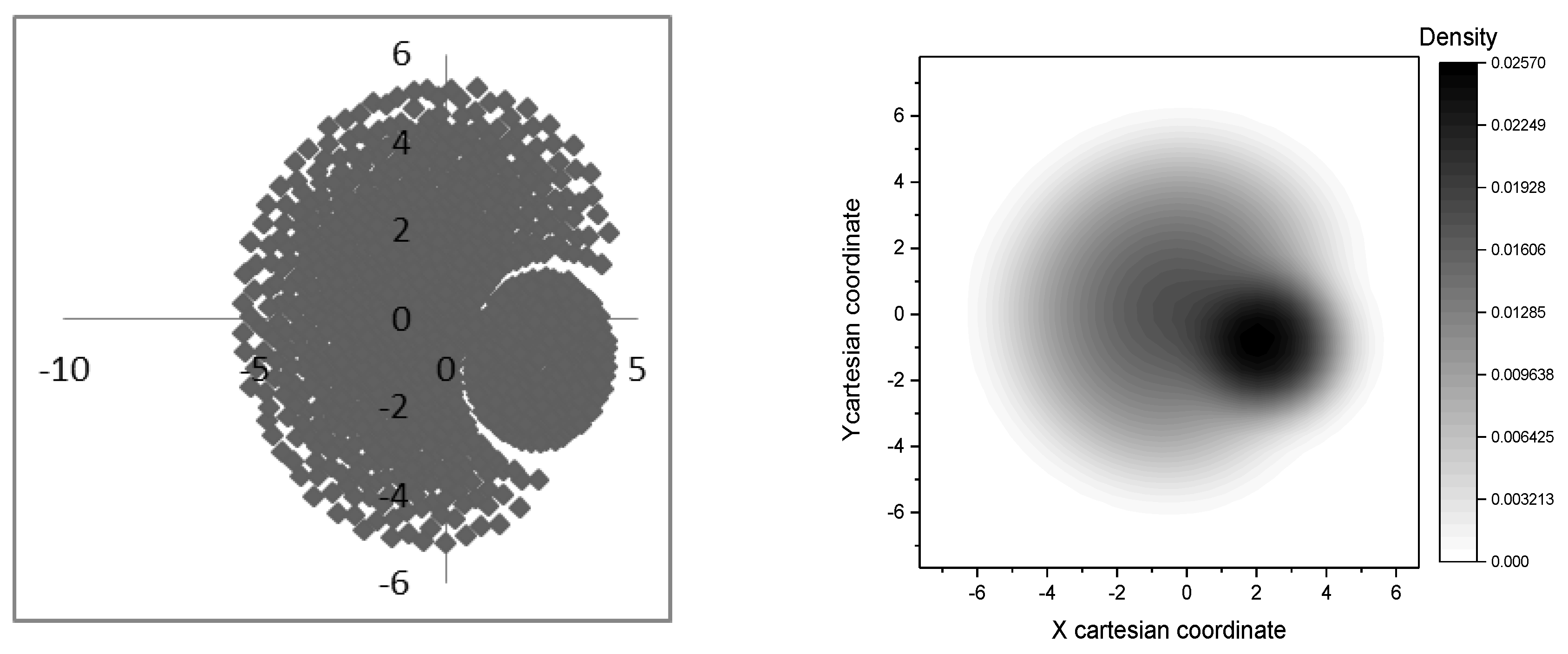
2.2.3. ADME Properties Evaluation
2.2.4. RECAP Analysis
3. Methods
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sun, Q.F.; Iwasa, J.; Ogawa, D.; Ishido, Y.; Sato, S.; Ozeki, T.; Sei, Y.; Yamaguchi, K.; Fujita, M. Self-assembled M24L48 polyhedra and their sharp structural switch upon subtle ligand variation. Science 2010, 328, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Bunzen, J.; Iwasa, J.; Bonakdarzadeh, P.; Numata, E.; Rissanen, K.; Sato, S.; Fujita, M. Self-assembly of M24L48 polyhedra based on empirical prediction. Angew. Chem. Int. Ed. 2012, 51, 3161–3163. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Fujita, D.; Fujita, M. Giant hollow MnL2n spherical complexes: Structure, functionalisation and applications. Chem. Commun. 2013, 49, 6703–6712. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Ueda, Y.; Sato, S.; Mizuno, N.; Kumasaka, T.; Fujita, M. Self-assembly of tetravalent Goldberg polyhedra from 144 small components. Nature 2016, 540, 563–566. [Google Scholar] [CrossRef]
- Fujita, D.; Ueda, Y.; Sato, S.; Yokoyama, H.; Mizuno, N.; Kumasaka, T.; Fujita, M. Self-assembly of M30L60 icosidodecahedron. Chem 2016, 1, 91–101. [Google Scholar] [CrossRef]
- Suzuki, K.; Tominaga, M.; Kawano, M.; Fujita, M. Self-assembly of an M6L12 coordination cube. Chem. Commun. 2009, 1638–1640. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Mondal, A.; Moulton, B.; Zaworotko, M.J. Polygons and faceted polyhedra and nanoporous networks. Angew. Chem. Int. Ed. 2001, 40, 2113–2116. [Google Scholar] [CrossRef]
- Abrahams, B.F.; Egan, S.J.; Robson, R. A very large metallosupramolecular capsule with cube−like 43 topology assembled from twelve Cu(II) centers and eight tri−bidentate tri−anionic ligands derived from 2,4,6−triphenylazo−1,3,5−trihydroxybenzene. J. Am. Chem. Soc. 1999, 121, 3535–3536. [Google Scholar] [CrossRef]
- Holden, A. Shapes, Space and Symmetry; Columbia University Press: New York, NY, USA, 1971. [Google Scholar]
- Cantrill, S.J.; Pease, A.R.; Stoddart, J.F. A molecular meccano kit. J. Chem. Soc. Dalton Trans. 2000, 3715–3734. [Google Scholar] [CrossRef]
- Sun, Q.-F.; Murase, T.; Sato, S.; Fujita, M. A sphere-in-sphere complex by orthogonal self-assembly. Angew. Chem. Int. Ed. 2011, 50, 10318–10321. [Google Scholar] [CrossRef] [PubMed]
- Diudea, M.V. Rhombellanes—A new class of nanostructures. In Proceedings of the Bio-Nano-Math-Chem International Conference 2017, Cluj, Romania, 26–30 June 2017. [Google Scholar]
