Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns

 ,
, 
 ,
,  and
and 
Abstract
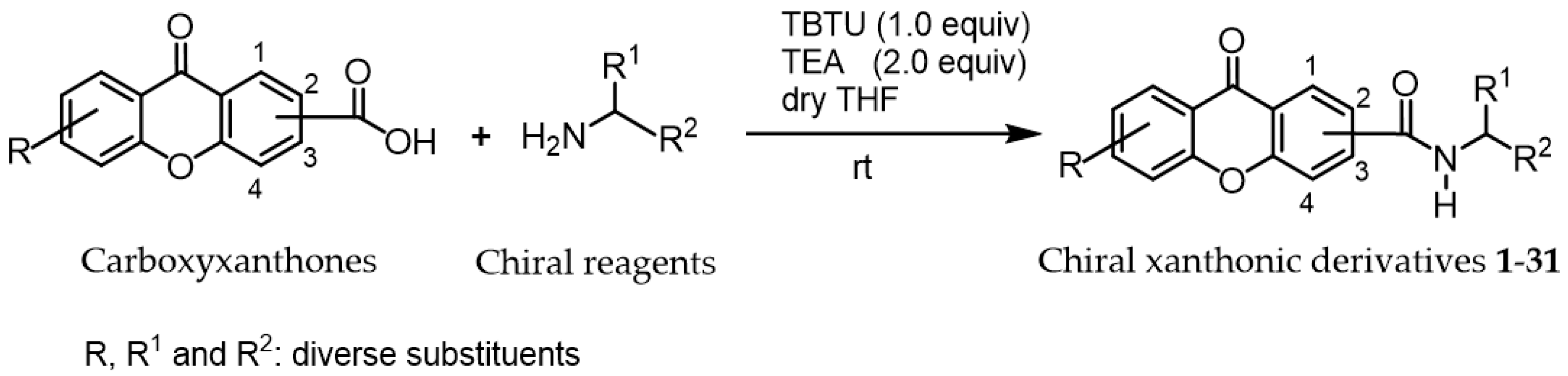
:1. Introduction
2. Results and Discussion
2.1. Performance of Chirobiotic T Column for Enantioresolution of Chiral Xanthonic Analytes
2.2. Performance of ChirobioticTM R Column for Enantioresolution of Chiral Xanthonic Analytes
2.3. Performance of ChirobioticTM V Column for Enantioresolution of Chiral Xanthonic Analytes
2.4. Performance of ChirobioticTM TAG Column for Enantioresolution of Chiral Xanthonic Analytes
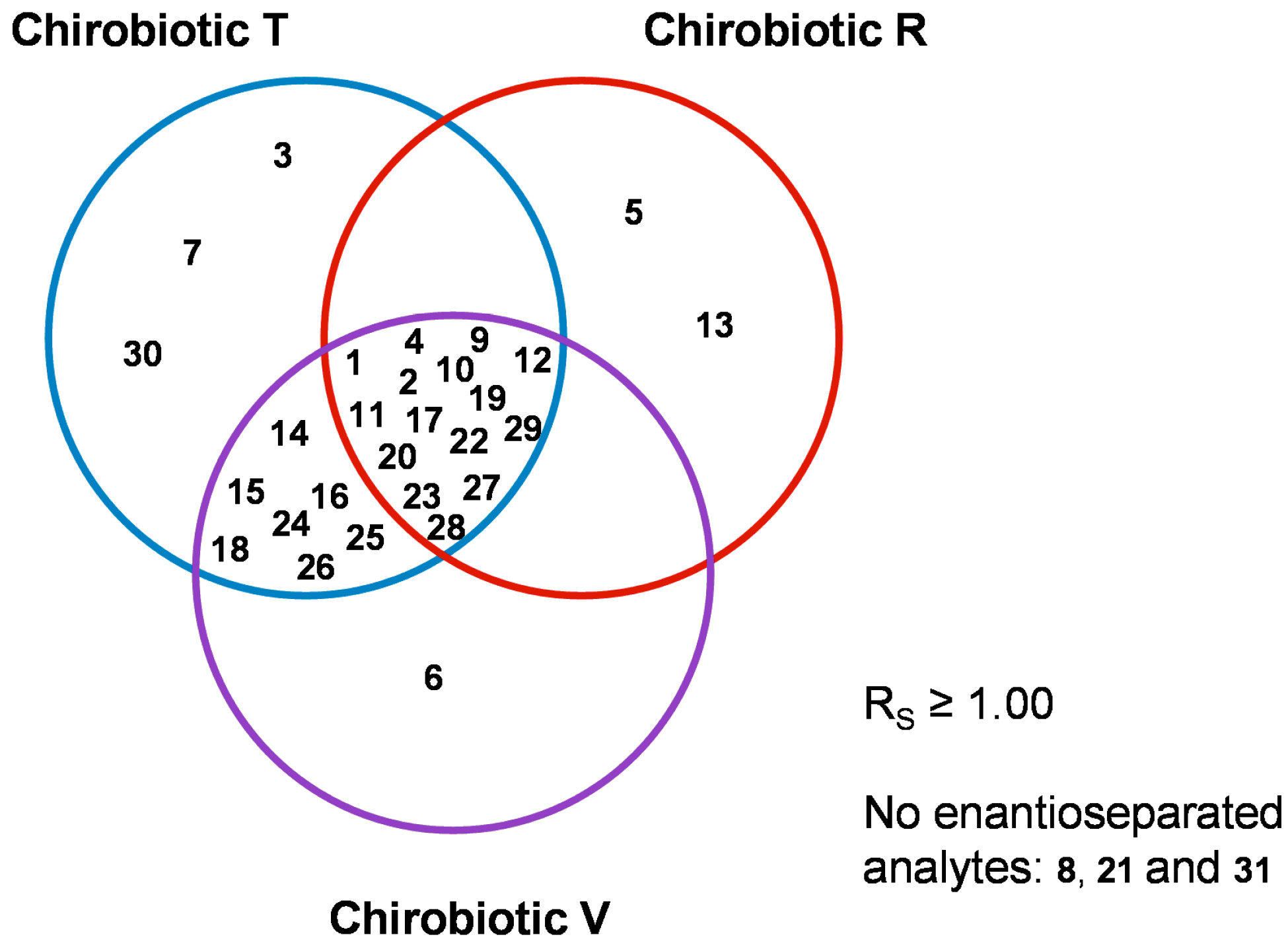
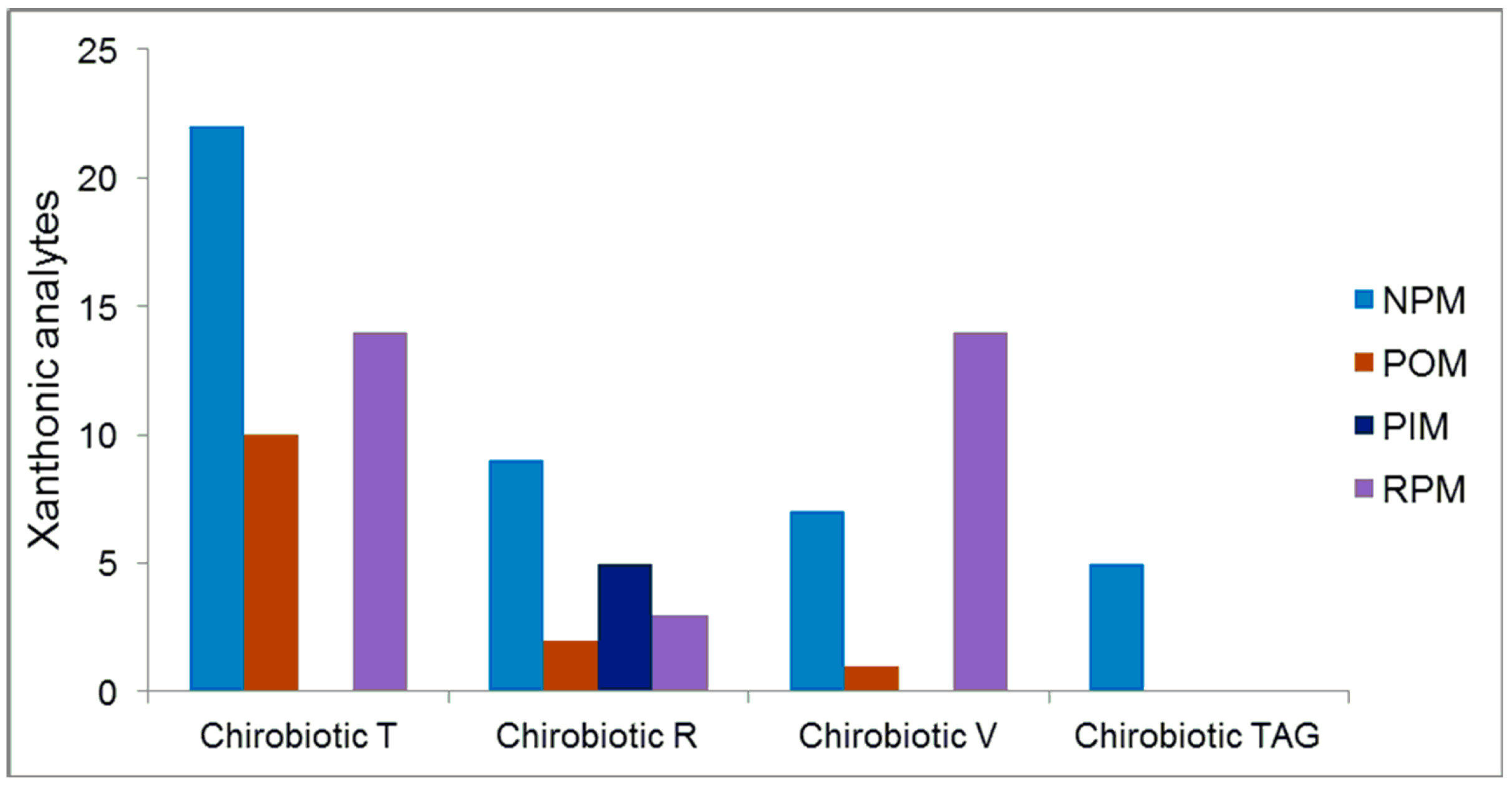
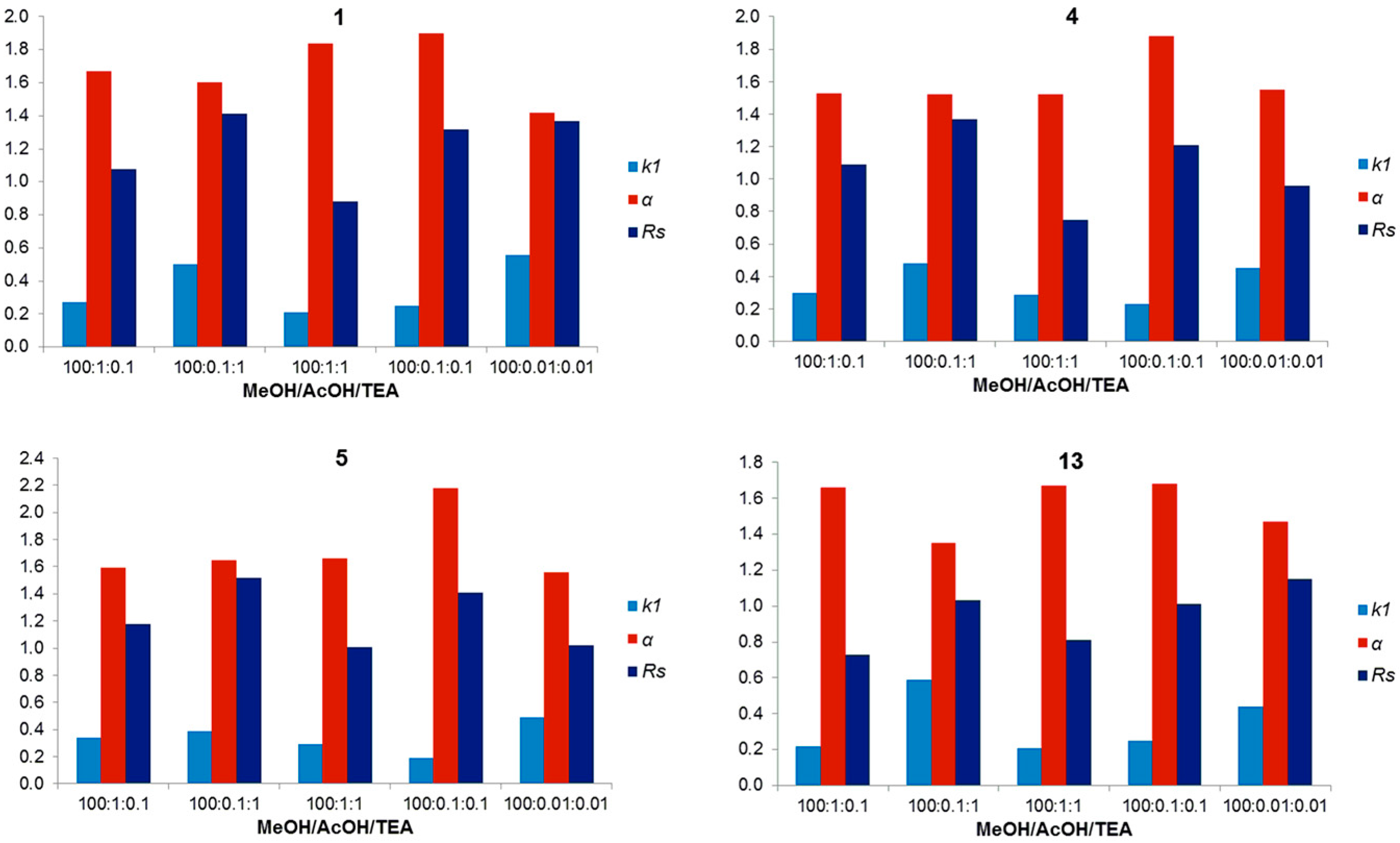
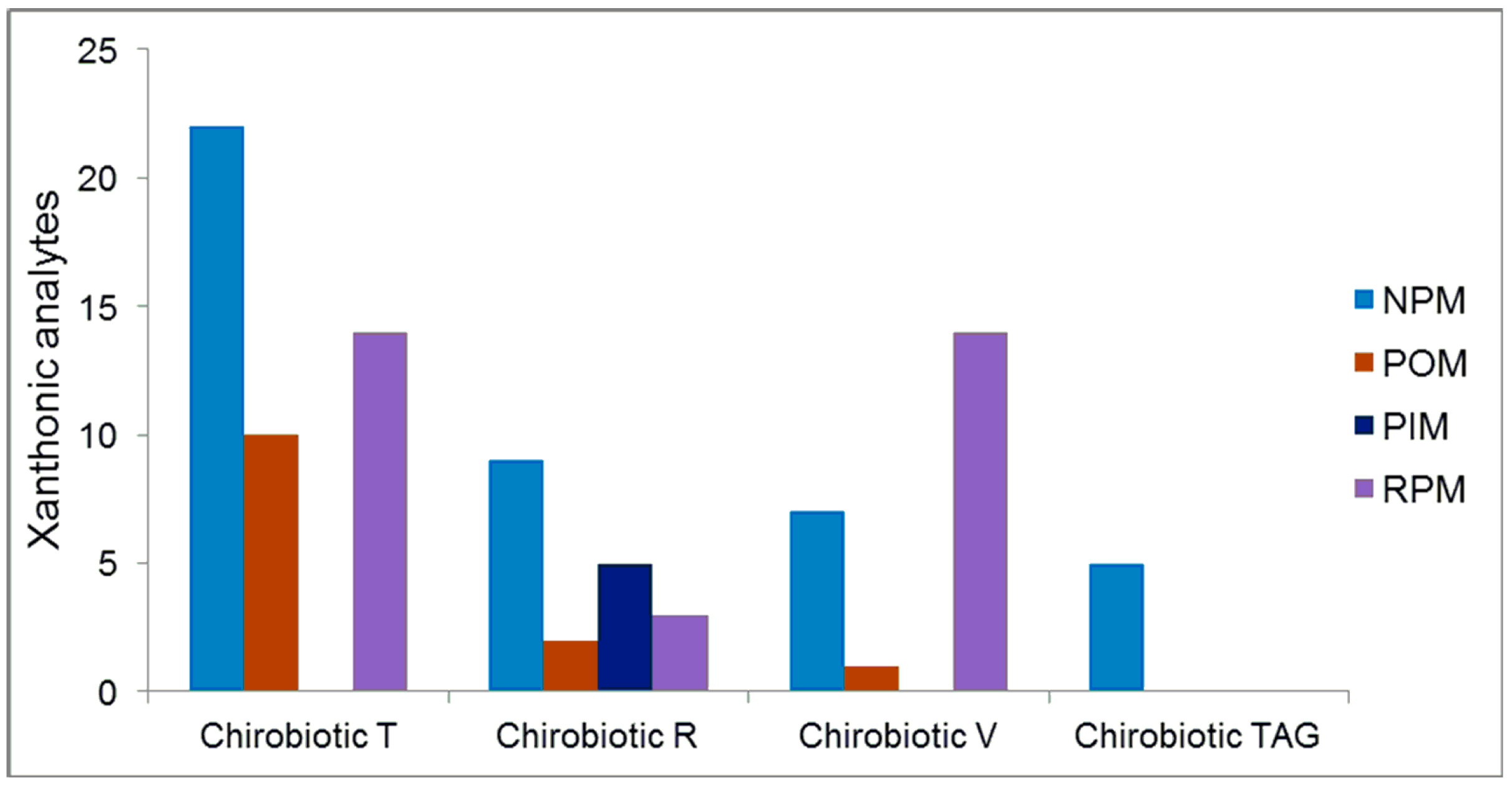
2.5. Overall Effectiveness of ChirobioticTM Columns for Enantioresolution of Chiral Xanthonic Compounds under Multimodal Elution Conditions
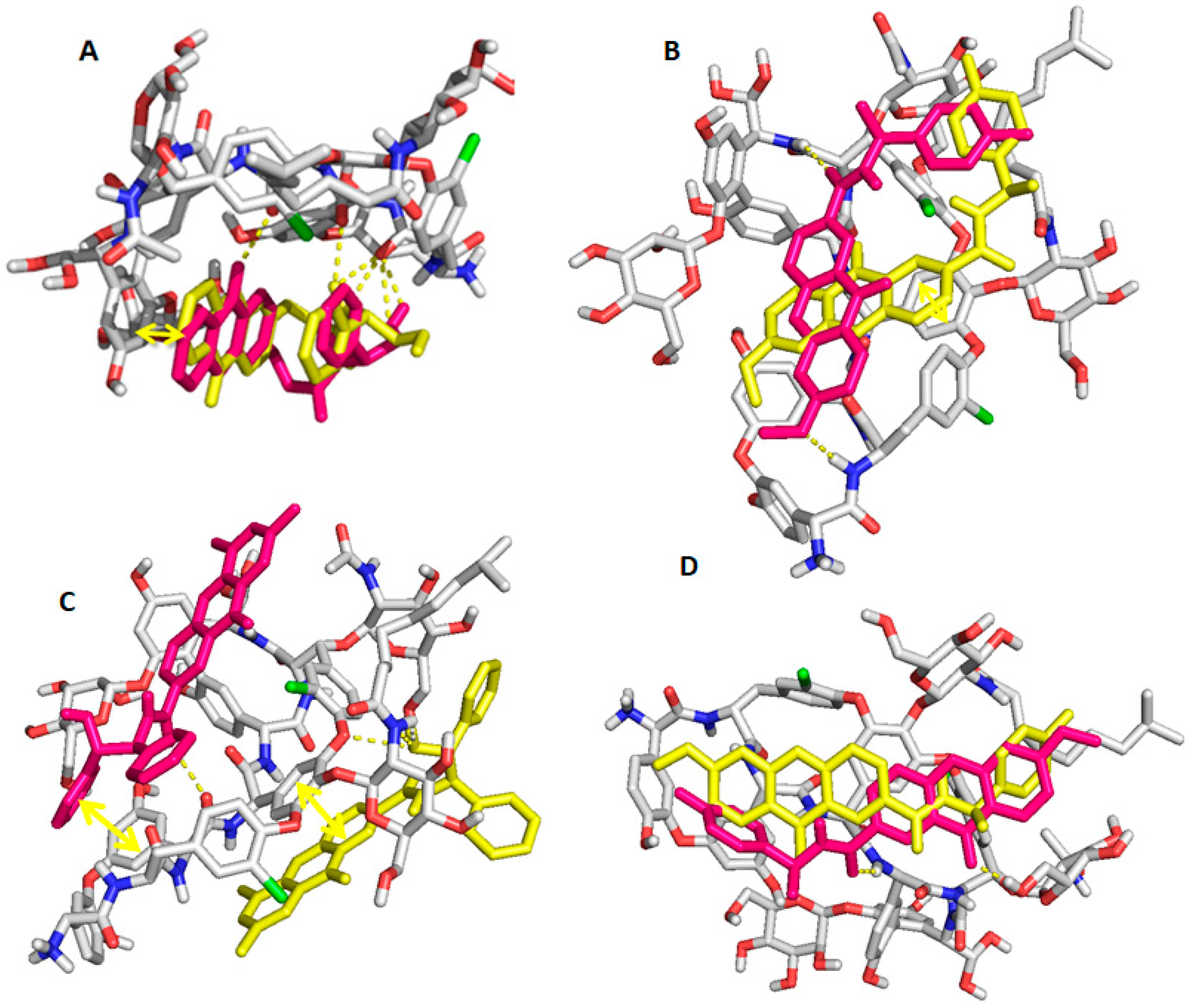
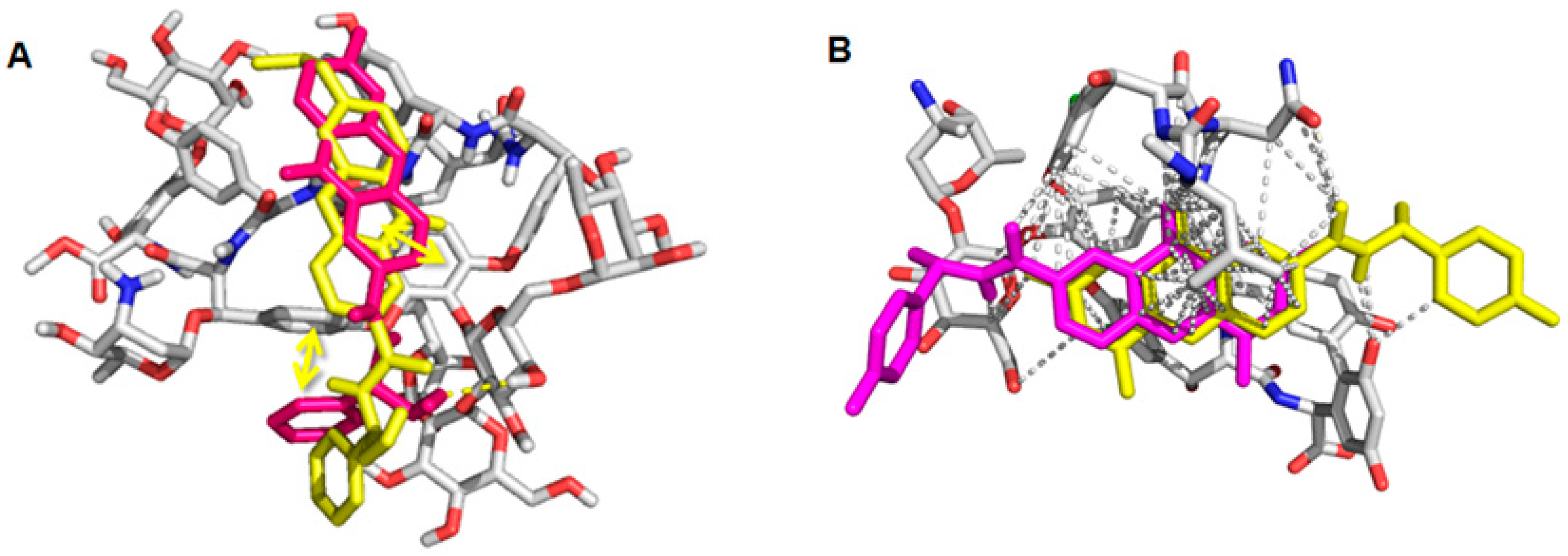
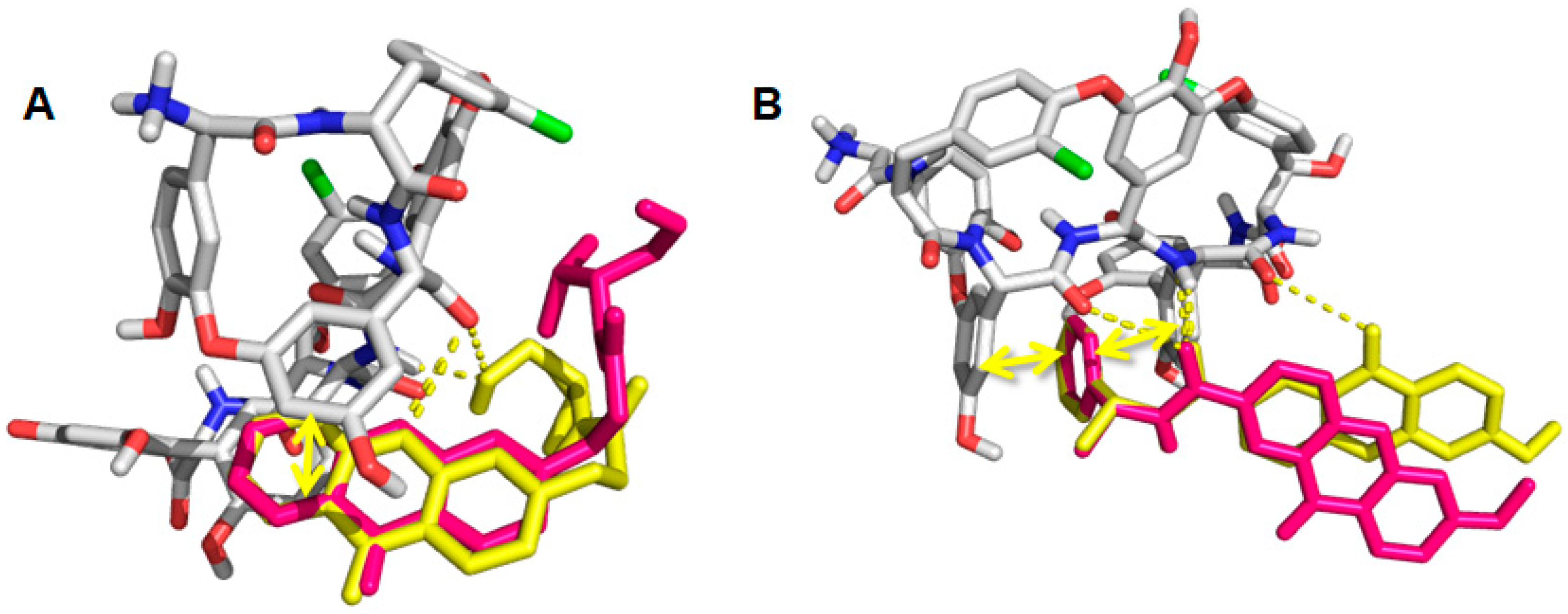
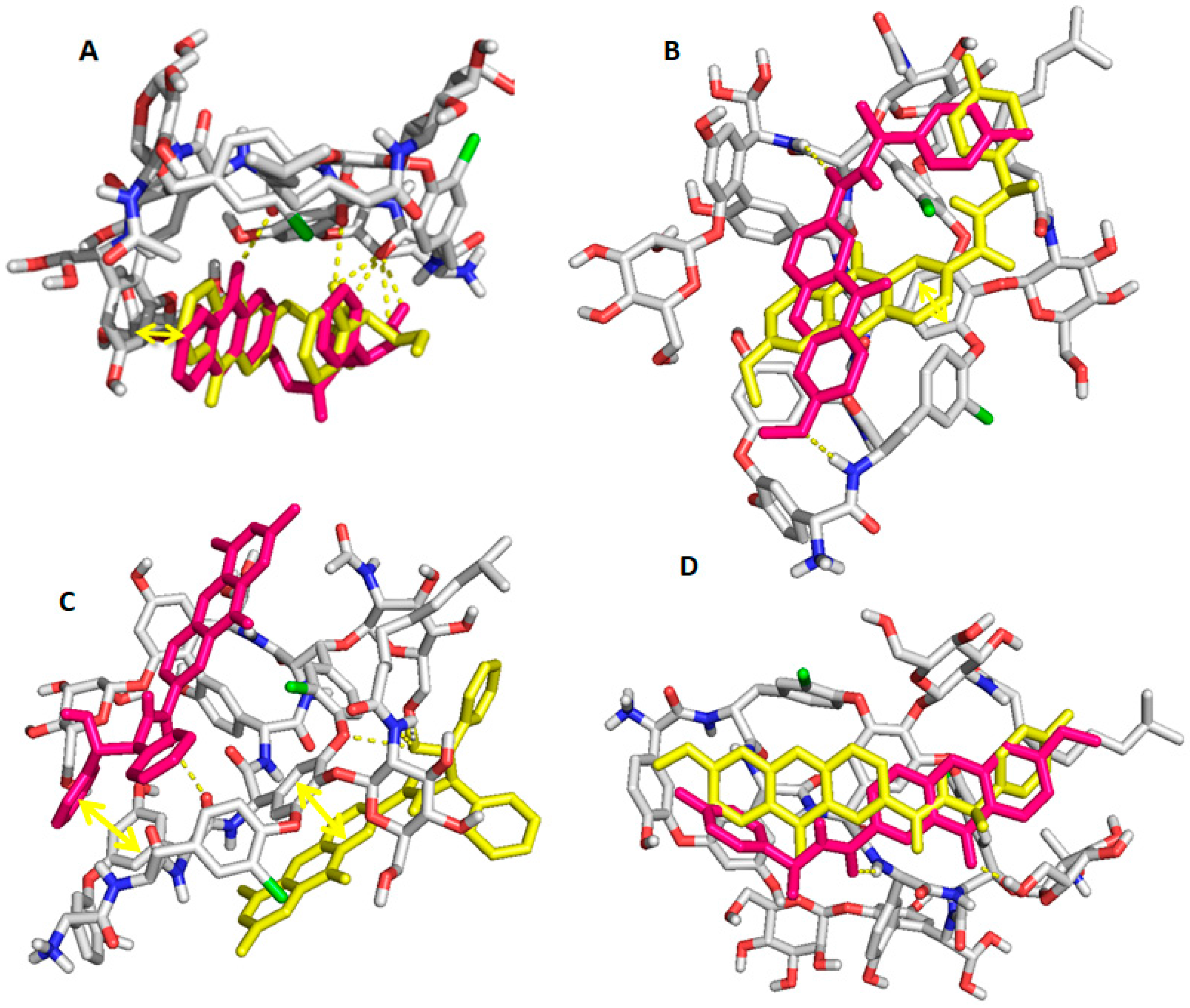
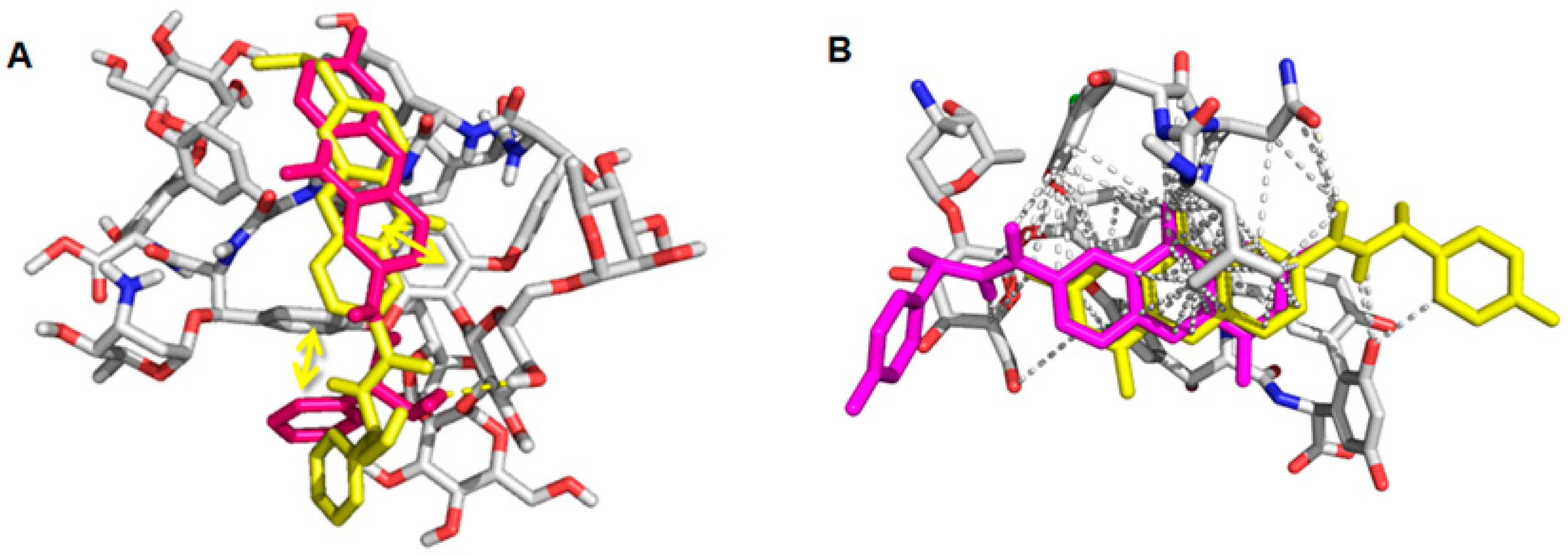
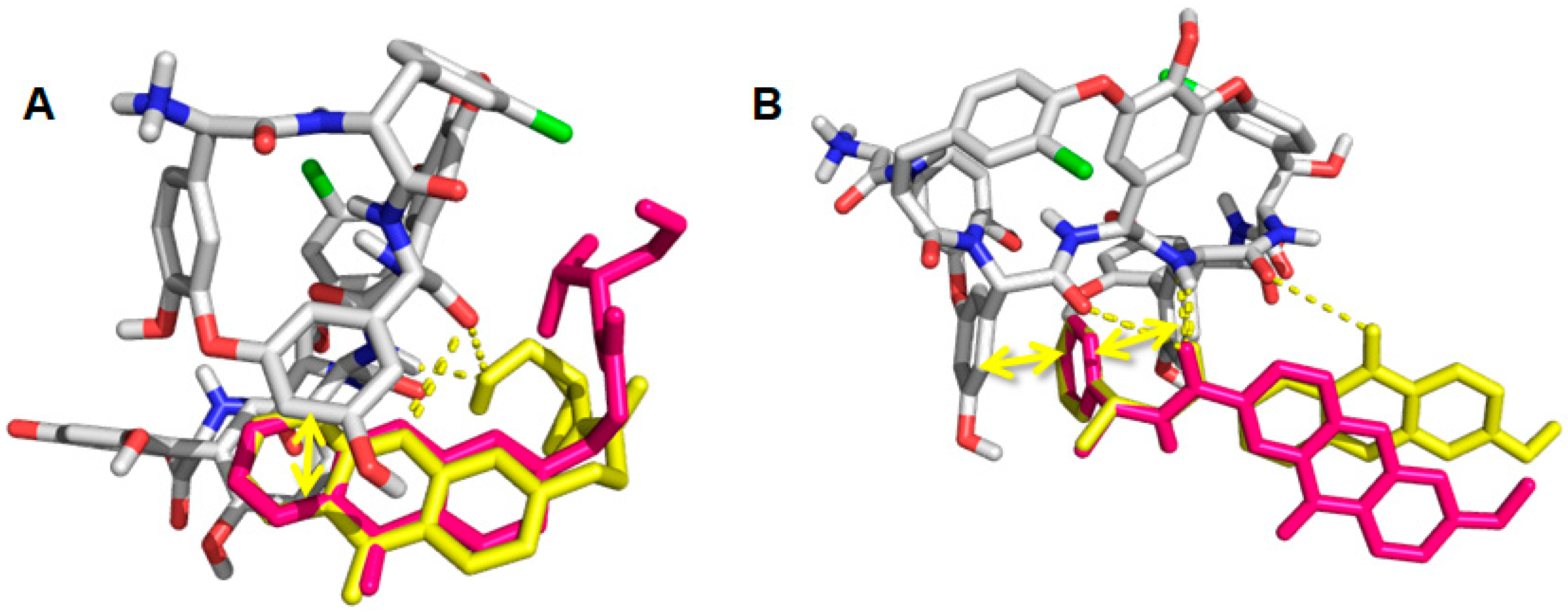
2.6. Computational Studies
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Apparatus and Chromatography
3.3. Computational
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nguyen, H.T.; Lallemand, M.C.; Boutefnouchet, S.; Michel, S.; Tillequin, F. Antitumor psoropermum xanthones and sarcomelicope acridones: Privileged structures implied in DNA alkylation. J. Nat. Prod. 2009, 72, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.M.; Castanheiro, R.A.P.; Kijjoa, A. Xanthones from marine-derived microorganisms: Isolation, structure elucidation, and biological activities. In Encyclopedia of Analytical Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Lesch, B.; Bräse, S. A short, atom-economical entry to tetrahydroxanthenones. Angew. Chem. Int. Ed. 2004, 43, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.I.; Cidade, H.; Pinto, M. Dual/multitargeted xanthone derivatives for alzheimer’s disease: Where do we stand? Future Med. Chem. 2017, 9, 1611–1630. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Sousa, D.; Paiva, A.M.; Palmeira, A.; Barbosa, J.; Pedro, M.; Pinto, M.M.; Sousa, E.; Vasconcelos, M.H. Modulation of autophagy by a thioxanthone decreases the viability of melanoma cells. Molecules 2016, 21, 1343. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.; Lima, R.T.; Sousa, D.; Gomes, A.S.; Palmeira, A.; Seca, H.; Choosang, K.; Pakkong, P.; Bousbaa, H.; Pinto, M.M.; et al. Screening a small library of xanthones for antitumor activity and identification of a hit compound which induces apoptosis. Molecules 2016, 21, 81. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Sousa, E.; Carmo, H.; Palmeira, A.; Barbosa, D.J.; Gameiro, M.; Pinto, M.; Bastos, M.D.; Remiao, F. Induction and activation of p-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the p-glycoprotein substrate paraquat. Arch. Toxicol. 2014, 88, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.M.G.; Afonso, C.M.M.; Soares, J.X.; Reis, S.; Sousa, D.; Lima, R.T.; Vasconcelos, M.H.; Pedro, M.; Barbosa, J.; Gales, L.; et al. Pyranoxanthones: Synthesis, growth inhibitory activity on human tumor cell lines and determination of their lipophilicity in two membrane models. Eur. J. Med. Chem. 2013, 69, 798–816. [Google Scholar] [CrossRef] [PubMed]
