
Practical Enantioselective Reduction of Ketones Using Oxazaborolidine Catalysts Generated In Situ from Chiral Lactam Alcohols


Abstract
:
1. Introduction
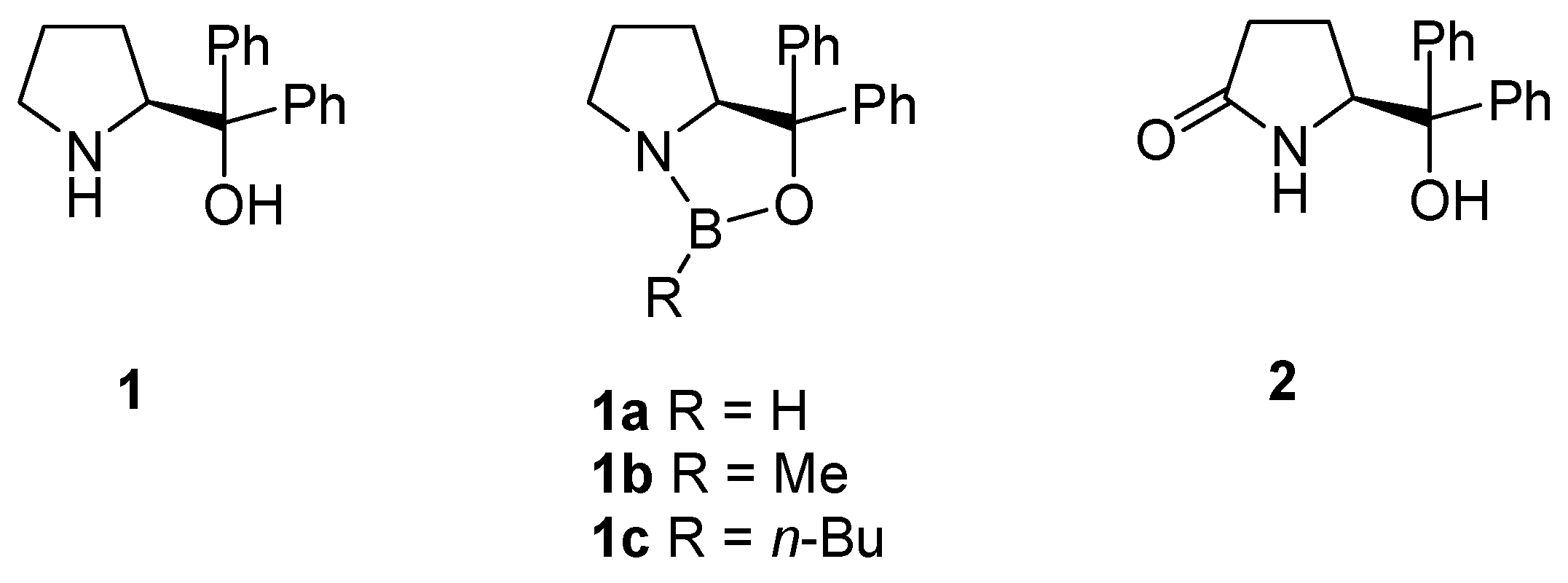
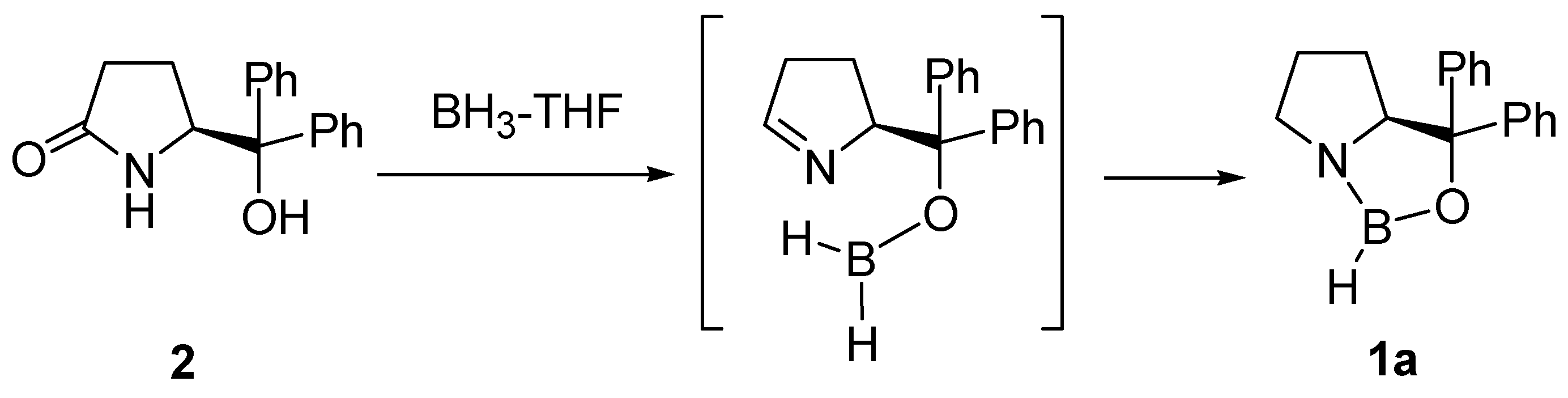
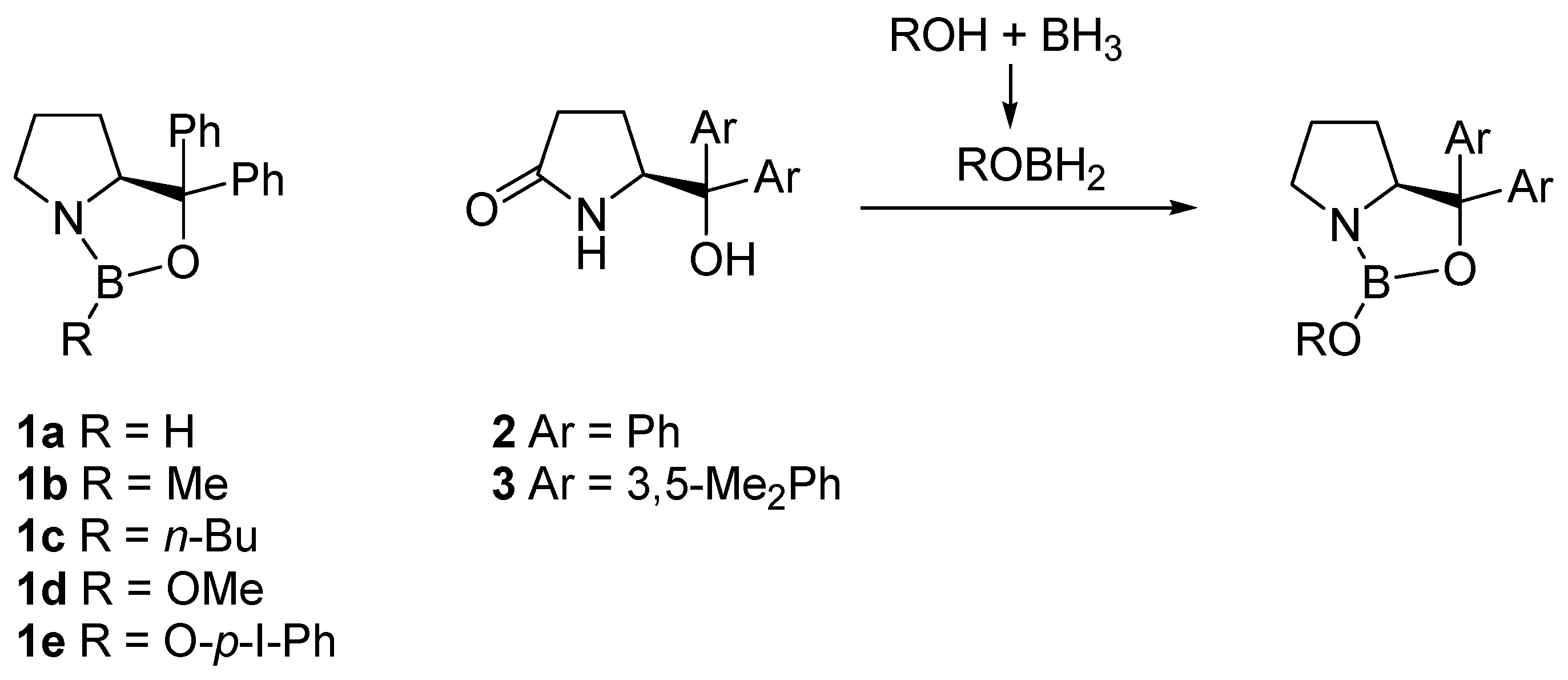
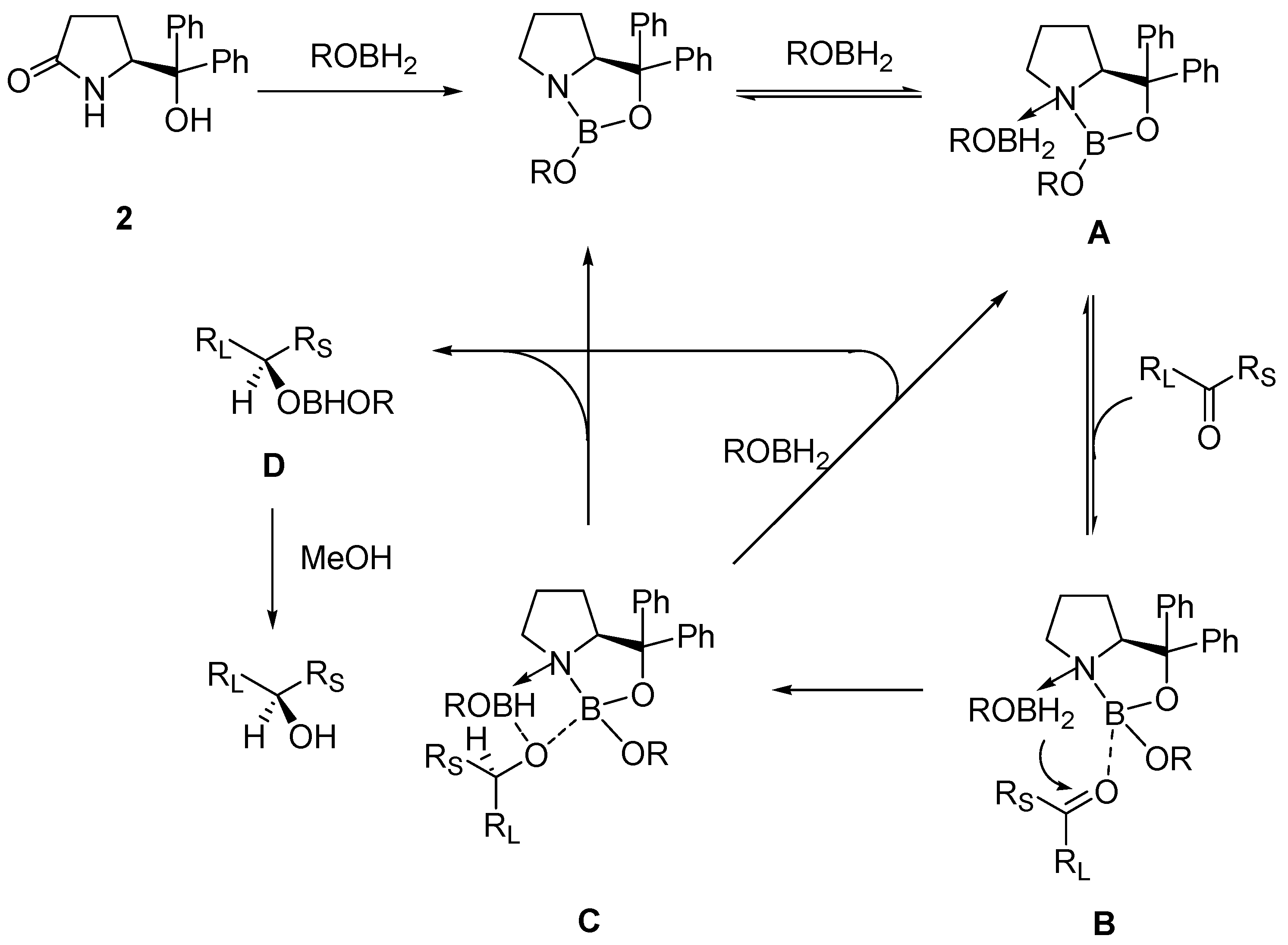
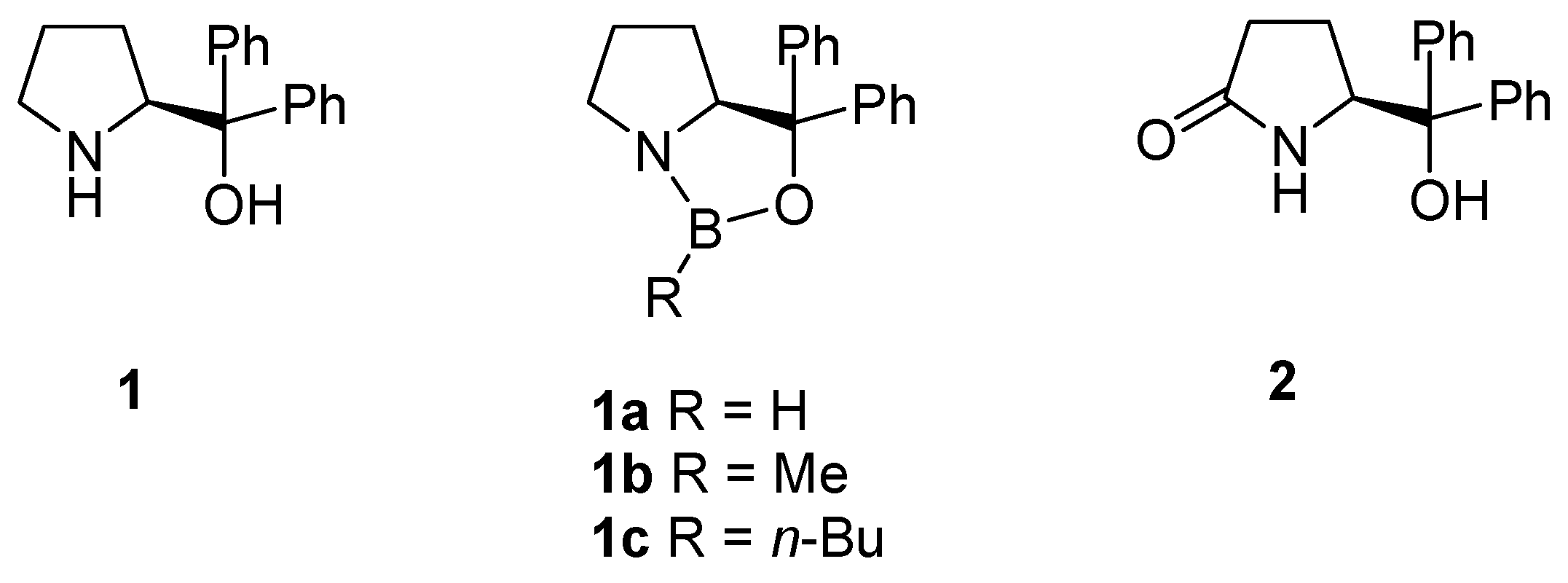
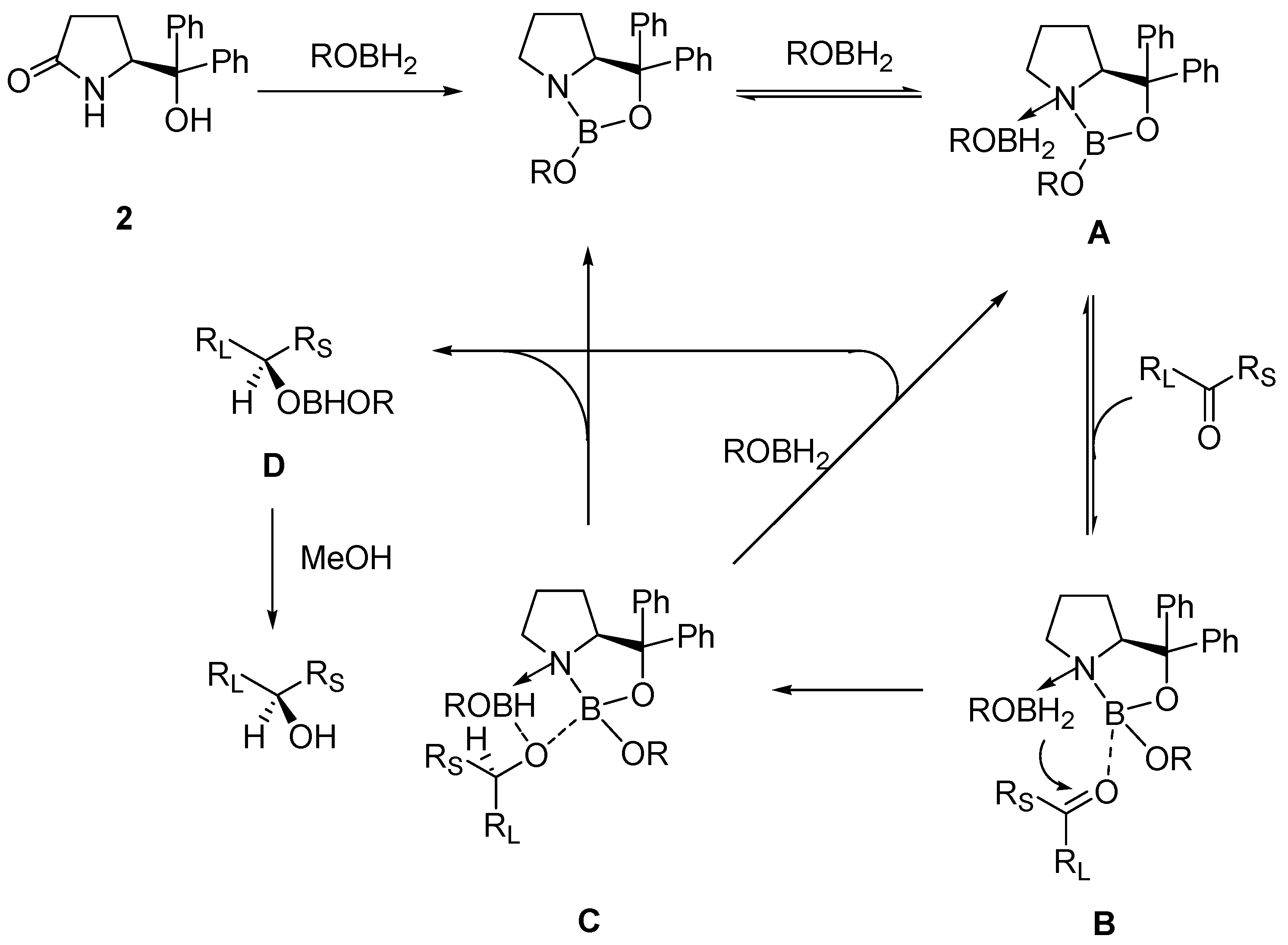
2. Oxazaborolidine Catalyst Generated In Situ from a Chiral Lactam Alcohol and Borane
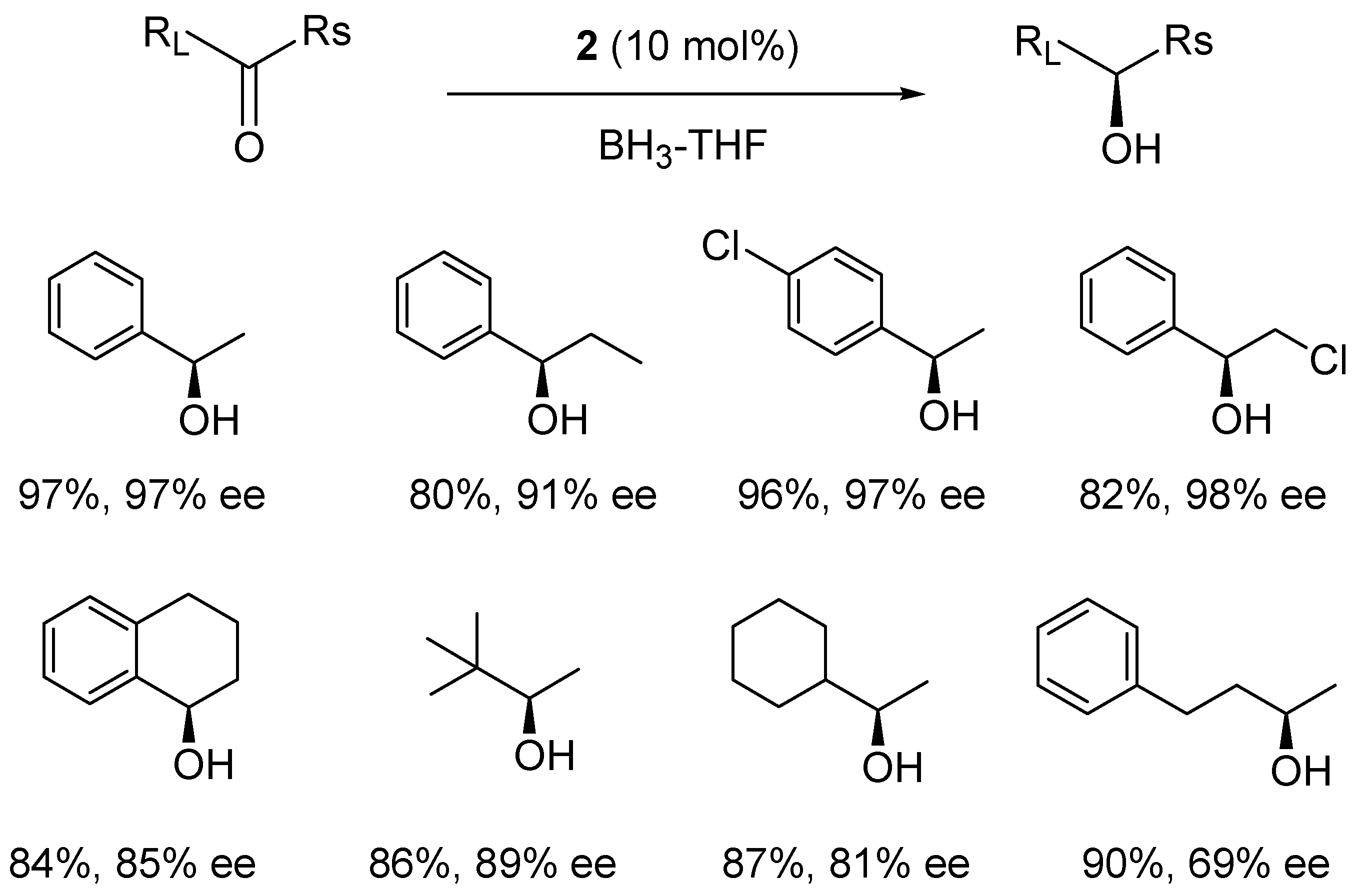
3. Asymmetric Reduction of Ketones
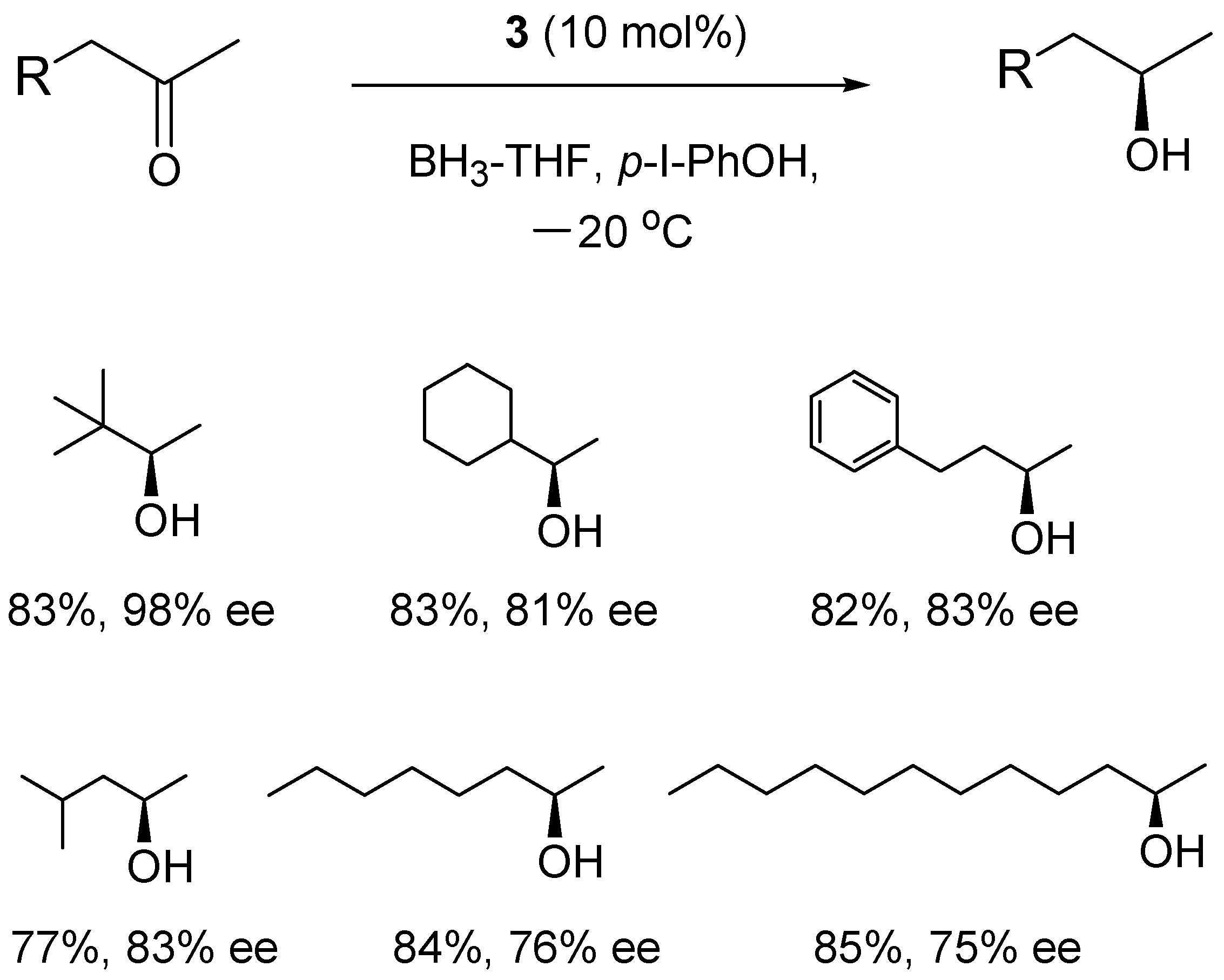
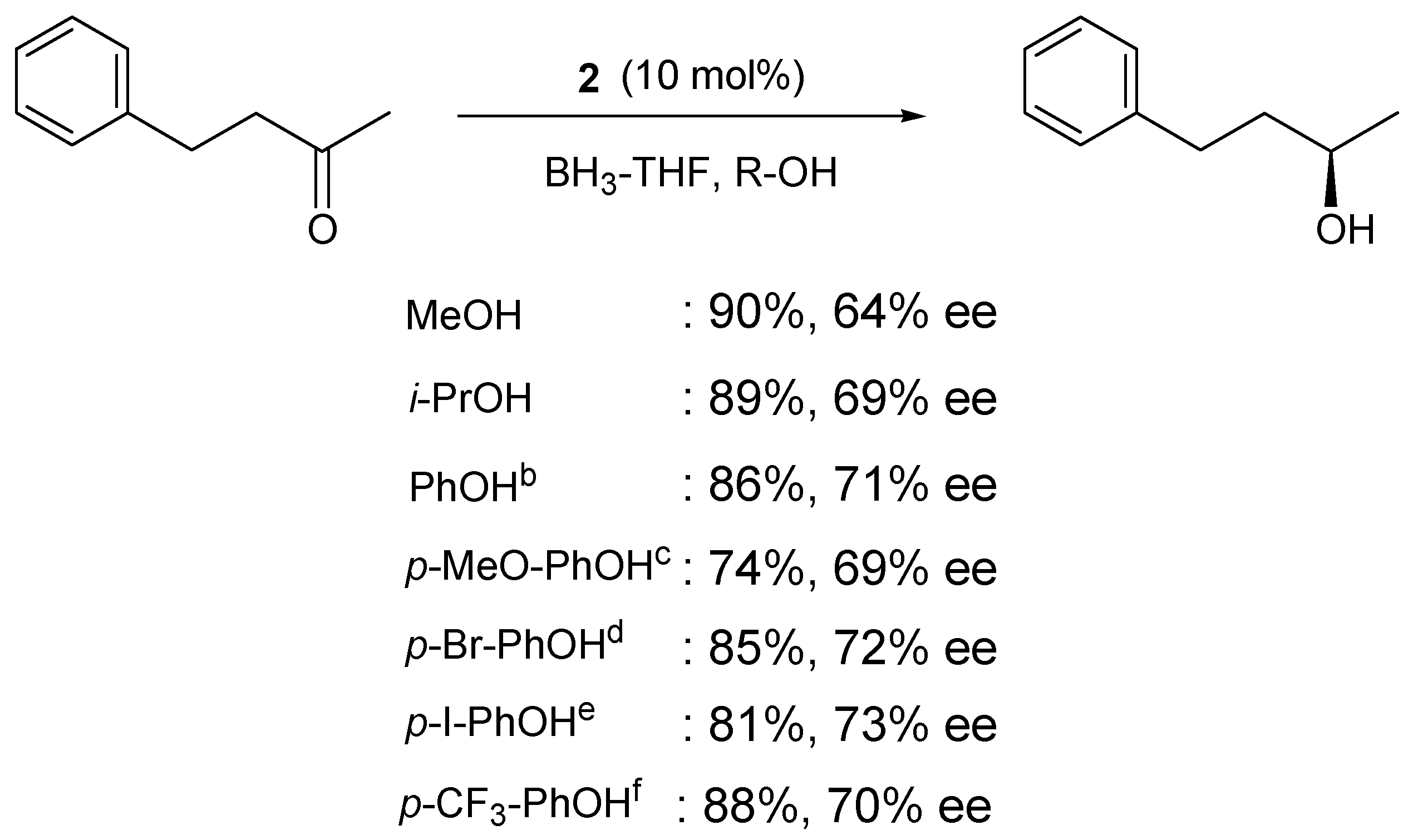
4. Asymmetric Reduction of Aliphatic Ketones
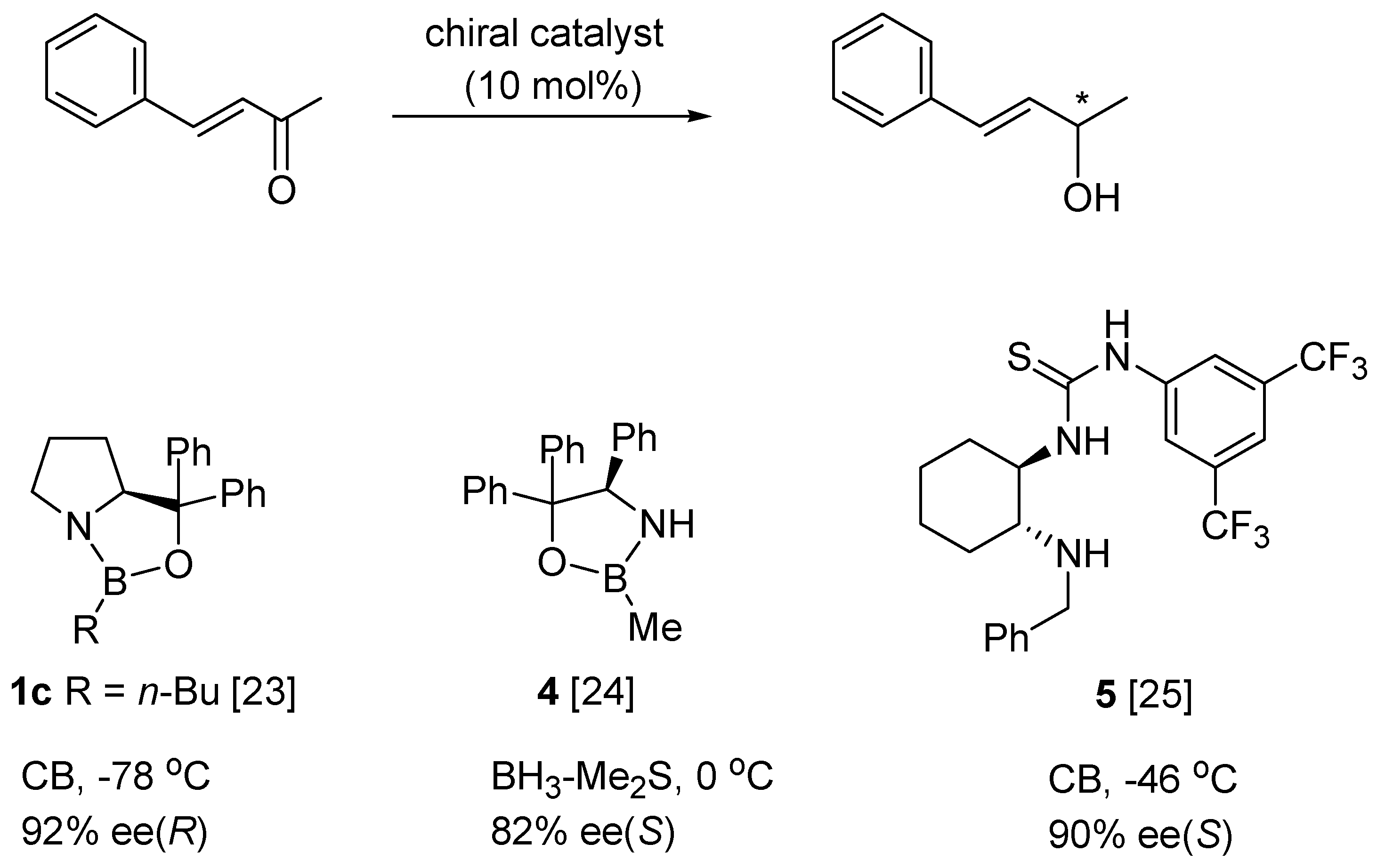
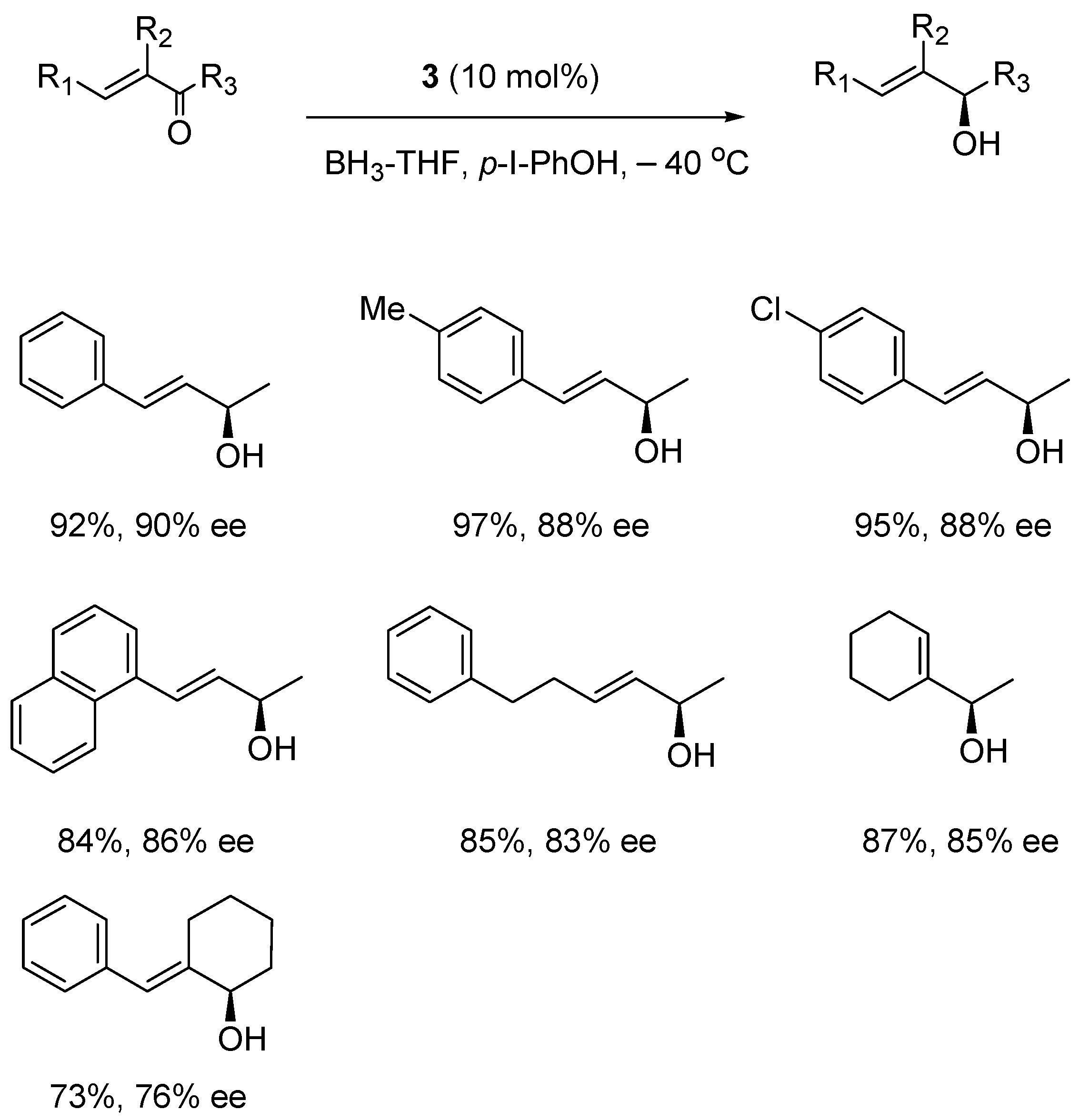
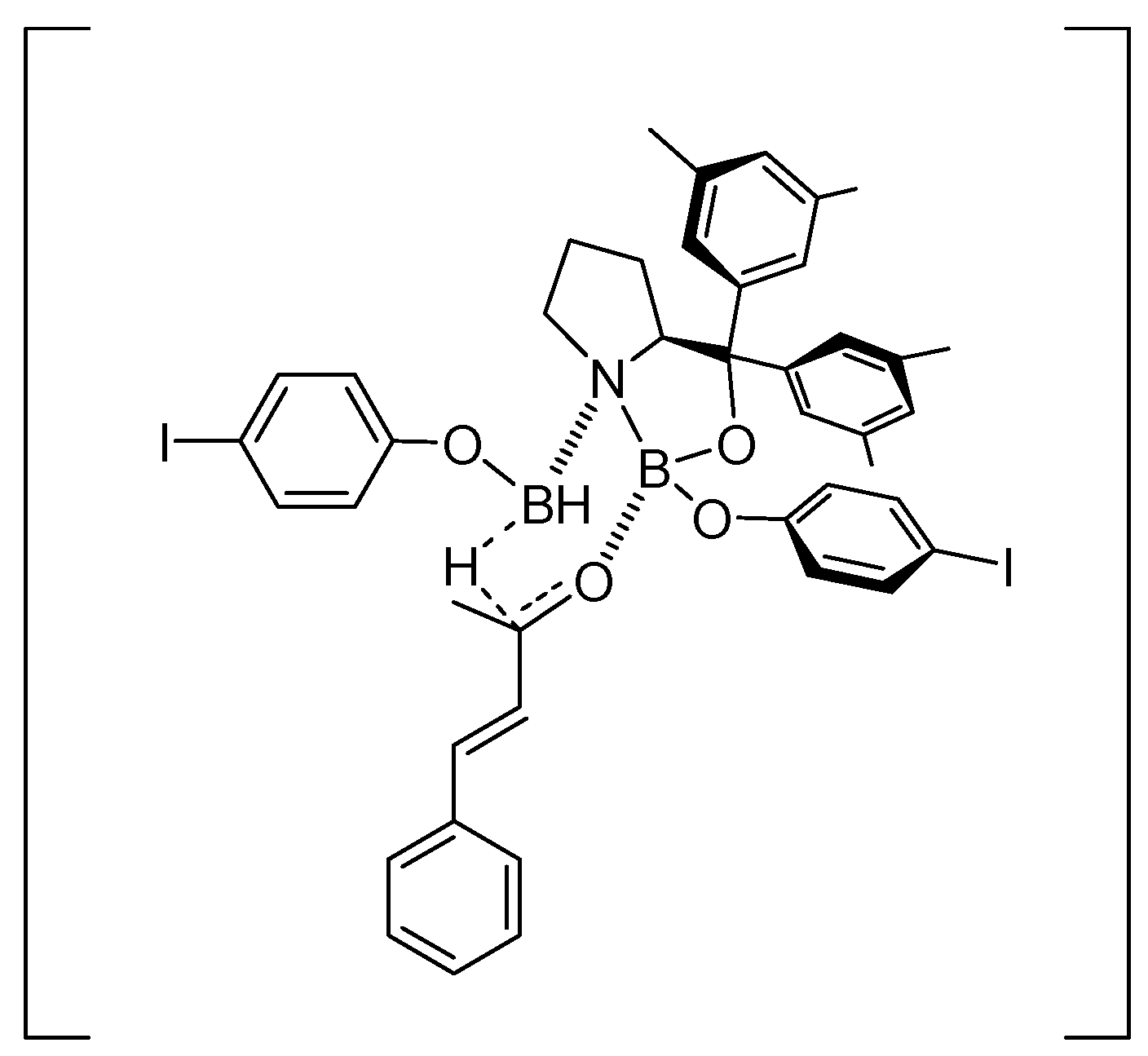
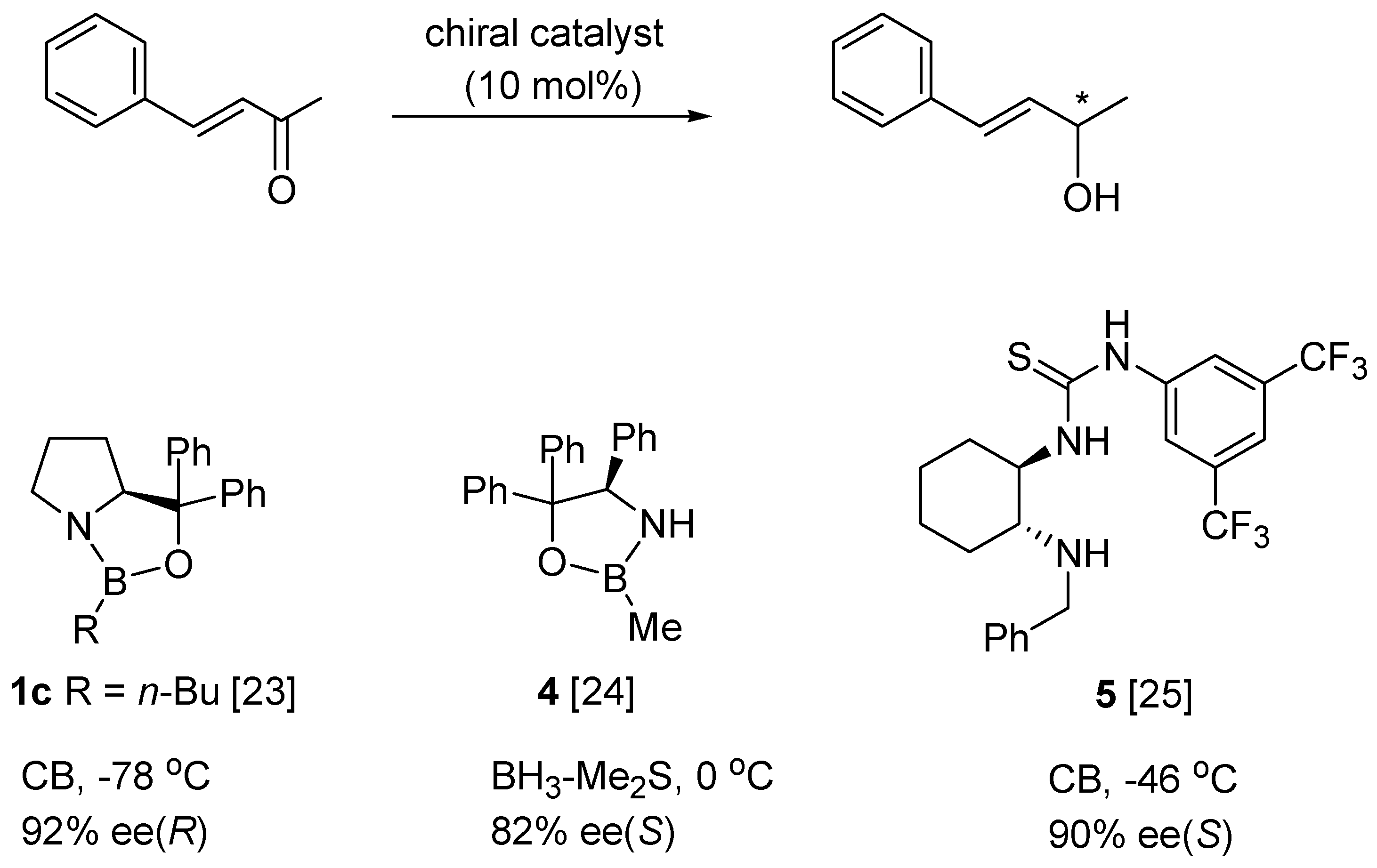
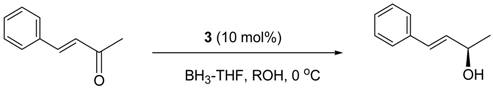
5. Asymmetric Reduction of α,β-Enones
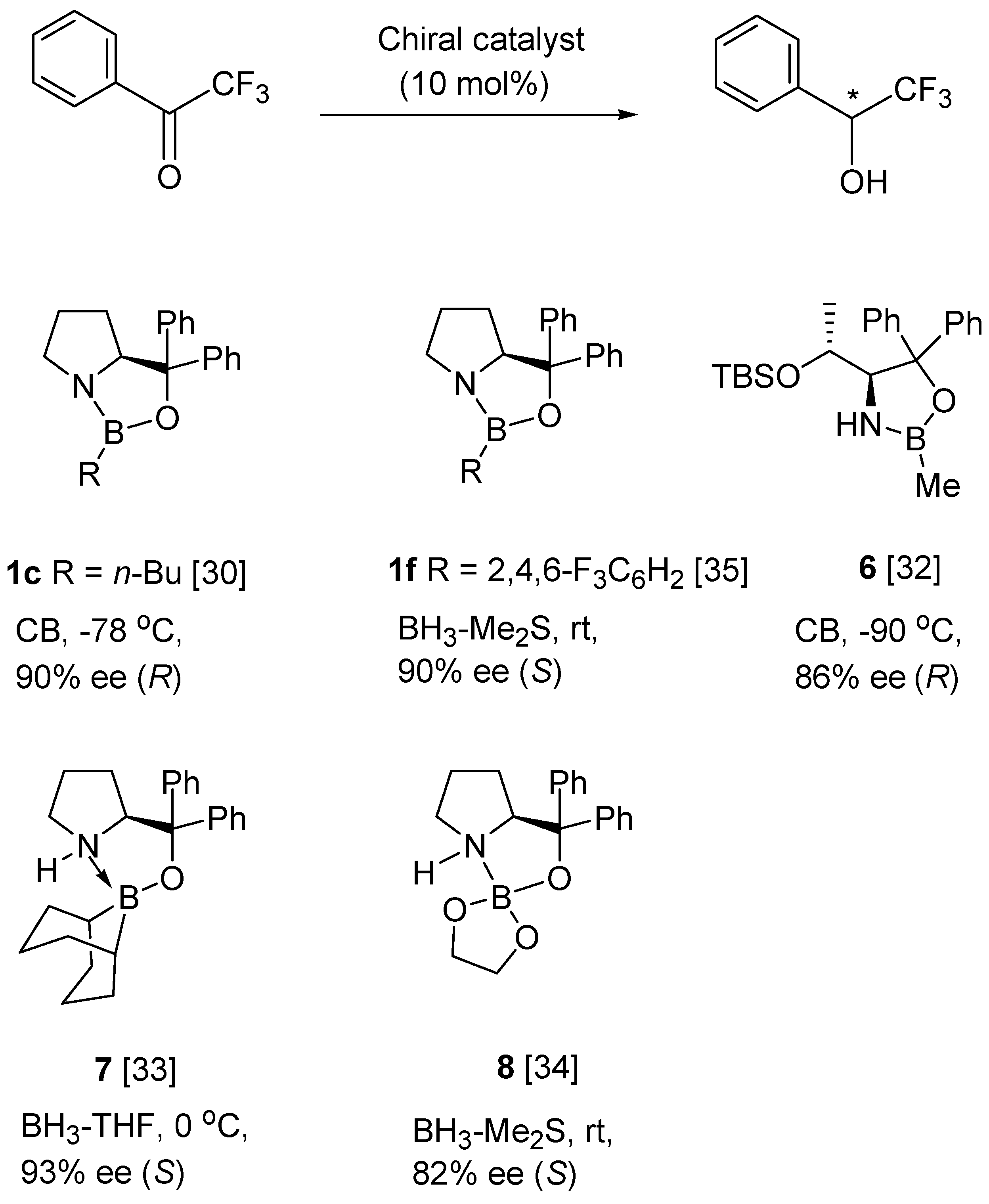
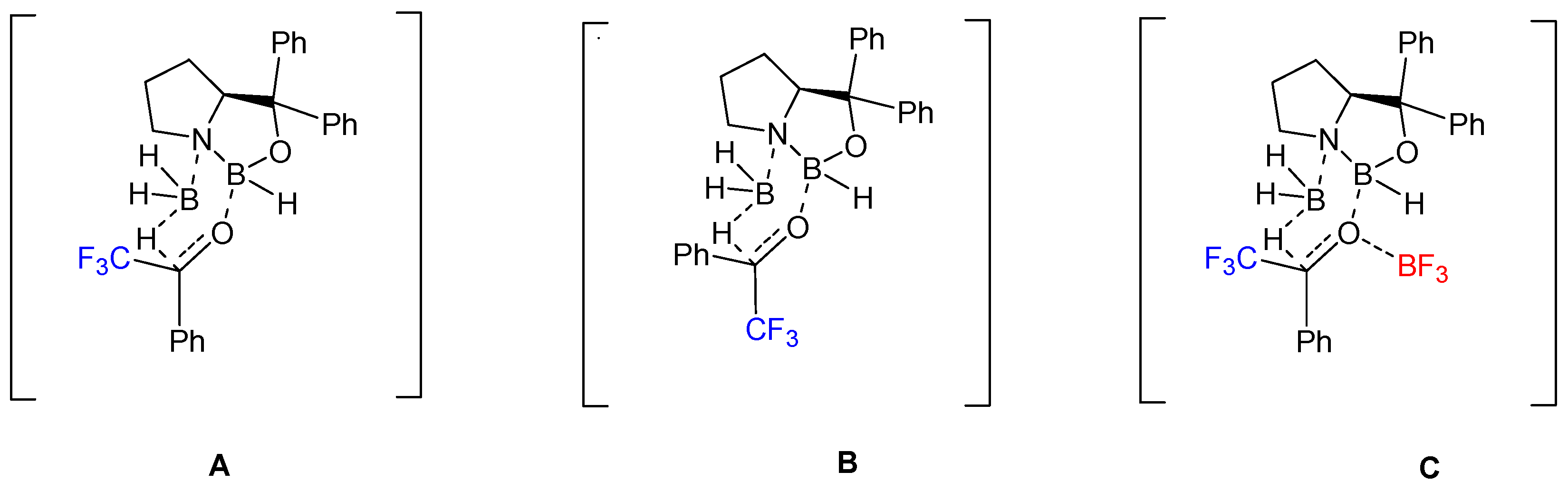
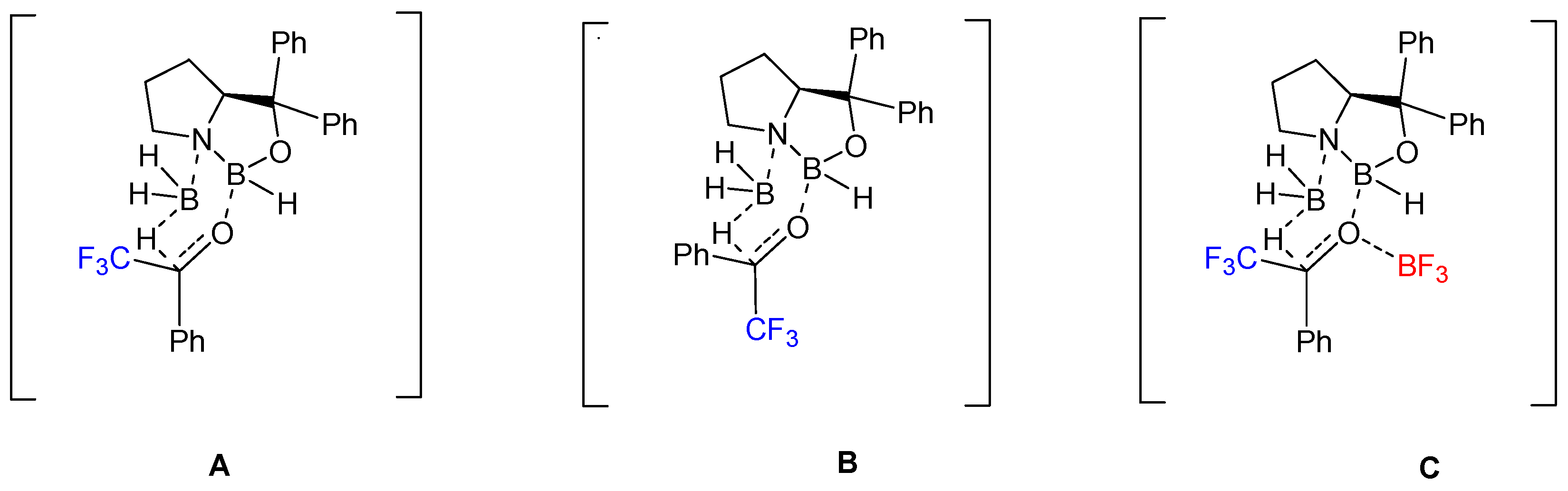
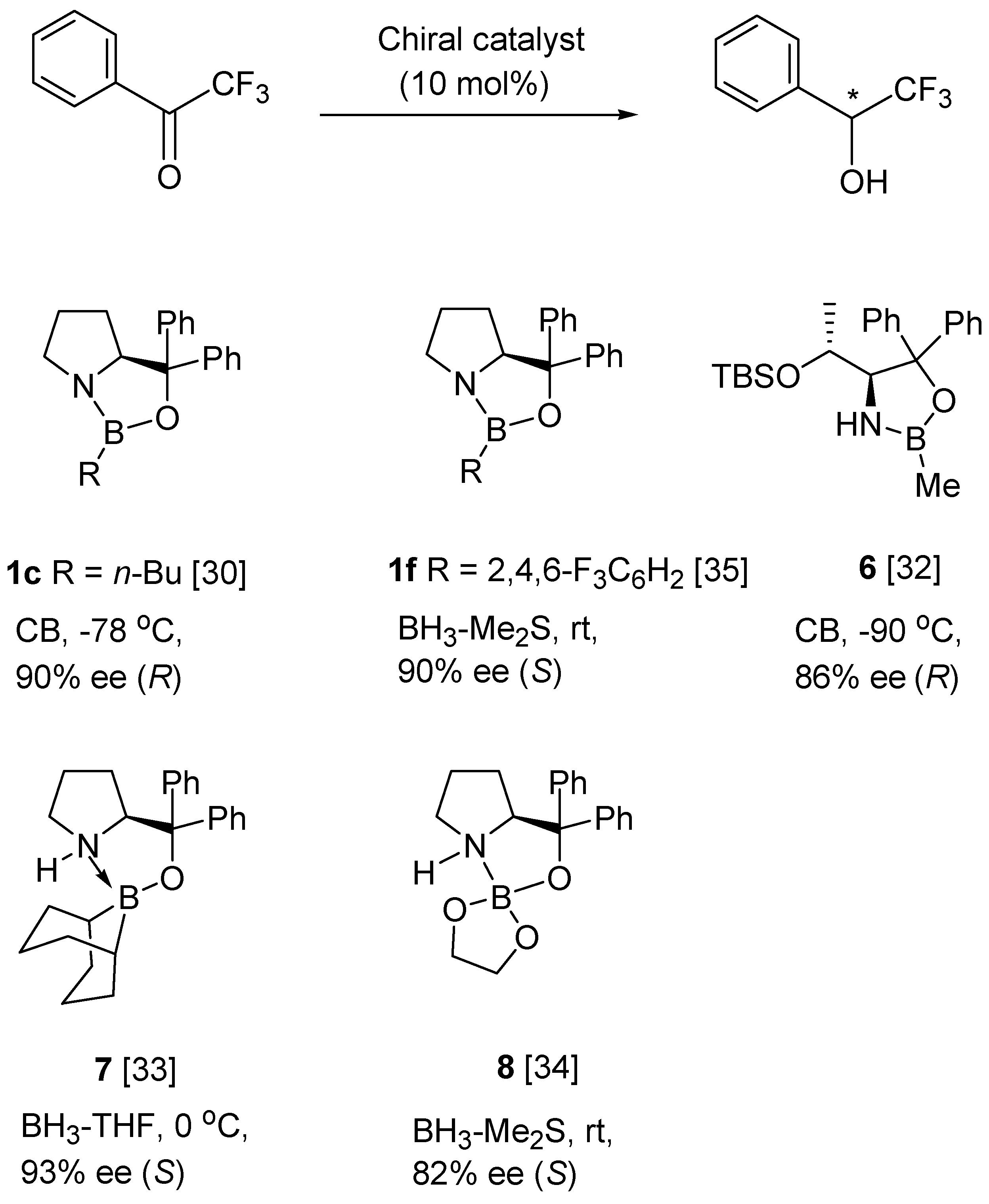
6. Asymmetric Reduction of Trifluoromethyl Ketones
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wallbaum, S.; Martens, J. Asymmetric syntheses with chiral oxazaborolidines. Tetrahedron Asymmetry 1992, 3, 1475–1504. [Google Scholar] [CrossRef]
- Singh, V.K. Practical and useful methods for the enantioselective reduction of unsymmetrical ketones. Synthesis 1992, 605–620. [Google Scholar] [CrossRef]