- Wiberg, K.B.; Walker, F.H. [1.1.1]Propellane. J. Am. Chem. Soc. 1982, 104, 5239–5240. [Google Scholar] [CrossRef]
- Kazynsky, P.; Michl, J. Staffanes: A molecular−size tinkertoy construction set for nanotechnology. Preparation of end−functionalized telomers and a polymer of [1.1.1]propellane. J. Am. Chem. Soc. 1988, 110, 5225–5226. [Google Scholar] [CrossRef]
- Diudea, M.V. Hypercube related polytopes. Iran. J. Math. Chem. 2018, 9, 1–8. [Google Scholar]
- Diudea, M.V. Rhombellanic crystals and quasicrystals. Iran. J. Math. Chem. 2018, 9, 167–178. [Google Scholar]
- Diudea, M.V. Rhombellanes—A new class of structures. Intl. J. Chem. Model. 2018, 9. in press. [Google Scholar]
- Blatov, V.A.; O’Keeffe, M.; Proserpio, D.M. Vertex-, face-, point-, Schläfli-, and Delaney-symbols in nets, polyhedra and tilings: Recommended terminology. Cryst. Eng. Comm. 2010, 12, 44–48. [Google Scholar] [CrossRef]
- Diudea, M.V. Multi-Shell Polyhedral Clusters; Springer: Dordrecht, The Netherlands, 2018. [Google Scholar]
- Steinitz, E. Polyeder und Raumeinteilungen. Enzykl. Math. Wiss. 1922, 3, 1–139. [Google Scholar]
- Diudea, M.V. Cube-rhombellane: From graph to molecule. Intl. J. Chem. Model. 2018, 9. in press. [Google Scholar]
- Do Carmo, D.R.; Paim, L.L.; Dias Filho, N.L.; Stradiotto, N.R. Preparation, characterization and application of a nanostructured composite: Octackis (cyanopropyldimetylsiloxy) octasilsesquioxane. Appl. Surface Sci. 2007, 253, 3683–3689. [Google Scholar] [CrossRef]
- Do Carmo, D.R.; Bicalho, U.O.; Silveira, T.F.; Dias Filho, N.L.; Paim, L.L. Determination of copper in different ethanolic matrices using a chloropropyl silica gel modified with a nanostructured cubic octa(3−aminopropyl)octasilsesquioxane. J. Chem. 2013, 1–11. [Google Scholar] [CrossRef]
- Eaton, P.E.; Cole, T.W. The cubane system. J. Am. Chem. Soc. 1964, 86, 962–964. [Google Scholar] [CrossRef]
- Pichierri, F. Hypercubane: DFT-based prediction of an Oh-symmetric double-shell hydrocarbon. Chem. Phys. Lett. 2014, 612, 198–202. [Google Scholar] [CrossRef]
- Hopf, H.; Liebman, J.F.; Perks, H.M. Cubanes, fenestranes, ladderanes, prismanes, staffanes and other oligocyclobutanoids. In PATAI’s Chemistry of Functional Groups; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 2009. [Google Scholar]
- Nagy, C.L.; Diudea, M.V. Nano-Studio Software; Babes-Bolyai University: Cluj, Romania, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Schrödinger; Release 2009-1; LLC: New York, NY, USA, 2009.