- Genovese, S.; Fiorito, S.; Taddeo, V.A.; Epifano, F. Recent developments in the pharmacology of prenylated xanthones. Drug Discov. Today 2016, 21, 1814–1819. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.; Sousa, M.E.; Nascimento, M.S. Xanthone derivatives: New insights in biological activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef] [PubMed]
- Shagufta; Ahmad, I. Recent insight into the biological activities of synthetic xanthone derivatives. Eur. J. Med. Chem. 2016, 116, 267–280. [Google Scholar]
- Masters, K.S.; Bräse, S. Xanthones from fungi, lichens, and bacteria: The natural products and their synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.M.M.; Kijjoa, A. Naturally-occurring xanthones: Recent developments. Curr. Med. Chem. 2005, 12, 2413–2446. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.M.G.; Afonso, C.M.M.; Pinto, M.M.M. Routes to xanthones: An update on the synthetic approaches. Curr. Org. Chem. 2012, 16, 2818–2867. [Google Scholar] [CrossRef]
- Sousa, M.E.; Pinto, M.M.M. Synthesis of xanthones: An overview. Curr. Med. Chem. 2005, 12, 2447–2479. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Palmeira, A.; Ramos, I.I.; Carneiro, C.; Afonso, C.; Tiritan, M.E.; Cidade, H.; Pinto, P.C.A.G.; Saraiva, M.L.M.F.S.; Reis, S.; et al. Chiral derivatives of xanthones: Investigation of the effect of enantioselectivity on inhibition of cyclooxygenases (Cox-1 and Cox-2) and binding interaction with human serum albumin. Pharmaceuticals 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; de Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M.M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Oliveira, L.; Tiritan, M.E.; Leitao, L.; Pozzi, A.; Noronha-Matos, J.B.; Correia-de-Sa, P.; Pinto, M.M. Synthesis of new chiral xanthone derivatives acting as nerve conduction blockers in the rat sciatic nerve. Eur. J. Med. Chem. 2012, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Cravo, S.; Phyo, Y.Z.; Kijjoa, A.; Silva, A.M.S.; Cass, Q.B.; Pinto, M.M.M. New chiral stationary phases based on xanthone derivatives for liquid chromatography. Chirality 2017. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.J.; Ward, K.D. Chiral separations: A review of current topics and trends. Anal. Chem. 2012, 84, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Cass, Q.; Kairys, V.; Fernandes, M.X.; Pinto, M. Enantioseparation and chiral recognition mechanism of new chiral derivatives of xanthones on macrocyclic antibiotic stationary phases. J. Chromatogr. A 2012, 1241, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.L.; Palmeira, A.; Tiritan, M.E.; Fernandes, C.; Pinto, M.M.M. Resolution, determination of enantiomeric purity and chiral recognition mechanism of new xanthone derivatives on (S,S)-whelk-O1 stationary phase. Chirality 2017, 29, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Palmeira, A.; Santos, A.; Tiritan, M.E.; Afonso, C.; Pinto, M.M. Enantioresolution of chiral derivatives of xanthones on (S,S)-Whelk-O1 and l-phenylglycine stationary phases and chiral recognition mechanism by docking approach for (S,S)-Whelk-O1. Chirality 2013, 25, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Brandão, P.; Santos, A.; Tiritan, M.E.; Afonso, C.; Cass, Q.B.; Pinto, M.M. Resolution and determination of enantiomeric purity of new chiral derivatives of xanthones using polysaccharide-based stationary phases. J. Chromatogr. A 2012, 1269, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Pinto, M.; Tiritan, M.E. Enantioresolution of chiral derivatives of xanthones on different types of liquid chromatography stationary phases: A comparative study. Curr. Chromatogr. 2014, 1, 139–150. [Google Scholar] [CrossRef]
- Lämmerhofer, M. Chiral recognition by enantioselective liquid chromatography: Mechanisms and modern chiral stationary phases. J. Chromatogr. A 2010, 1217, 814–856. [Google Scholar] [CrossRef] [PubMed]
- Ilisz, I.; Pataj, Z.; Aranyi, A.; Péter, A. Macrocyclic antibiotic selectors in direct hplc enantioseparations. Sep. Purif. Rev. 2012, 41, 207–249. [Google Scholar] [CrossRef]
- Orosz, T.; Grecsó, N.; Lajkó, G.; Szakonyi, Z.; Fülöp, F.; Armstrong, D.W.; Ilisz, I.; Péter, A. Liquid chromatographic enantioseparation of carbocyclic β-amino acids possessing limonene skeleton on macrocyclic glycopeptide-based chiral stationary phases. J. Pharm. Biomed. Anal. 2017, 145, 119–126. [Google Scholar] [CrossRef] [PubMed]
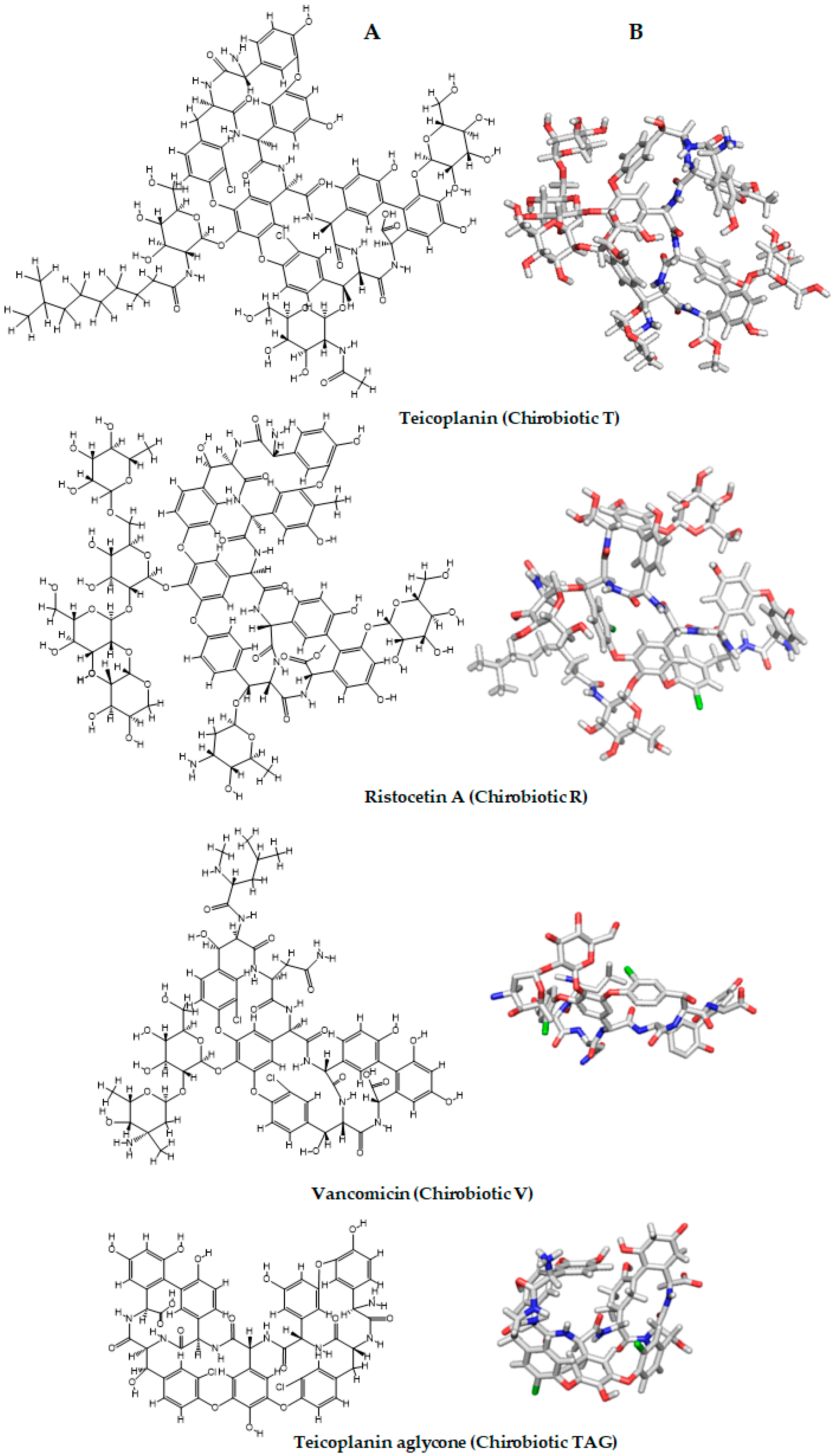
- Armstrong, D.W.; Tang, Y.; Chen, S.; Zhou, Y.; Bagwill, C.; Chen, J.R. Macrocyclic antibiotics as a new class of chiral selectors for liquid chromatography. Anal. Chem. 1994, 66, 1473–1484. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Liu, Y.; Ekborgott, K.H. A covalently bonded teicoplanin chiral stationary phase for hplc enantioseparations. Chirality 1995, 7, 474–497. [Google Scholar] [CrossRef]
- Flieger, J.; Feder-Kubis, J.; Tatarczak-Michalewska, M.; Płazińska, A.; Madejska, A.; Swatko-Ossor, M. Natural terpene derivatives as new structural task-specific ionic liquids to enhance the enantiorecognition of acidic enantiomers on teicoplanin-based stationary phase by high-performance liquid chromatography. J. Sep. Sci. 2017, 40, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Flieger, J. Improvement of chiral discrimination of acidic enantiomers on teicoplanin stationary phase by the use of chaotropic effect. J. Liq. Chromatogr. Relat. Technol. 2009, 32, 948–963. [Google Scholar] [CrossRef]
- Feder-Kubis, J.; Flieger, J.; Tatarczak-Michalewska, M.; Płazińska, A.; Madejska, A.; Swatko-Ossor, M. Renewable sources from plants as the starting material for designing new terpene chiral ionic liquids used for the chromatographic separation of acidic enantiomers. RSC Adv. 2017, 7, 32344–32356. [Google Scholar] [CrossRef]
- Ward, T.J.; Farris Iii, A.B. Chiral separations using the macrocyclic antibiotics: A review. J. Chromatogr. A 2001, 906, 73–89. [Google Scholar] [CrossRef]
- Scriba, G.K.E. Chiral recognition mechanisms in analytical separation sciences. Chromatographia 2012, 75, 815–838. [Google Scholar] [CrossRef]
- Scriba, G.K.E. Chiral recognition in separation science—An update. J. Chromatogr. A 2016, 1467, 56–78. [Google Scholar] [CrossRef] [PubMed]