- Deloux, L.; Srebnik, M. Asymmetric boron-catalyzed reactions. Chem. Rev. 1993, 93, 763–777. [Google Scholar] [CrossRef]
- Corey, E.J.; Helal, C.J. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: A new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Ed. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
- Hirao, A.; Itsuno, S.; Nakahama, S.; Yamazaki, N. Asymmetric reduction of aromatic ketones with chiral alkoxy-amine-borane complexes. J. Chem. Soc. Chem. Commun. 1981, 0, 315–317. [Google Scholar] [CrossRef]
- Itsuno, S.; Hirao, A.; Nakahama, S.; Yamazaki, N. Asymmetric synthesis using chirally modified borohydrides. Part 1. Enantioselective reduction of aromatic ketones with the reagent prepared from borane and (S)-valinol. J. Chem. Soc. Perkin Trans. 1 1983, 0, 1673–1676. [Google Scholar] [CrossRef]
- Itsuno, S.; Ito, A.; Hirao, A.; Nakahama, S. Asymmetric reduction of aliphatic ketones with reagent prepared from (S)-(−)-2-amino-3-methyl-1,1-diphenylbutan-1-ol and borane. J. Org. Chem. 1984, 49, 555–557. [Google Scholar] [CrossRef]
- Corey, E.J.; Bakshi, R.K.; Shibata, S. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc. 1987, 109, 5551–5553. [Google Scholar] [CrossRef]
- Qualich, G.; Woodall, T.M. In situ oxazaborolidines, practical enantioselective hydride reagents. Synlett 1993, 12, 929–930. [Google Scholar] [CrossRef]