- Medeleanu, M.; Ciubotariu, D.; Ciubotariu, C. New shape descriptors for quantitative treatment of steric effect. III. A new globularity measure for QSPR/QSAR Studies. Chem. Bull. “POLITEHNICA” Univ. Timisoara 2006, 31, 1–2. [Google Scholar]
- Balani, S.K.; Devishree, V.S.; Miwa, G.T.; Gan, L.S.; Wu, J.T.; Lee, F.W. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top Med. Chem. 2005, 5, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Ntie−Kang, F. An in silico evaluation of the ADME profile of the Streptome DB database. Springer Plus 2013, 2, 353. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Kier, L.B. An index of flexibility from molecular shape descriptors. Prog. Clin. Biol. Res. 1989, 291, 105–109. [Google Scholar] [PubMed]
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. RECAP−retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [Google Scholar] [CrossRef] [PubMed]
- QikProp; Schrödinger, LLC: New York, NY, USA, 2009.
- Lungu, C.N.; Bratanovici, B.I.; Mirabela, G.M.; Antoci, V.; Mangalagiu, I.I. Hybrid imidazole−pyridine derivatives: An approach to novel anticancer DNA intercalators. Curr. Med. Chem. 2018, in press. [Google Scholar]
- Lungu, C.N.; Diudea, M.V.; Putz, M.V. Ligand shaping in induced fit docking of MraY inhibitors. Polynomial discriminant and laplacian operator as biological activity descriptors. Int. J. Mol. Sci. 2017, 18, 1377. [Google Scholar] [CrossRef] [PubMed]