- Bauvais, C.; Barbault, F.; Zhu, Y.; Petitjean, M.; Fan, B. Elucidation of chiral recognition processes of macrocyclic antibiotic vancomycin. SAR QSAR Environ. Res. 2006, 17, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Berthod, A.; Chen, X.; Kullman, J.P.; Armstrong, D.W.; Gasparrini, F.; D’Acquaric, I.; Villani, C.; Carotti, A. Role of the carbohydrate moieties in chiral recognition on teicoplanin-based LC stationary phases. Anal. Chem. 2000, 72, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Berthod, A.; Qiu, H.X.; Staroverov, S.M.; Kuznestov, M.A.; Armstrong, D.W. Chiral recognition with macrocyclic glycopeptides: Mechanisms and applications. In Chiral Recognition in Separation Methods: Mechanisms and Applications; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Berthod, A. Chiral recognition mechanisms with macrocyclic glycopeptide selectors. Chirality 2009, 21, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Sandra, P.; Vanhoenacker, G.; David, F.; Sandra, K.; Pereira, A. Green chromatography (part 1): Introduction and liquid chromatography. LC-GC Eur. 2010, 23, 242–259. [Google Scholar]
- Majors, R.E.; Raynie, D. The greening of the chromatography laboratory. LC-GC North. Am. 2011, 29, 118–134. [Google Scholar]
- Berkecz, R.; Ilisz, I.; Benedek, G.; Fülöp, F.; Armstrong, D.W.; Péter, A. High-performance liquid chromatographic enantioseparation of 2-aminomono- and dihydroxycyclopentanecarboxylic and 2-aminodihydroxycyclohexanecarboxylic acids on macrocyclic glycopeptide-based phases. J. Chromatogr. A 2009, 1216, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.L.; Rozhkov, R.V.; Larock, R.C.; Armstrong, D.W. Separation of the enantiomers of substituted dihydrofurocoumarins by hplc using macrocyclic glycopeptide chiral stationary phases. Anal. Bioanal. Chem. 2003, 377, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Gasper, M.P.; Berthod, A.; Nair, U.B.; Armstrong, D.W. Comparison and modeling study of vancomycin, ristocetin a, and teicoplanin for ce enantioseparations. Anal. Chem. 1996, 68, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Akhter, M. Challenges in docking: Mini review. JSM Chem. 2016, 4, 1–8. [Google Scholar]
- Zhou, P.; Zou, J.; Tian, F.; Shang, Z. Fluorine bonding—How does it work in protein-ligand interactions? J. Chem. Inf. Model. 2009, 49, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. Pubchem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. Am1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Mirzaei, H.; Zarbafian, S.; Villar, E.; Mottarella, S.; Beglov, D.; Vajda, S.; Paschalidis, I.; Vakili, P.; Kozakov, D. Energy minimization on manifolds for docking flexible molecules. J. Chem. Theory Comput. 2015, 11, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using pymol. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
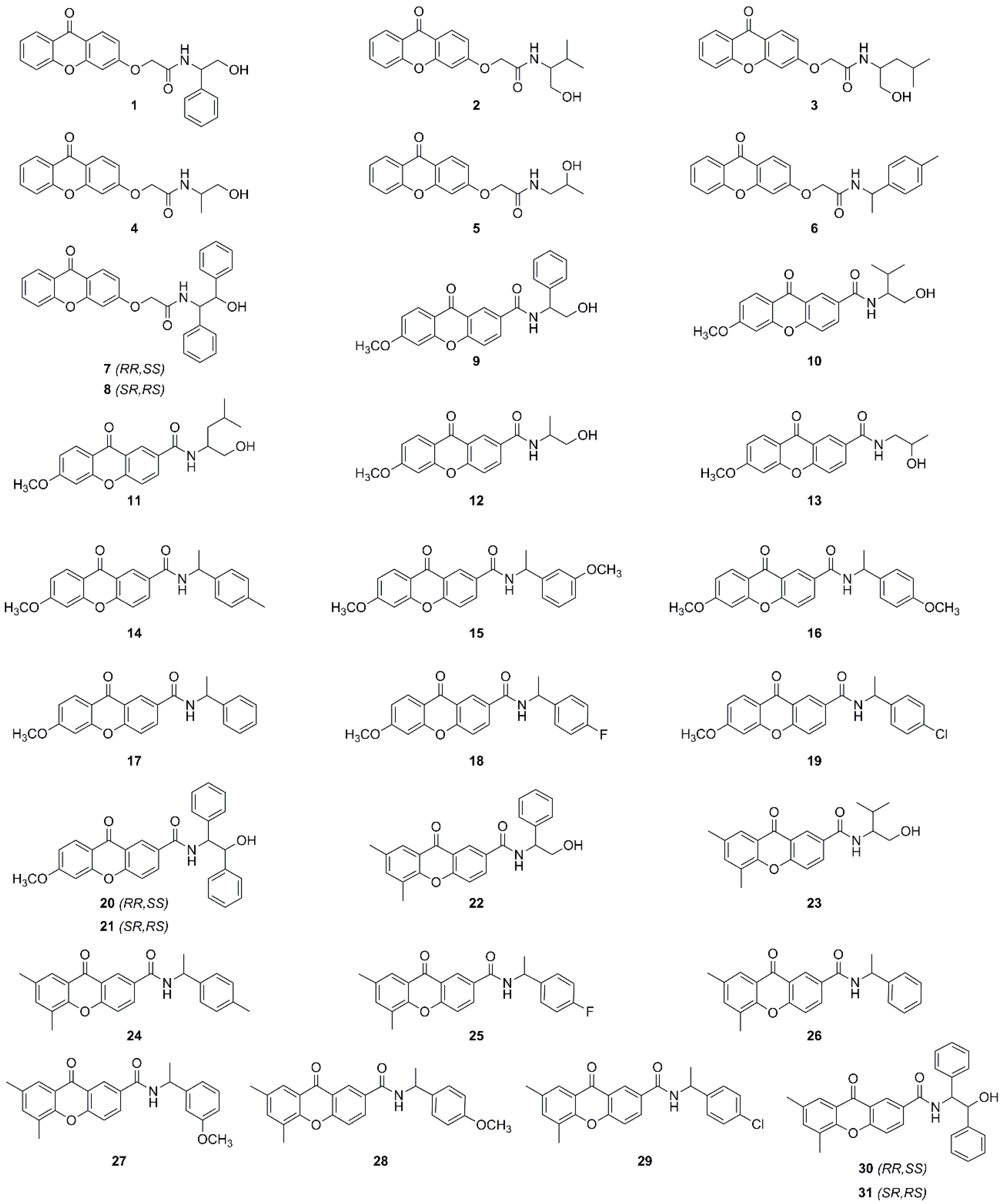
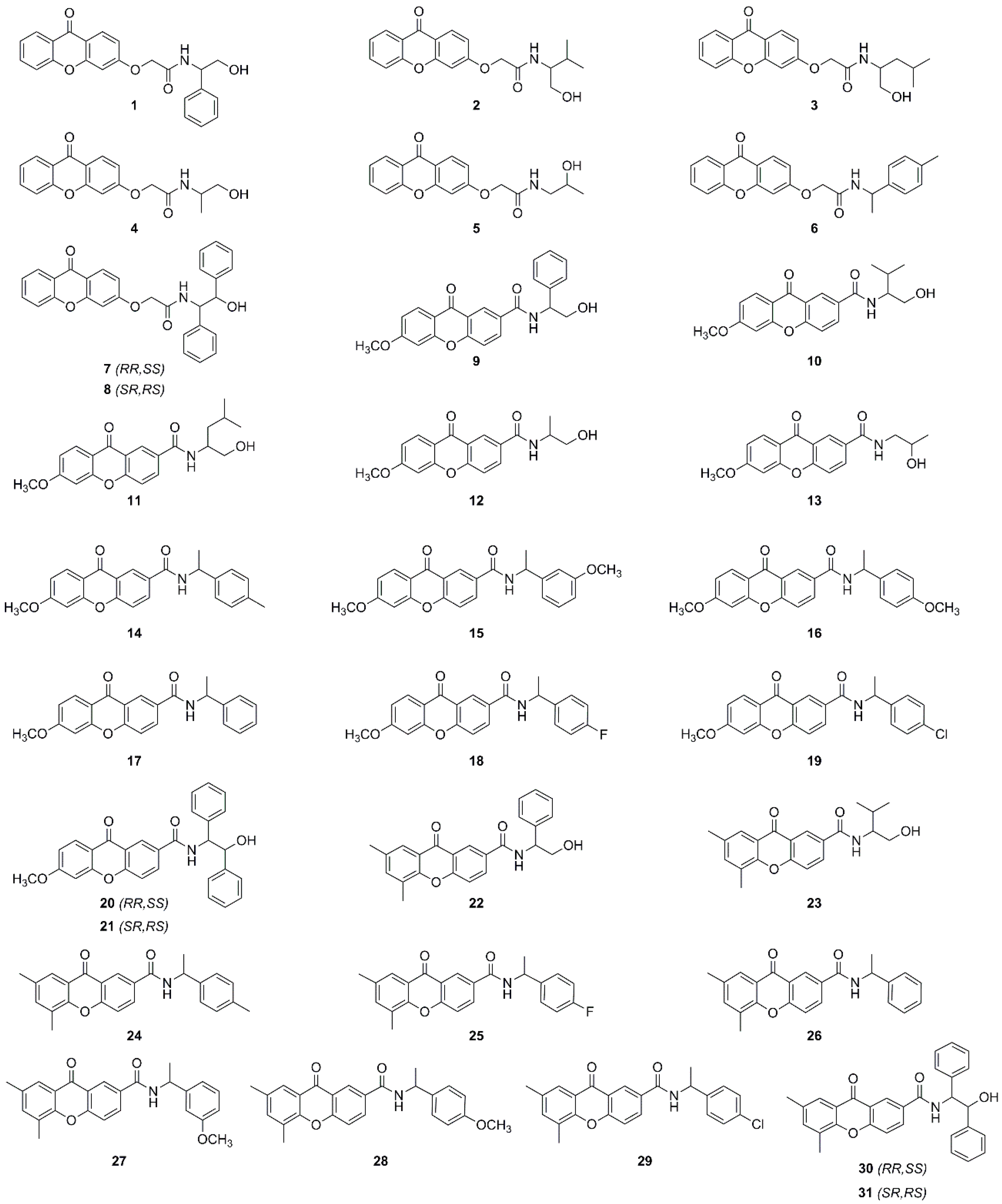
Sample Availability: Samples of the compounds 1–31 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Elution Mode | Mobile Phase | k1 | α | RS | First Eluted Enantiomer |
|---|---|---|---|---|---|---|
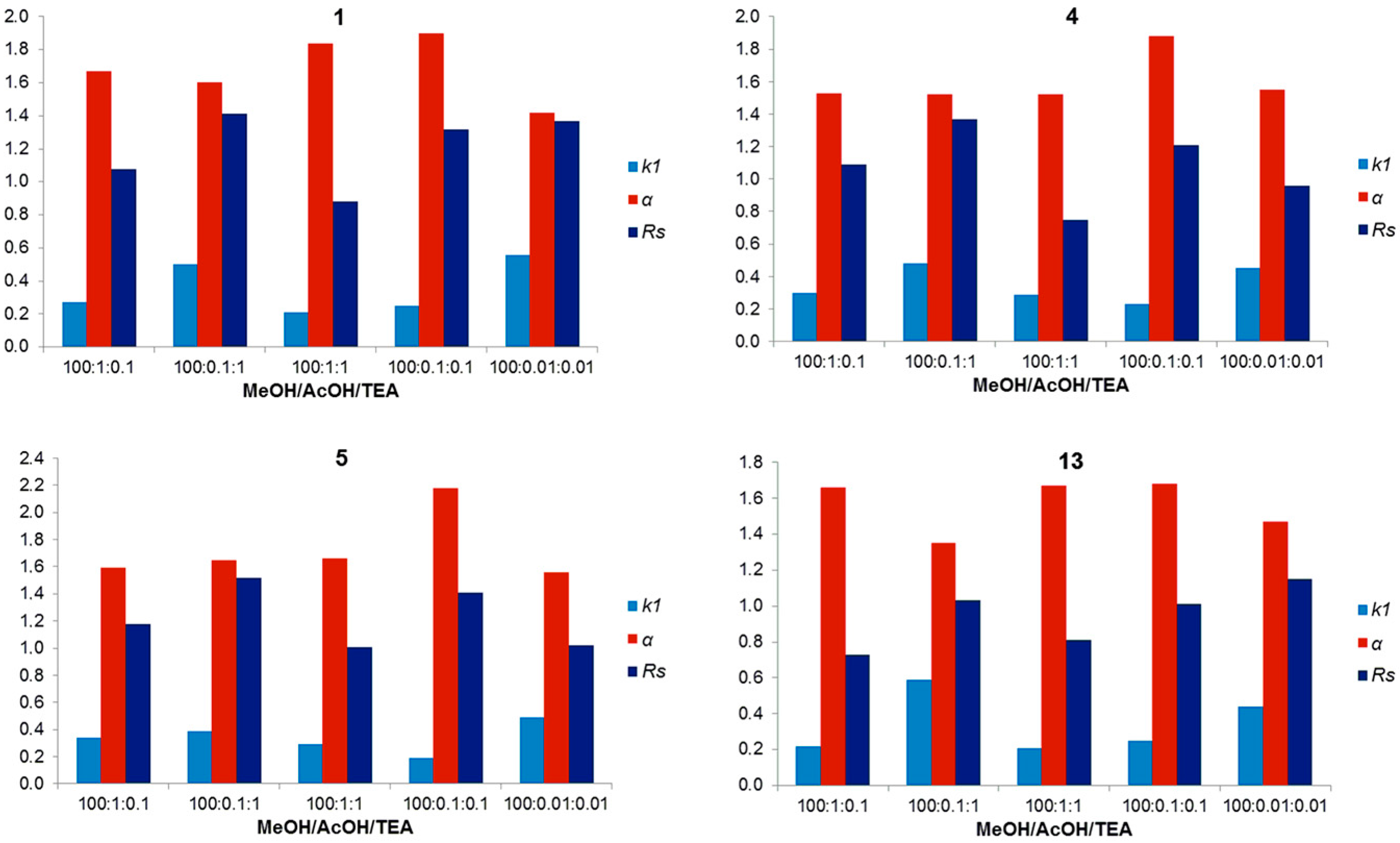
| 1 | NPM | Hex:EtOH (80:20 v/v) | 18.13 | 1.16 | 2.26 | (R) |
| NPM | Hex:2-PrOH (70:30 v/v) | 33.19 | 1.47 | 1.93 | (R) | |
| POM | 100% EtOH | 1.19 | 1.14 | 0.70 | (R) | |
| 2 | NPM | Hex:EtOH (40:60 v/v) | 1.92 | 1.26 | 1.28 | (R) |