- Yanagi, T.; Kikuchi, K.; Takeuchi, H.; Ishikawa, T.; Nishihara, T.; Kubota, M.; Yamamoto, I. Asymmetric borane reduction of prochiral ketone using chiral bis (α,α-diphenyl-2-pyrrolidinemethanol) carbonate. Chem. Pharm. Bull. 2003, 51, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Bakshi, R.K.; Shibata, S.; Chen, C.-P.; Singh, V.K. A stable and easily prepared catalyst for the enantioselective reduction of ketones. Application to multistep syntheses. J. Am. Chem. Soc. 1987, 109, 7925–7926. [Google Scholar] [CrossRef]
- Mathre, D.J.; Jones, T.K.; Xavier, L.C.; Blacklock, T.J.; Reamer, R.A.; Mohan, J.J.; Jones, E.T.T.; Hoogsteen, K.; Baum, M.W.; Grabowski, E.J.J. A practical enantioselective synthesis of α,α-diaryl-2-pyrrolidinemethanol. Preparation and chemistry of the corresponding oxazaborolidines. J. Org. Chem. 1991, 56, 751–762. [Google Scholar] [CrossRef]
- Fujihara, H.; Tomioka, K. Asymmetric protonation of lithium enolate using 5-substituted pyrrolidin-2-one as a chiral proton source. J. Chem. Soc. Perkin Trans. 1 1999, 0, 2377–2381. [Google Scholar] [CrossRef]
- Harauchi, Y.; Takakura, C.; Furumoto, T.; Yanagita, R.C.; Kawanami, Y. Effect of BF3 on the enantioselective reduction of trifluoromethyl ketones using a chiral lactam alcohol with borane. Tetrahedron Asymmetry 2015, 26, 333–337. [Google Scholar] [CrossRef]
- Kawanami, Y.; Murao, S.; Ohga, T.; Kobayashi, N. Practical enantioselective reduction ketones using oxazaborolidine catalyst generated in situ from chiral lactam alcohol and borane. Tetrahedron 2003, 59, 8411–8414. [Google Scholar] [CrossRef]
- Kawanami, Y.; Mikami, Y.; Hoshino, K.; Suzue, M.; Kajihara, I. Enantioselective reduction of aliphatic ketones using oxazaborolidine catalyst generated in situ from chiral lactam alcohol and phenoxyborane. Chem. Lett. 2009, 38, 722–723. [Google Scholar] [CrossRef]