- Lungu, C.N. C−C Chemokine Receptor Type 3 Inhibitors: Bioactivity prediction using local vertex invariants based on thermal conductivity layer matrix. Studia Univ. “Babes-Bolyai” Chemia 2018, 63, 177–188. [Google Scholar] [CrossRef]
- Delepine, B.; Duigou, T.; Carbonell, P.; Faulon, J.L. RetroPath2.0: A retrosynthesis workflow for metabolic engineers. Metab. Eng. 2018, 45, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Lungu, C.N.; Diudea, M.V. Binding site and potency of teixobactin and other lipid II ligands by statistical base scoring of conformational space maps. Curr. Comp.-Aided Drug Des. 2018, 14, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Lungu, C.N.; Diudea, M.V.; Putz, M.V.; Grudzińsk, P.I. Linear and branched PEIs (polyethylenimines) and their property space. Int. J. Mol. Sci. 2016, 17, 555. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, H.P.; Peer, J.; Sander, J.; Zimek, A. Density-based clustering. Wires. Data Min. Knowl. 2011, 1, 231–240. [Google Scholar] [CrossRef]
Sample Availability: Not applicable. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | v | C | H | N | O | Table | Type I | Type II |
|---|---|---|---|---|---|---|---|---|
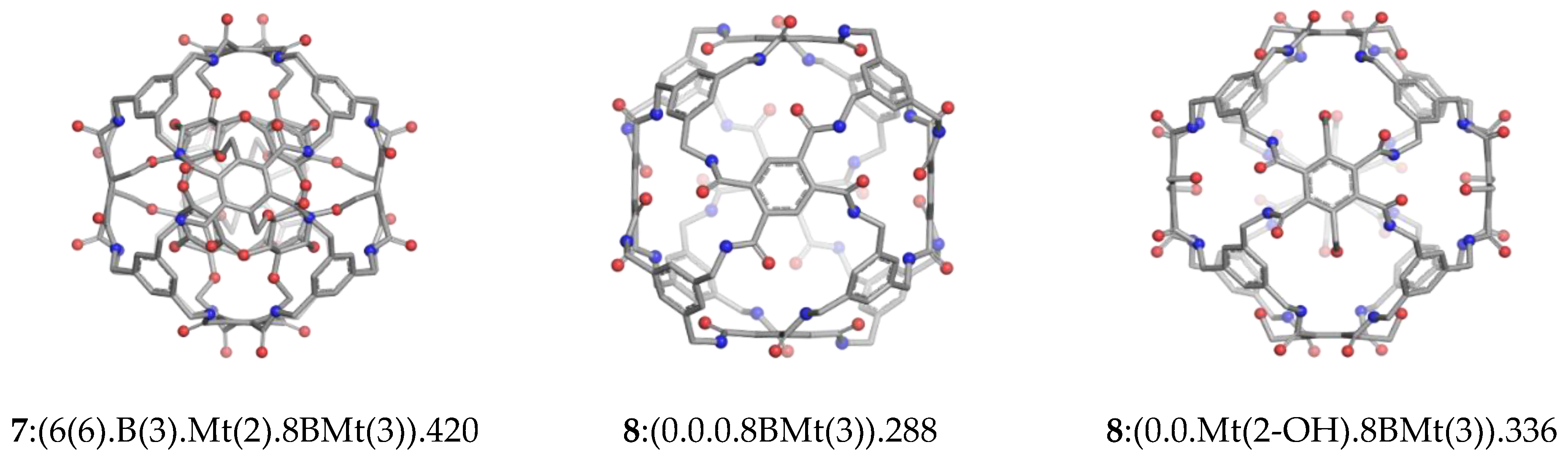
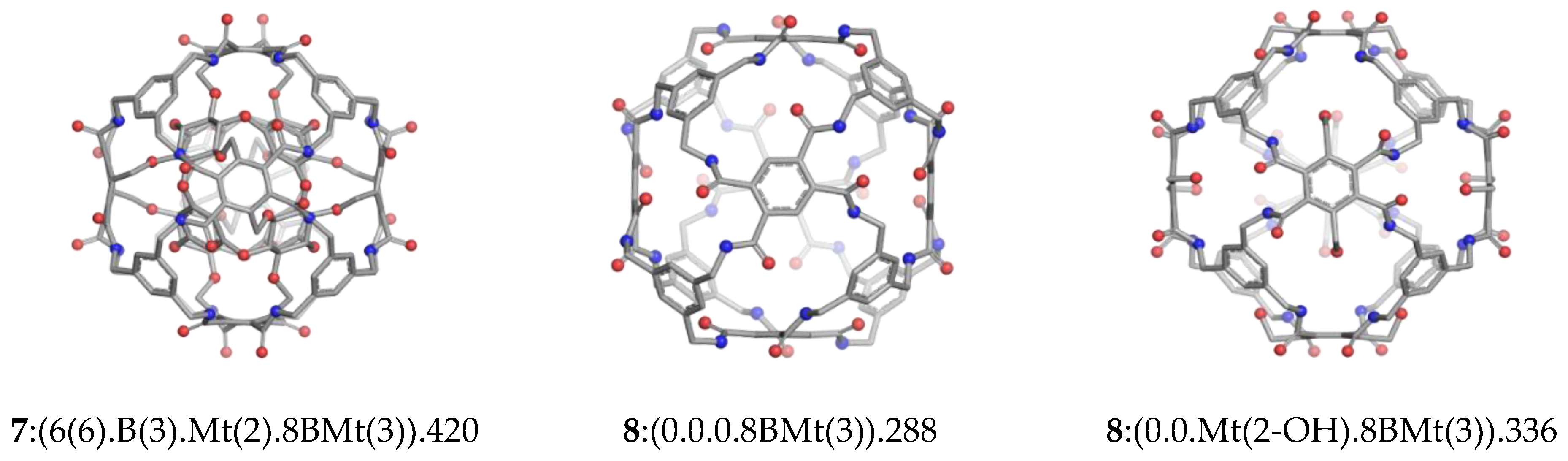
| C-rbl | 420 | 192 | 156 | 24 | 48 | 2(1) | Ether; Amide | 7:(6(6).B(3).Mt(2).8BMt(3)) |
| Env (420) | 288 | 132 | 108 | 24 | 24 | 2(2) | Amide | 8:(0.0.0.8BMt(3)) |
| Env (420) | 336 | 144 | 132 | 24 | 36 | 2(3) | Amide | 8:(0.0. Mt(2-OH).8BMt(3)) |
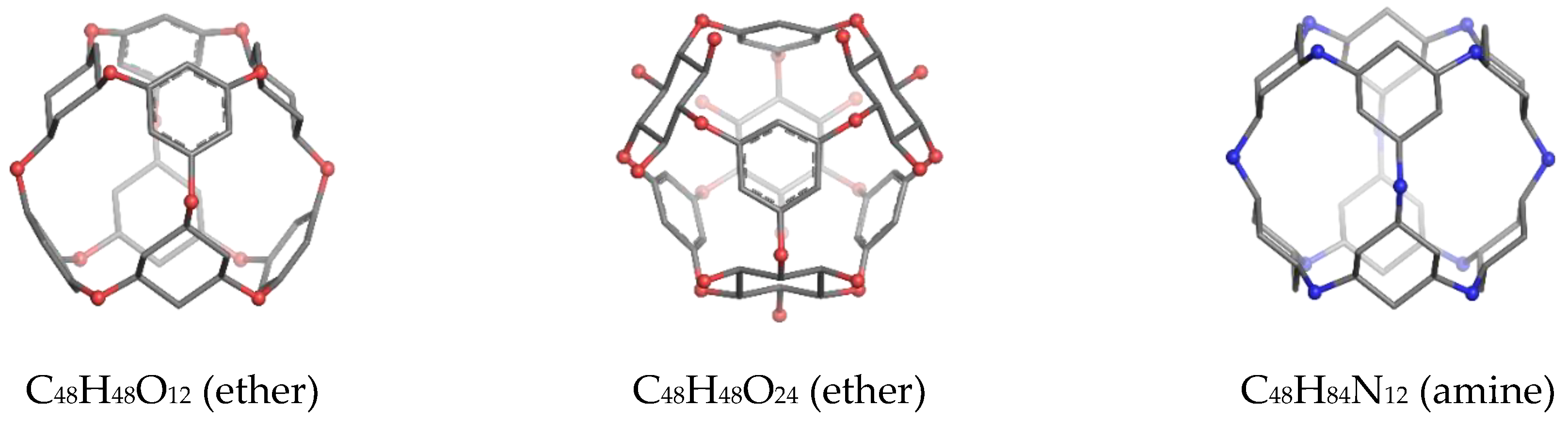
| Core | 120 | 48 | 48 | 0 | 24 | 2(4) | Ether | 4:(6(6).B(3).0.0) |