| NPM | Hex:2-PrOH (70:30 v/v) | 15.98 | 1.62 | 1.49 | (R) | |
| POM | 100% EtOH | 0.80 | 1.20 | 0.86 | (R) | |
| 3 | NPM | Hex:EtOH (80:20 v/v) | 32.34 | 1.09 | 1.09 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 2.93 | 1.32 | 1.00 | (R) | |
| 4 | NPM | Hex:EtOH (80:20 v/v) | 19.56 | 1.42 | 2.95 | (R) |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 1.13 | 1.09 | 0.80 | (R) | |
| POM | 100% EtOH | 1.09 | 1.33 | 1.28 | (R) | |
| 7 | NPM | Hex:EtOH (90:10 v/v) | 23.91 | 1.29 | 2.83 | (R) |
| 9 | NPM | Hex:EtOH (70:30 v/v) | 9.21 | 1.47 | 2.48 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 7.11 | 1.46 | 1.07 | (R) | |
| PIM | MeOH:AcOH:TEA (100:0.1:0.1 v/v/v) | 1.34 | 1.08 | 0.60 | (R) | |
| POM | 100% EtOH | 1.13 | 1.42 | 1.73 | (R) | |
| POM | 100% 2-PrOH | 1.63 | 1.63 | 1.32 | (R) | |
| 10 | NPM | Hex:EtOH (70:30 v/v) | 5.04 | 1.54 | 3.30 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 4.05 | 1.61 | 1.56 | (R) | |
| POM | 100% EtOH | 0.80 | 1.49 | 1.90 | (R) | |
| POM | 100% 2-PrOH | 1.01 | 1.74 | 1.61 | (R) | |
| 11 | NPM | Hex:EtOH (80:20 v/v) | 6.93 | 1.29 | 2.43 | (R) |
| NPM | Hex:2-PrOH (80:20 v/v) | 8.35 | 1.33 | 0.84 | (R) | |
| POM | 100% EtOH | 0.63 | 1.25 | 1.00 | (R) | |
| POM | 100% 2-PrOH | 0.69 | 1.41 | 1.14 | (R) | |
| 12 | NPM | Hex:EtOH (80:20 v/v) | 15.31 | 1.32 | 2.31 | (R) |
| NPM | Hex:2-PrOH (70:30 v/v) | 22.72 | 1.38 | 0.50 | (R) | |
| POM | 100% EtOH | 1.03 | 1.25 | 1.00 | (R) | |
| 14 | NPM | Hex:EtOH (90:10 v/v) | 9.78 | 1.29 | 1.55 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 1.53 | 1.15 | 0.78 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (40:60 v/v) | 10.54 | 1.19 | 1.53 | (R) | |
| 15 | NPM | Hex:EtOH (90:10 v/v) | 16.67 | 1.26 | 1.97 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 2.33 | 1.23 | 0.89 | (R) | |
| POM | 100% EtOH | 0.67 | 1.15 | 0.70 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 20.59 | 1.33 | 2.46 | (R) | |
| 16 | NPM | Hex:EtOH (90:10 v/v) | 17.86 | 1.18 | 1.51 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 2.48 | 1.15 | 0.40 | (R) | |
| POM | 100% EtOH | 0.70 | 1.08 | 0.50 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 22.32 | 1.24 | 1.92 | (R) | |
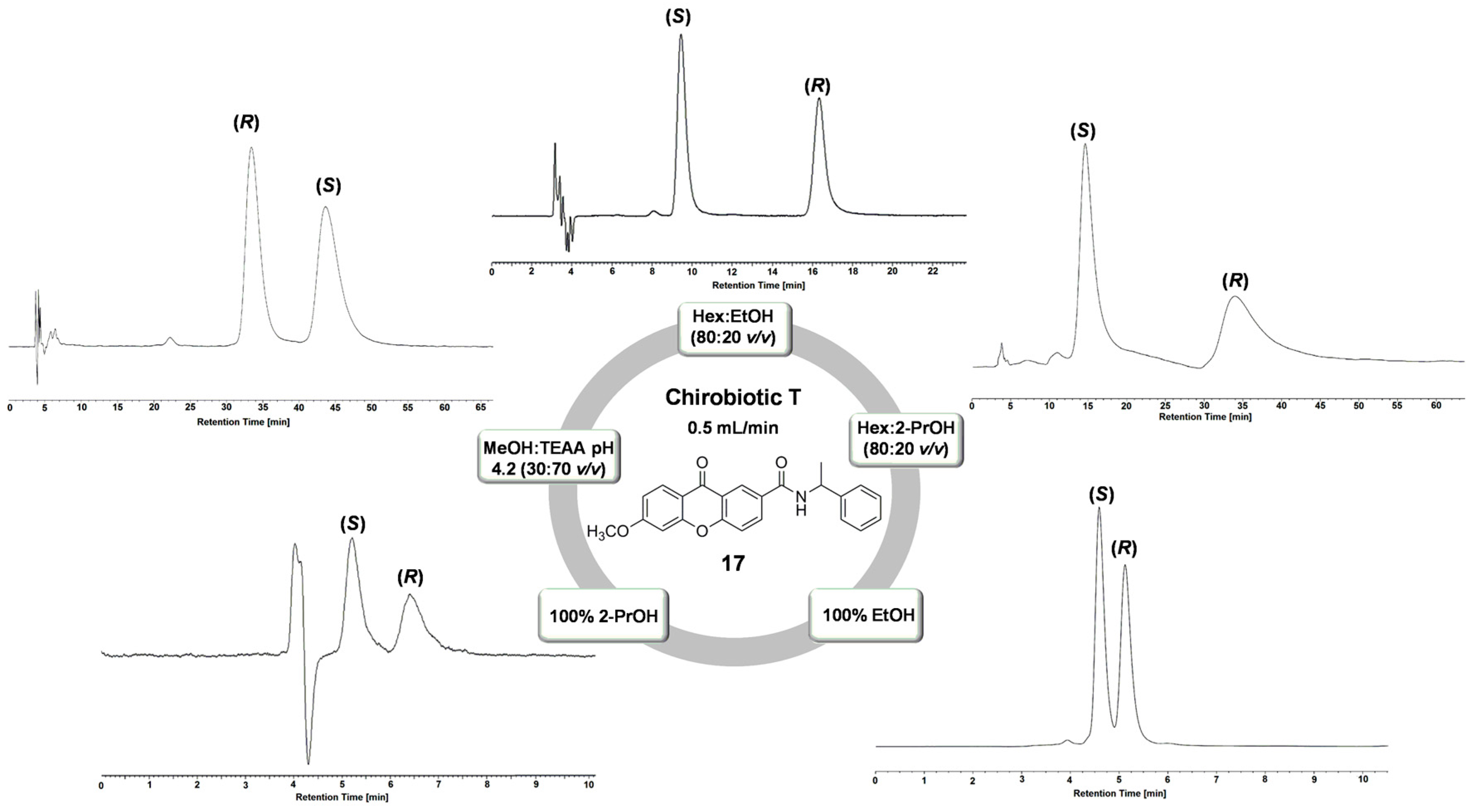
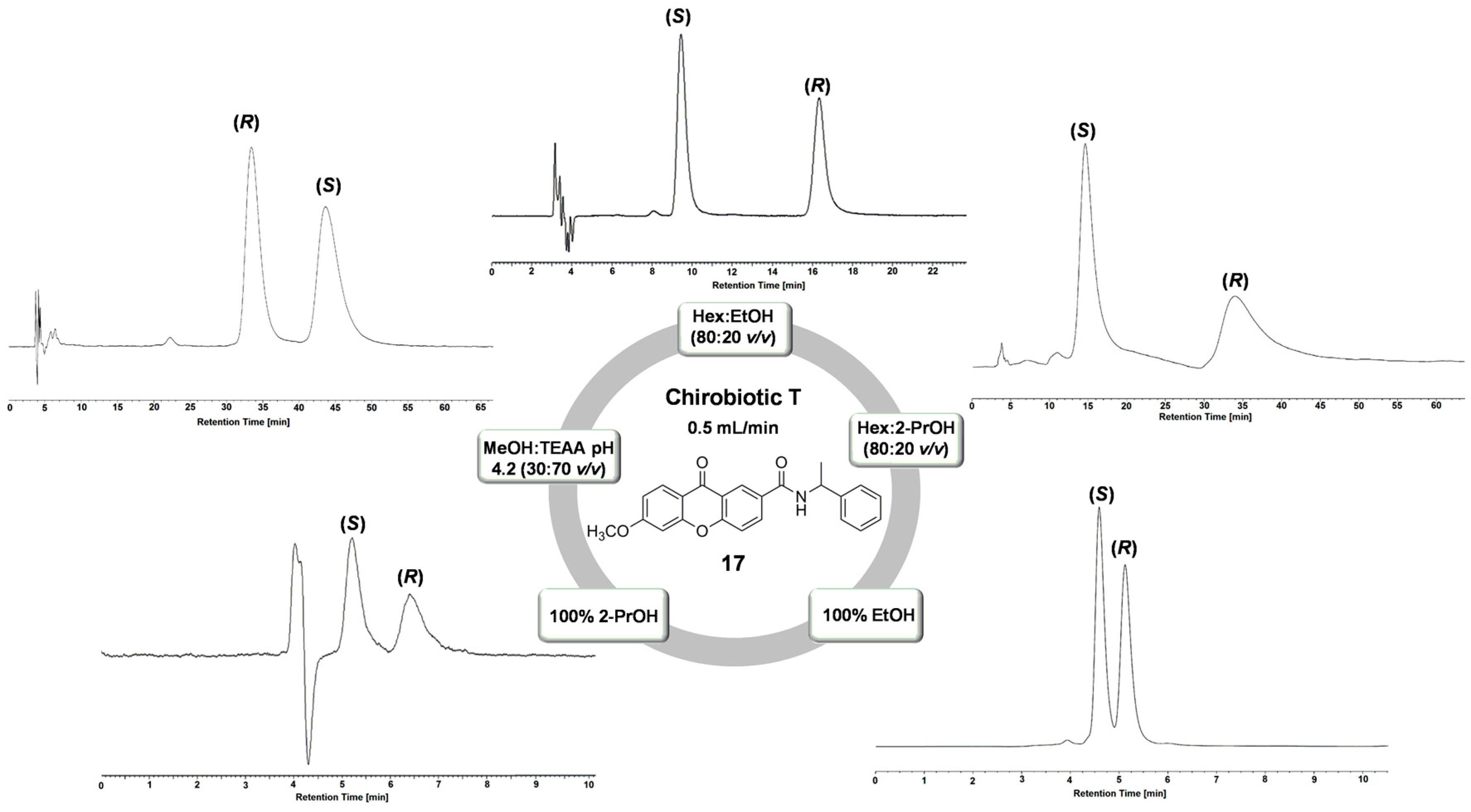
| 17 * | NPM | Hex:EtOH (80:20 v/v) | 2.03 | 2.09 | 7.37 | (S) |
| NPM | Hex:2-PrOH (80:20 v/v) | 3.33 | 2.71 | 3.31 | (S) | |
| POM | 100% EtOH | 0.42 | 1.39 | 1.45 | (S) | |
| POM | 100% 2-PrOH | 0.30 | 1.99 | 1.78 | (S) | |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 21.13 | 1.49 | 3.20 | (R) | |
| 18 | NPM | Hex:EtOH (90:10 v/v) | 12.05 | 1.16 | 1.43 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 1.67 | 1.13 | 0.30 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 18.89 | 1.18 | 1.37 | (R) | |
| 19 | NPM | Hex:EtOH (90:10 v/v) | 10.68 | 1.13 | 1.28 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 1.59 | 1.10 | 0.30 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 29.61 | 1.17 | 1.30 | (R) | |
| 20 | NPM | Hex:EtOH (80:20 v/v) | 11.20 | 1.31 | 2.24 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 3.45 | 1.30 | 1.11 | (R) | |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 1.10 | 1.11 | 0.74 | (R) | |