- Masui, M.; Shioiri, T. A practical method for asymmetric borane reduction of prochiral ketones using amino alcohols and trimethylborate. Synlett 1997, 3, 273–274. [Google Scholar] [CrossRef]
- Tehan, B.G.; Lloyd, E.J.; Wong, M.G.; Pitt, W.R.; Montana, J.G.; Manallack, D.T.; Gancia, E. Estimation of pKa using semiempirical molecular orbital methods. Part 1: Application to phenols and carboxylic acids. Mol. Inf. 2002, 21, 457–472. [Google Scholar] [CrossRef]
- Stone, G. Oxazaborolidine catalyzed borane reductions of ketones: A significant effect of temperature on selectivity. Tetrahedron Asymmetry 1994, 5, 465–472. [Google Scholar] [CrossRef]
- Gunes, Y.; Polat, M.F.; Sahin, E.; Fleming, F.F.; Altundas, R. Enantioselective synthesis of cyclic, quaternary oxonitriles. J. Org. Chem. 2010, 75, 7092–7098. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Su, W.-G. Enantioselective total synthesis of bilobalide, A C15 ginkgolide. Tetrahedron Lett. 1988, 29, 3423–3426. [Google Scholar] [CrossRef]
- Calaza, M.I.; Hupe, E.; Knochel, P. Highly anti-selective SN2′ substitutions of chiral cyclic 2-iodo-allylic alcohol derivatives with mixed zinc-copper reagents. Org. Lett. 2003, 5, 1059–1091. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Bakshi, R.K. A new system for catalytic enantioselective reduction of achiral ketones to chiral alcohols. Synthesis of chiral α-hydroxy acids. Tetrahedron Lett. 1990, 31, 611–614. [Google Scholar] [CrossRef]