| Core | 108 | 48 | 48 | 0 | 12 | 2(5) | Ether | 4:(6(3).B(3).0.0) |
| Core | 132 | 60 | 60 | 0 | 12 | 2(6) | Ether | 4:(6(3).6(3).0.0) |
| C-rbl (4) | 264 | 132 | 84 | 0 | 48 | 2(7) | Ether; Ester | 6:((6(6).Mt(2).4B(3)) |
| C-rbl (4) | 300 | 144 | 84 | 0 | 72 | 2(8) | Ether; Ester | 6:((6(6).Mt(2-COOH).4B(3)) |
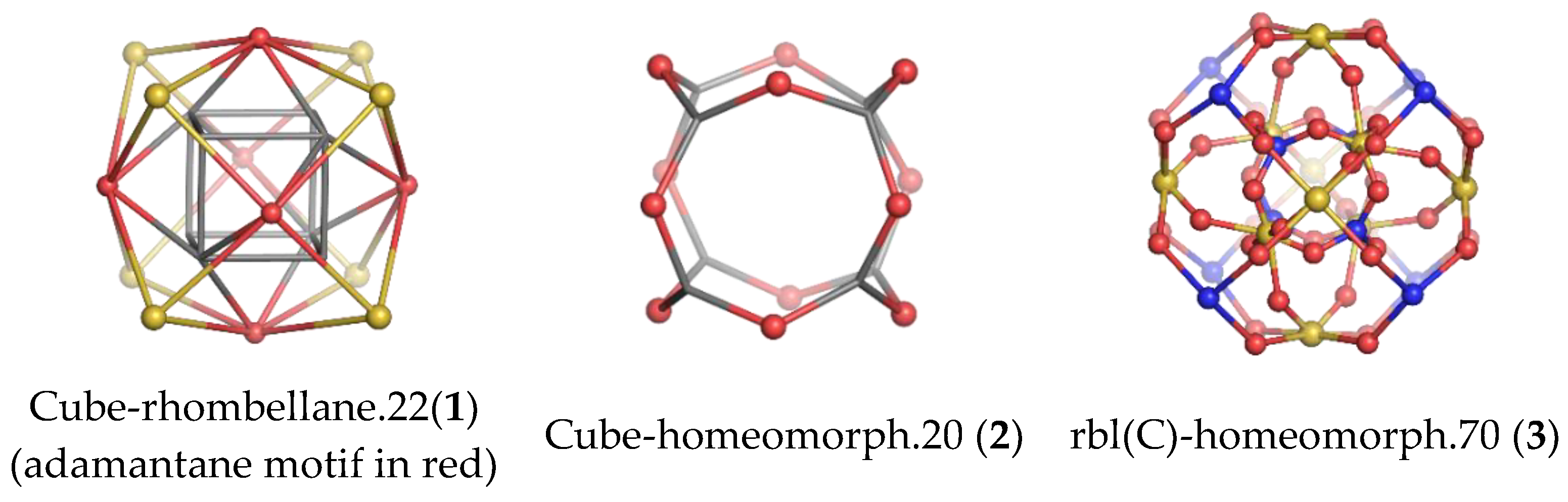
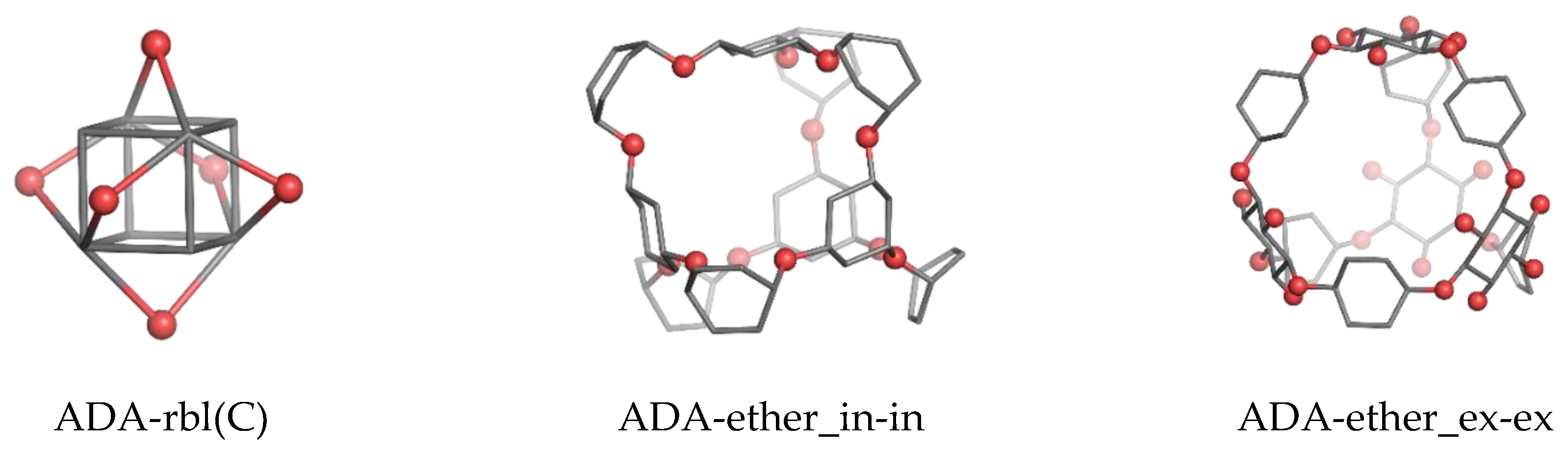
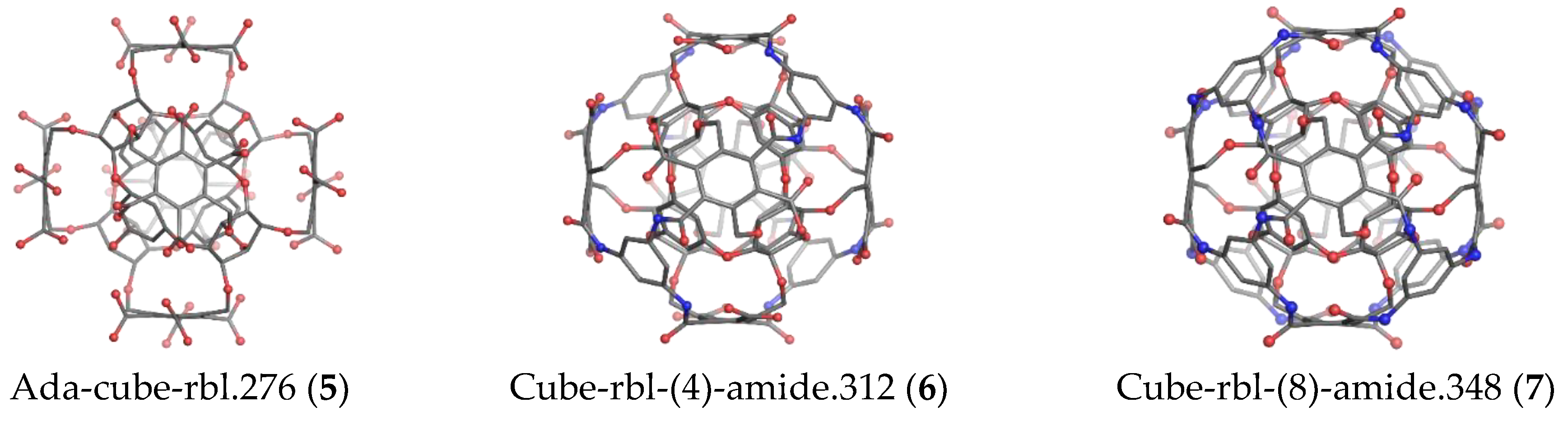
| Ada-C-rbl | 276 | 120 | 84 | 0 | 72 | 2(9); 3(1) | Ether | 5:(6(6).B(3).Mt(2).0) |
| C-rbl | 348 | 168 | 108 | 24 | 48 | 3(2) | Ether; Amide | 7:(6(6).B(3).Mt(2).8B(3)) |
| Env (348) | 264 | 120 | 84 | 24 | 36 | 3(3) | Amide | 8:(0.0.0.8B(3)) |
| C-rbl | 444 | 192 | 180 | 24 | 48 | 3(4) | Ether; Amide | 7:(6(6).6(3).Mt(2).8BMt(3)) |
| C-rbl | 456 | 156 | 192 | 24 | 84 | 3(5) | Ether; Amide | 7:(6(6).6(6).Mt(2).8BMt(3)) |
| Formula | Total Atoms | PG | Egap (eV) | Ebind (a.u.) | Ebind/N (kcal/mol) | Etot (a.u.) | |
|---|---|---|---|---|---|---|---|
| 1 | C192H156N24O48 | 420 | T | 4.390 | −93.497 | −222.236 | −12334.939 |
| 2 | C132H108N24O24 | 288 | T | 4.451 | −65.811 | −229.427 | −8215.224 |
| 3 | C144H132N24O36 | 336 | T | 4.965 | −73.981 | −227.568 | −9589.441 |
| 4 | C48H48O24 | 120 | T | 6.104 | −24.528 | −236.370 | −3663.244 |
| 5 | C48H48O12 | 108 | Td | 6.200 | −22.601 | −213.768 | −2760.590 |
| 6 | C48H72O12 | 132 | Oh | 6.139 | −24.906 | −260.481 | −2774.902 |
| 7 | C132H84O48 | 264 | T | 4.020 | −59.334 | −206.849 | −8690.700 |
| 8 | C144H84O72 | 300 | T | 3.692 | −67.266 | −195.418 | −10953.399 |