| POM | 100% EtOH | 0.85 | 1.30 | 1.32 | (R) | |
| POM | 100% 2-PrOH | 1.09 | 1.46 | 0.99 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 8.55 | 2.26 | 1.66 | (R) | |
| 22 | NPM | Hex:EtOH (80:20 v/v) | 5.50 | 1.36 | 2.45 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 2.62 | 1.39 | 1.18 | (R) | |
| POM | 100% EtOH | 0.72 | 1.34 | 1.38 | (R) | |
| POM | 100% 2-PrOH | 0.77 | 1.52 | 1.10 | (R) | |
| 23 * | NPM | Hex:EtOH (50:50 v/v) | 1.22 | 1.25 | 1.28 | (S) |
| NPM | Hex:2-PrOH (50:50 v/v) | 1.57 | 1.19 | 0.87 | (S) | |
| POM | 100% EtOH | 0.56 | 1.25 | 1.00 | (S) | |
| POM | 100% 2-PrOH | 0.48 | 1.54 | 1.08 | (S) | |
| RPM | MeOH:TEAA pH 4.2 (40:60 v/v) | 4.68 | 2.83 | 6.79 | (R) | |
| 24 | NPM | Hex:EtOH (90:10 v/v) | 3.57 | 1.11 | 0.79 | (R) |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 29.11 | 1.23 | 1.55 | (R) | |
| 25 | NPM | Hex:EtOH (90:10 v/v) | 4.09 | 1.09 | 0.92 | (R) |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 22.38 | 1.16 | 1.08 | (R) | |
| 26 | NPM | Hex:EtOH (90:10 v/v) | 4.53 | 1.12 | 1.16 | (R) |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 21.58 | 1.22 | 1.49 | (R) | |
| 27 | NPM | Hex:EtOH (90:10 v/v) | 5.91 | 1.16 | 1.40 | (R) |
| RPM | MeOH:TEAA pH 4.2 (40:60 v/v) | 10.87 | 1.26 | 1.97 | (R) | |
| 28 | NPM | Hex:EtOH (90:10 v/v) | 6.41 | 1.11 | 1.13 | (R) |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 27.13 | 1.26 | 1.87 | (R) | |
| 29 | NPM | Hex:EtOH (90:10 v/v) | 4.67 | 1.07 | 0.89 | (R) |
| RPM | MeOH:TEAA pH 4.2 (40:60 v/v) | 13.04 | 1.16 | 1.18 | (R) | |
| 30 | NPM | Hex:EtOH (90:10 v/v) | 15.16 | 1.24 | 1.93 | (R) |
| NPM | Hex:2-PrOH (80:20 v/v) | 9.76 | 1.33 | 0.88 | (R) | |
| POM | 100% EtOH | 0.56 | 1.17 | 0.70 | (R) | |
| POM | 100% 2-PrOH | 0.49 | 1.43 | 1.05 | (R) |
| Analyte | Elution Mode | Mobile Phase | k1 | α | RS | First Eluted Enantiomer |
|---|---|---|---|---|---|---|
| 1 | POM | 100% MeOH | 0.27 | 1.72 | 1.23 | (S) |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 0.50 | 1.60 | 1.41 | (S) | |
| 2 | POM | 100% MeOH | 0.27 | 1.44 | 0.45 | (R) |
| PIM | MeOH:AcOH:TEA (100:0.01:0.01 v/v/v) | 0.48 | 1.32 | 0.81 | (R) | |
| 4 | POM | 100% MeOH | 0.23 | 1.88 | 1.00 | (S) |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 0.48 | 1.52 | 1.37 | (S) | |
| 5 | POM | 100% MeOH | 0.28 | 1.80 | 1.02 | (R) |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 0.39 | 1.65 | 1.52 | (R) | |
| 9 | NPM | Hex:EtOH (80:20 v/v) | 9.32 | 1.46 | 1.99 | (R) |
| NPM | Hex:2-PrOH (70:30 v/v) | 7.59 | 1.88 | 1.49 | (R) | |
| POM | 100% 2-PrOH | 1.44 | 1.85 | 1.32 | (R) | |
| PIM | MeOH:AcOH:TEA (100:0.1:0.1 v/v/v) | 0.22 | 1.72 | 0.87 | (R) | |
| 10 * | NPM | Hex:EtOH (90:10 v/v) | 15.78 | 1.24 | 1.77 | (R) |
| NPM | Hex:2-PrOH (80:20 v/v) | 8.41 | 1.69 | 1.73 | (R) | |
| 11 | NPM | Hex:EtOH (90:10 v/v) | 10.83 | 1.16 | 1.68 | (R) |
| 12 | NPM | Hex:EtOH (90:10 v/v) | 28.07 | 1.16 | 1.37 | (R) |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 0.67 | 1.26 | 0.84 | (R) | |
| 13 * | PIM | MeOH:AcOH:TEA (100:0.01:0.01 v/v/v) | 0.44 | 1.47 | 1.15 | (S) |
| 17 | NPM | Hex:EtOH (90:10 v/v) | 2.32 | 2.91 | 3.95 | (S) |
| NPM | Hex:2-PrOH (70:30 v/v) | 0.39 | 4.65 | 2.05 | (S) | |
| POM | 100% 2-PrOH | 0.29 | 2.05 | 0.91 | (S) | |