- Bach, J.; Berenguer, R.; Farràs, J.; Garcia, J.; Meseguer, J.; Vilarrasa, J. Allylic alcohols of unexpected configuration by oxazaborolidine-catalyzed reduction of α,β-unsaturated ketones. An explanation based on MO calculations. Tetrahedron Asymmetry 1995, 6, 2683–2686. [Google Scholar] [CrossRef]
- Li, D.R.; He, A.; Falck, J.R. Enantioselective, organocatalytic reduction of ketones using bifunctional thiourea-amine catalysts. Org. Lett. 2010, 12, 1756–1759. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, Y.; Mikami, Y.; Kiguchi, K.; Harauchi, Y.; Yanagita, R.C. Enantioselective reduction α,β-enones using an oxazaborolidine catalyst generated in situ from chiral lactam alcohol. Tetrahedron Asymmetry 2011, 22, 1891–1894. [Google Scholar] [CrossRef]
- Shi, Y.; Cai, D.; Dolling, U.-H.; Douglas, A.W.; Tschaen, D.M.; Verhoeven, T.R. An improved method for chiral oxazaborolidine-catalyzed reduction of 4-chromanone analogs and MK-0499. Tetrahedron Lett. 1994, 35, 6409–6412. [Google Scholar] [CrossRef]
- Tschaen, D.M.; Abramson, L.; Cai, D.; Desmond, R.; Dolling, U.-H.; Frey, L.; Karady, S.; Shi, Y.; Verhoeven, T.R. Asymmetric synthesis of MK-0499. J. Org. Chem. 1995, 60, 4324–4330. [Google Scholar] [CrossRef]
- Dolbier, W.R., Jr. Fluorine chemistry at the millennium. J. Fluorine Chem. 2005, 126, 157–163. [Google Scholar] [CrossRef]
- Corey, E.J.; Cheng, X.; Cimprich, K.A.; Sarshar, S. Remarkably effective and simple syntheses of enantiomerically pure secondary carbinol from achiral ketones. Tetrahedron Lett. 1991, 32, 6835–6838. [Google Scholar] [CrossRef]