| 9 | C120H84O72 | 276 | T | 4.276 | −59.767 | −195.335 | −10039.276 |
| 10 | C12H15N3O3 | 33 | C3 | 6.282 | −6.854 | −238.962 | −856.290 |
| 11 | C16H22N4O6 | 48 | C2 | 5.220 | −9.666 | −233.289 | −1293.368 |
| Ada−C−rbl.276 (1) | C_rbl.348 (2) | Env.264 (3) | C_rbl.444 (4) | C_rbl.456 (5) | |
|---|---|---|---|---|---|
| 1 | 412.152 | 1876.9 | 133.294 | 754.677 | 1334.91 |
| 2 | 411.509 | 1888.4 | 133.281 | 770.938 | 1275.35 |
| 3 | 407.547 | 1872.15 | 133.086 | 776.412 | 1301.15 |
| 4 | 445.824 | 1931.21 | 105.310 | 793.852 | 1354.78 |
| 5 | 387.843 | 1916.52 | 150.839 | 778.074 | 1305.25 |
| 6 | 392.251 | 1858.42 | 141.293 | 820.662 | 1303.09 |
| 7 | 413403 | 1866.64 | 136.255 | 774.167 | 1283.52 |
| 8 | 421.086 | 1857.53 | 108.876 | 779.648 | 1301.57 |
| 9 | 435.597 | 1907.43 | 125.237 | 821.927 | 1293.61 |
| 10 | 439.588 | 1867.08 | 155.056 | 816.659 | 1336.80 |
| Molecule | QPpolrz | QPlogPC16 | QPlogPoct | QPlogPw | QPlogKhsa | QPlogPo/w |
|---|---|---|---|---|---|---|
| Nanotube (5,5) | 172.56 | 40.958 | 70.161 | 14.096 | 10.553 | 23.631 |
| Nanotube (3,3) | 102.772 | 25.308 | 41.65 | 6.671 | 5.881 | 13.667 |
| C70 | 58.28 | 16.258 | 22.272 | 3.89 | 2.853 | 8.587 |
| C60 | 50.017 | 14.398 | 18.716 | 3.444 | 2.292 | 7.599 |
| C_rbl_456 (3(5) | 196.72 | 59.101 | 191.881 | 189.665 | −17.697 | −26.217 |
| C_rbl_444 (3(4)) | 192.178 | 54.083 | 166.025 | 152.579 | −14.823 | −18.859 |
| C_rbl_348 (3(2)) | 180.498 | 46.315 | 169.989 | 151.045 | −4.982 | −11.232 |
| Ada−C−rbl_276 (3(1)) | 155.706 | 58.948 | 155.222 | 120.044 | −11.895 | −5.558 |