| RPM | MeOH:TEAA pH 4.2 (50:50 v/v) | 1.09 | 1.35 | 0.70 | (S) | |
| 19 | RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 1.90 | 1.21 | 1.00 | (R) |
| 20 | NPM | Hex:EtOH (90:10 v/v) | 20.33 | 1.11 | 1.18 | (R) |
| NPM | Hex:2-PrOH (50:50 v/v) | 2.13 | 1.28 | 0.91 | (R) | |
| PIM | MeOH:AcOH:TEA (100:0.1:1 v/v/v) | 1.10 | 1.11 | 0.74 | (R) | |
| 22 | NPM | Hex:EtOH (90:10 v/v) | 9.69 | 1.58 | 2.81 | (R) |
| NPM | Hex:2-PrOH (70:30 v/v) | 2.06 | 2.21 | 1.83 | (R) | |
| POM | 100% 2-PrOH | 0.61 | 1.97 | 1.00 | (R) | |
| 23 * | NPM | Hex:2-PrOH (80:20 v/v) | 2.42 | 1.52 | 1.12 | (R) |
| POM | 100% 2-PrOH | 0.41 | 1.36 | 0.50 | (R) | |
| RPM | MeOH:TEAA pH 4.2 (40:60 v/v) | 0.64 | 1.70 | 1.51 | (S) | |
| 27 | RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 15.54 | 1.09 | 0.70 | (S) |
| 28 | NPM | Hex:EtOH (90:10 v/v) | 6.41 | 1.11 | 1.13 | (S) |
| RPM | MeOH:TEAA pH 4.2 (30:70 v/v) | 15.45 | 1.25 | 1.12 | (S) | |
| 29 | RPM | MeOH:TEAA pH 4.2 (40:60 v/v) | 13.04 | 1.16 | 1.18 | (S) |
| Analyte | Elution Mode | Mobile Phase | k1 | α | RS | First Eluted Enantiomer |
|---|---|---|---|---|---|---|
| 1 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 2.32 | 1.12 | 0.92 | (R) |
| 2 | NPM | Hex:EtOH (80:20 v/v) | 8.72 | 1.09 | 0.88 | (R) |
| 4 | NPM | Hex:EtOH (50:50 v/v) | 3.37 | 1.08 | 0.66 | (R) |
| 6 | NPM | Hex:EtOH (80:20 v/v) | 4.69 | 1.09 | 0.89 | (R) |
| RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.21 | 1.39 | 2.29 | (R) | |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.68 | 1.39 | 2.67 | (R) | |
| 9 * | NPM | Hex:EtOH (80:20 v/v) | 15.72 | 1.19 | 1.16 | (S) |
| POM | 100% EtOH | 0.72 | 1.17 | 0.90 | (S) | |
| 10 | NPM | Hex:EtOH (80:20 v/v) | 8.45 | 1.24 | 1.89 | (R) |
| POM | 100% EtOH | 0.39 | 1.23 | 0.60 | (R) | |
| POM | 100% 2-PrOH | 1.24 | 1.29 | 0.60 | (R) | |
| 11 | NPM | Hex:EtOH (80:20 v/v) | 6.14 | 1.15 | 1.39 | (R) |
| 12 | NPM | Hex:EtOH (80:20 v/v) | 13.56 | 1.13 | 1.35 | (R) |
| POM | 100% 2-PrOH | 1.79 | 1.11 | 0.60 | (R) | |
| 14 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.52 | 1.43 | 1.56 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.98 | 1.47 | 2.24 | (R) | |
| 15 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.41 | 1.42 | 2.32 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.87 | 1.44 | 2.49 | (R) | |
| 16 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.49 | 1.35 | 2.08 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.98 | 1.36 | 1.85 | (R) | |
| 17 * | NPM | Hex:EtOH (80:20 v/v) | 1.85 | 2.31 | 7.71 | (S) |
| POM | 100% EtOH | 0.13 | 2.20 | 1.29 | (S) | |
| POM | 100% 2-PrOH | 0.20 | 2.00 | 1.71 | (S) | |
| RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.11 | 1.21 | 1.18 | (R) | |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.61 | 1.21 | 1.05 | (R) | |
| 18 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 4.26 | 1.04 | 0.90 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.66 | 1.37 | 1.89 | (R) | |
| 19 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 4.09 | 1.53 | 2.79 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 3.03 | 1.55 | 2.47 | (R) | |
| 20 | NPM | Hex:EtOH (80:20 v/v) | 3.39 | 1.21 | 1.32 | (R) |
| POM | 100% EtOH | 0.51 | 1.25 | 0.60 | (R) | |