- Corey, E.J.; Link, J.O.; Bakshi, R.K. A mechanistic and structural analysis of the basis for high enantioselectivity in the oxazaborolidine-catalyzed reduction of trihalomethyl ketones by catecholborane. Tetrahedron Lett. 1992, 33, 7107–7110. [Google Scholar] [CrossRef]
- Fujisawa, T.; Onogawa, Y.; Shimizu, M. Structural effect of the oxazaborolidine derived from L-threonine in the reduction of (trifluoroacetyl)biphenyl derivatives with catecholborane. Tetrahedron Lett. 1998, 39, 6019–6022. [Google Scholar] [CrossRef]
- Kanth, J.V.B.; Brown, H.C. A new catalytic enantioselective reducing reagent system from (−)-α,α-diphenylpyrrolidinemethanol and 9-borabicyclo[3.3.1]nonane, especially effective for hindered and substituted aralkylketones. Tetrahedron 2002, 58, 1069–1074. [Google Scholar] [CrossRef]
- Stepanenko, V.; Jesus, M.D.; Correa, W.; Guzman, I.; Vazquez, C.; Cruz, W.; Ortiz-Marciales, M.; Barnes, C.L. Enantioselective reduction of prochiral ketones using spiroborate esters as catalysts. Tetrahedron Lett. 2007, 48, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, T.; Nomura, K.; Onoue, K.; Sakai, T. Rational electronic tuning of CBS catalyst for highly enantioselective borane reduction of trifluoroacetophenone. J. Chem. Soc. Chem. Commun. 2010, 46, 8624–8626. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, Y.; Hoshino, K.; Tsunoi, W. Enantioselective reduction of trifluoromethyl ketones using an oxazaborolidine catalyst generated in situ from chiral lactam alcohol. Tetrahedron Asymmetry 2011, 22, 1464–1466. [Google Scholar] [CrossRef]