| Env_264 (3(3)) | 152.497 | 49.581 | 157.638 | 151.739 | −12.271 | −25.591 |
| Molecule | QPlogS | CIQPlogS | QPlogHERG | QPPCaco | QPlogBB | QPPMDCK | QPlogKp |
|---|---|---|---|---|---|---|---|
| Nanotube (5,5) | −55.716 | −55.716 | −11.271 | 9906.038 | 0.192 | 5899.293 | 3.712 |
| Nanotube (3,3) | −32.425 | −32.425 | −6.455 | 9906.038 | 0.192 | 5899.293 | 1.726 |
| C70 | −17.329 | −17.329 | −4.863 | 9906.038 | 0.192 | 5899.293 | 0.714 |
| C60 | −14.534 | −14.534 | −4.634 | 9906.038 | 0.192 | 5899.293 | 0.517 |
| C_rbl_456 (Tb3(5) | 2 | −6.18 | 29.537 | 0 | −13.489 | 0.002 | 0.76 |
| C_rbl_444 (Tb3(4)) | 2 | −12.722 | 29.742 | 0 | −12.202 | 0.007 | 0.52 |
| C_rbl_348 (Tb3(2)) | 2 | −17.995 | 16.156 | 0 | −6.523 | 0.003 | −6.921 |
| Ada−C−rbl_276(Tb3(1)) | 2 | −14.584 | 37.724 | 0 | −24.685 | 0 | −24.438 |
| Env_264 (Tb3(3)) | 2 | 3.158 | 29.921 | 0 | −11.775 | 0 | −12.728 |
| Molecule | CNS | Human Oral Absorption | Rule of Five Violation | Rule of Three Violation |
|---|---|---|---|---|
| Nanotube (5,5) | 1 | 1 | 2 | 1 |
| Nanotube (3,3) | 1 | 1 | 2 | 1 |
| C70 | 1 | 1 | 2 | 1 |
| C60 | 1 | 1 | 2 | 1 |
| C_rbl_456 (Tb3(5) | −2 | 1 | 3 | 2 |
| C_rbl_444 (Tb3(4)) | −2 | 1 | 3 | 2 |
| C_rbl_348 (Tb3(2)) | −2 | 1 | 3 | 2 |
| Ada−C−rbl _276 (Tb3(1)) | −2 | 1 | 3 | 2 |
| Env_264 (Tb3(3)) | −2 | 1 | 3 | 2 |
| Structure | Frequency (%) | Structure | Frequency (%) |
|---|---|---|---|
 M = 31.295 | 1.212 |  M = 30.820 | 1.212 |
 M = 29.809 | 1.212 |  M = 251.659 | 3.773 |
 M = 31.034 | 4.692 |  M = 30.612 | 8.493 |
 M = 10.646 | 2.877 |  M =14.352 | 4.721 |
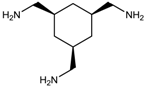 M =10.492 | 14.352 | 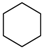 M =14.358 | 6.556 |
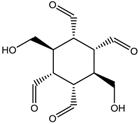 | 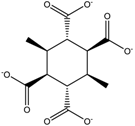 | 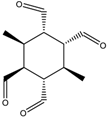 |
| M =256.254 | M = 284.22 | M = 224.256 |
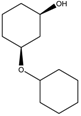 |  | 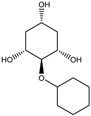 |
| M = 198.306 | M = 246.303 | M = 230.304 |
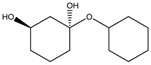 | 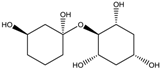 | 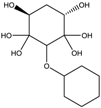 |
| M = 214.305 | M = 262.302 | M = 278.301 |
 | 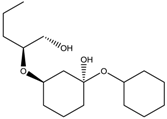 |  |
| M = 376.446 | M = 312.45 | M = 294.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diudea, M.V.; Lungu, C.N.; Nagy, C.L. Cube-Rhombellane Related Structures: A Drug Perspective. Molecules 2018, 23, 2533. https://doi.org/10.3390/molecules23102533
Diudea MV, Lungu CN, Nagy CL. Cube-Rhombellane Related Structures: A Drug Perspective. Molecules. 2018; 23(10):2533. https://doi.org/10.3390/molecules23102533
Chicago/Turabian StyleDiudea, Mircea Vasile, Claudiu Nicolae Lungu, and Csaba Levente Nagy. 2018. "Cube-Rhombellane Related Structures: A Drug Perspective" Molecules 23, no. 10: 2533. https://doi.org/10.3390/molecules23102533
APA StyleDiudea, M. V., Lungu, C. N., & Nagy, C. L. (2018). Cube-Rhombellane Related Structures: A Drug Perspective. Molecules, 23(10), 2533. https://doi.org/10.3390/molecules23102533