- Nettles, A.M.; Matos, K.; Burkhardt, E.R.; Rouda, D.R.; Corella, J.A. Role of NaBH4 stabilizer in the oxazaborolidine-catalyzed asymmetric reduction of ketones with BH3-THF. J. Org. Chem. 2002, 67, 2970–2976. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; McAllister, T.L.; Thiruvengadam, T.K.; Tann, C.-H.; Su, D. Process for preparing ezetimibe intermediate by an acid enhanced chemo- and enantioselective CBS catalyzed ketone reduction. Tetrahedron Lett. 2003, 44, 801–804. [Google Scholar] [CrossRef]
- Anwar, S.; Periasamy, M. A convenient method for the preparation of oxazaborolidine catalyst in situ using (S)-α,α-diphenylpyrrolidinemethanol, tetrabutylammonium borohydride, and methyl iodide for the asymmetric reduction of prochiral ketones. Tetrahedron Asymmetry 2006, 17, 3244–3247. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
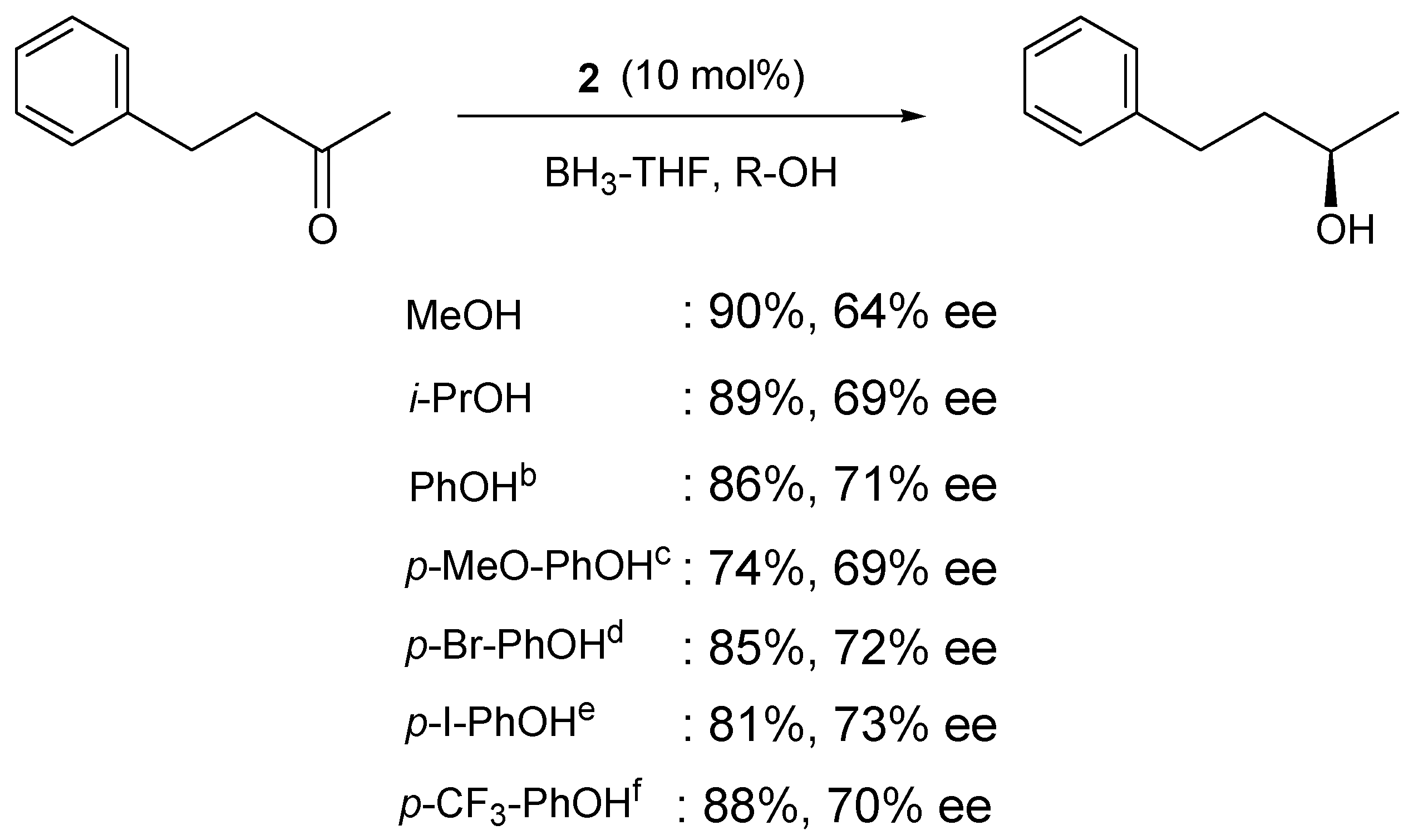
| ROH | Solvent | Yield | ee (%) |
|---|---|---|---|
| i-Pr | THF | 3 | 7 |
| Ph | THF | 7 | 8 |
| p-Cl-Ph | THF | 20 | 46 |
| p-Br-Ph | THF | 30 | 55 |
| p-I-Ph | THF | 48 | 73 |
| p-I-Ph | toloene | 61 | 73 |
| p-I-Ph | CH2Cl2 | 13 | 56 |
| p-I-Ph | CH2Cl2 | 43 | 66 |
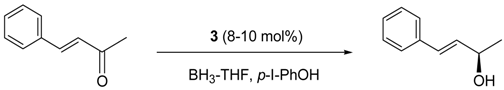
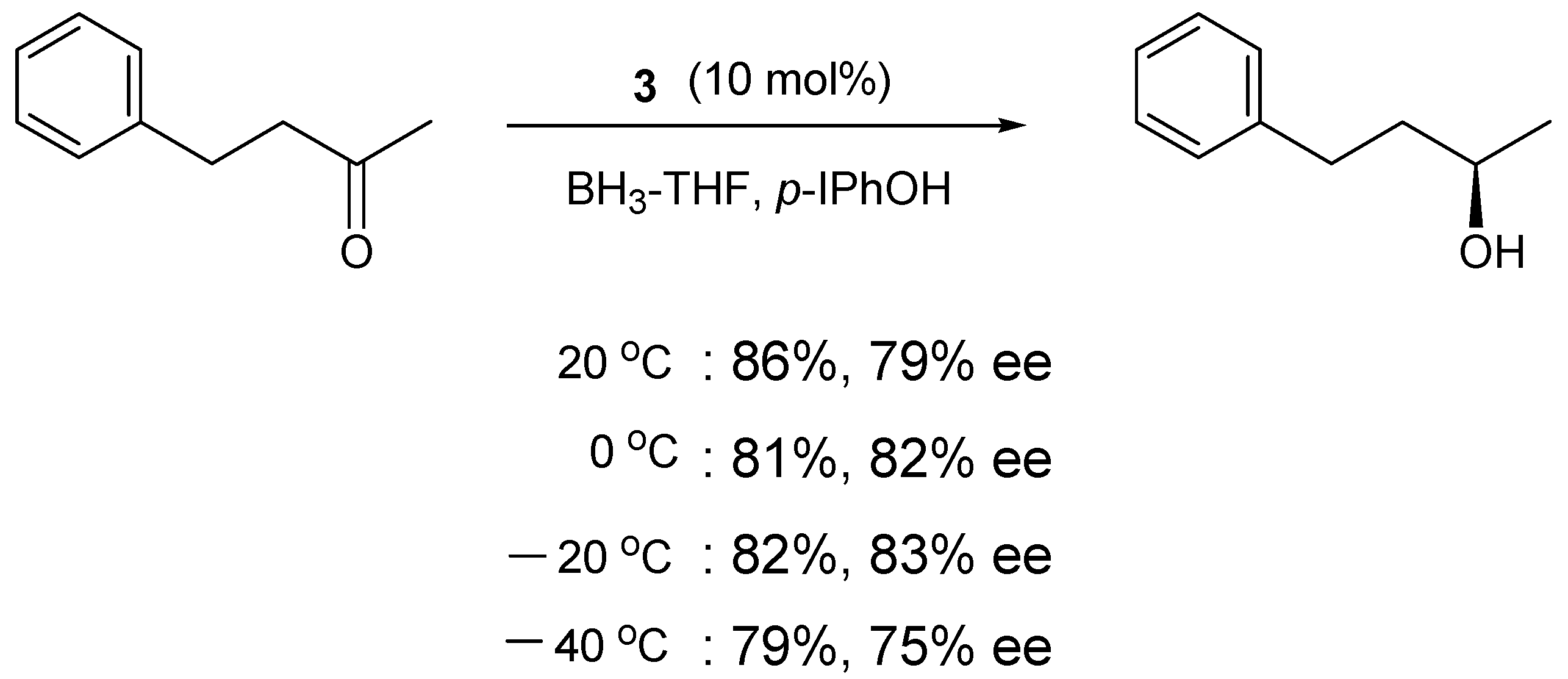
| Temp. (°C) | 3 (mol%) | p-I-PhOBH2 (equiv) | Yield (%) | ee (%) |
|---|---|---|---|---|
| 0 | 10 | 1.2 | 61 | 73 |
| −20 | 10 | 1.2 | 66 | 81 |
| −40 | 10 | 1.2 | 87 | 84 |
| −60 | 10 | 1.2 | 66 | 63 |
| −40 | 8 | 1.0 | 92 | 90 |
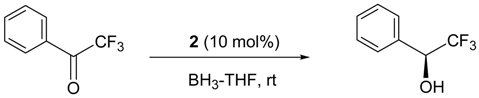
| Reducing Agent | Solvent | BF3 (mol%) | Yield (%) | ee (%) |
|---|---|---|---|---|
| BH3-THF | THF | – | 89 | 52 |
| BH3-Me2S | THF | – | 77 | 3 |
| CB | THF | – | 36 | 2 |
| p-I-PhOBH2 | THF | – | 67 | 42 |
| BH3-THF | toluene | – | 82 | 56 |
| BH3-THF | CH2Cl2 | – | 97 | 78 |
| BH3-THF | CHCl3 | – | 90 | 80 |
| BH3-THF b | CHCl3 | – | 94 | 55 |
| BH3-THF b | CHCl3 | 8 | 85 | 60 |
| BH3-THF b | CHCl3 | 160 | 91 | 80 |
| TBAB/MeI c | CHCl3 | 8 | 94 | 67 |
| TBAB/MeI c | CHCl3 | 160 | 97 | 81 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawanami, Y.; Yanagita, R.C. Practical Enantioselective Reduction of Ketones Using Oxazaborolidine Catalysts Generated In Situ from Chiral Lactam Alcohols. Molecules 2018, 23, 2408. https://doi.org/10.3390/molecules23102408
Kawanami Y, Yanagita RC. Practical Enantioselective Reduction of Ketones Using Oxazaborolidine Catalysts Generated In Situ from Chiral Lactam Alcohols. Molecules. 2018; 23(10):2408. https://doi.org/10.3390/molecules23102408
Chicago/Turabian StyleKawanami, Yasuhiro, and Ryo C. Yanagita. 2018. "Practical Enantioselective Reduction of Ketones Using Oxazaborolidine Catalysts Generated In Situ from Chiral Lactam Alcohols" Molecules 23, no. 10: 2408. https://doi.org/10.3390/molecules23102408
APA StyleKawanami, Y., & Yanagita, R. C. (2018). Practical Enantioselective Reduction of Ketones Using Oxazaborolidine Catalysts Generated In Situ from Chiral Lactam Alcohols. Molecules, 23(10), 2408. https://doi.org/10.3390/molecules23102408