| POM | 100% 2-PrOH | 1.77 | 1.36 | 1.23 | (R) | |
| 22 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 2.38 | 1.13 | 0.75 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.51 | 1.15 | 0.78 | (R) | |
| 23 | NPM | Hex:EtOH (80:20 v/v) | 3.45 | 1.31 | 2.68 | (R) |
| RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 1.34 | 2.34 | 4.94 | (R) | |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 1.35 | 3.32 | 4.65 | (R) | |
| 24 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.39 | 1.43 | 1.17 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 3.17 | 1.32 | 1.80 | (R) | |
| 25 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.00 | 1.10 | 1.45 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.55 | 1.35 | 1.66 | (R) | |
| 26 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 2.94 | 1.27 | 1.32 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.47 | 1.28 | 1.39 | (R) | |
| 27 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.14 | 1.38 | 1.91 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.29 | 1.41 | 1.79 | (R) | |
| 28 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 2.88 | 1.32 | 1.28 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.39 | 1.34 | 1.50 | (R) | |
| 29 | RPM | MeOH:NH4OAc pH 6 (50:50 v/v) | 3.31 | 1.49 | 1.97 | (R) |
| RPM | MeOH:NH4OAc pH 4 (50:50 v/v) | 2.99 | 1.54 | 2.08 | (R) |
| Analyte | Elution Mode | Mobile Phase | k1 | α | RS | First Eluted Enantiomer |
|---|---|---|---|---|---|---|
| 1 | POM | 100% EtOH | 1.26 | 1.24 | 0.60 | (R) |
| 2 | NPM | Hex:EtOH (70:30 v/v) | 5.01 | 1.42 | 1.45 | (R) |
| POM | 100% EtOH | 0.73 | 1.39 | 0.80 | (R) | |
| 4 | NPM | Hex:EtOH (70:30 v/v) | 10.66 | 1.38 | 1.76 | (R) |
| POM | 100% EtOH | 1.21 | 1.36 | 0.92 | (R) | |
| 5 | NPM | Hex:EtOH (70:30 v/v) | 8.29 | 1.37 | 1.02 | (R) |
| 17 | NPM | Hex:EtOH (70:30 v/v) | 0.80 | 2.77 | 3.87 | (S) |
| 30 | NPM | Hex:EtOH (70:30 v/v) | 2.01 | 1.45 | 1.16 | (R) |
| 31 | NPM | Hex:EtOH (70:30 v/v) | 1.57 | 1.27 | 0.50 | (R) |
| I | II | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analytes | RS Value | Docking Score | Docking Score (R)–(S) (Delta) δ | |||||||||
| T | R | V | TAG | T | R | V | TAG | T | R | V | TAG | |
| (R)-1 (S)-1 | 2.26 | 1.41 | <1.00 | <1.00 | −6.6 −7.1 | −7.1 −6.9 | * | * | −0.5 | −0.2 | * | * |
| (R)-2 (S)-2 | 1.49 | <1.00 | <1.00 | 1.45 | −5.9 −5.4 | * | * | −4.9 −5.1 | −0.5 | * | * | −0.2 |
| (R)-3 (S)-3 | 1.09 | <1.00 | <1.00 | <1.00 | −5.8 −5.8 | * | * | * | 0.0 | * | * | * |
| (R)-4 (S)-4 | 2.95 | 1.37 | <1.00 | 1.76 | −5.8 −5.9 | −6.2 −6.0 | * | −4.9 −4.8 | −0.1 | −0.2 | * | −0.1 |
| (R)-5 (S)-5 | <1.00 | 1.52 | <1.00 | 1.02 | * | −6.3 −6.1 | * | −5.1 −5.2 | * | −0.2 | * | −0.1 |
| (R)-6 (S)-6 | <1.00 | <1.00 | 2.67 | <1.00 | * | * | −6.9 −6.6 | * | * | * | −0.3 | * |
| (R,R)-7 (S,S)-7 | 2.83 | <1.00 | <1.00 | <1.00 | −7.0 −6.7 | * | * | * | −0.3 | * | * | * |
| (S,R)-8 (R,S)-8 | <1.00 | <1.00 | <1.00 | <1.00 | * | * | * | * | * | * | * | * |
| (R)-9 (S)-9 | 2.48 | 1.99 | 1.16 | <1.00 | −6.2 −6.1 | −6.2 −6.7 | ** | * | −0.1 | −0.5 | ** | * |
| (R)-10 (S)-10 | 3.30 | 1.77 | 1.89 | <1.00 | −5.7 −5.7 | ** | −6.7 −6.6 | * | 0.0 | ** | −0.1 | * |
| (R)-11 (S)-11 | 2.43 | 1.68 | 1.39 | <1.00 | −5.7 −5.8 | −6.3 −6.1 | −6.1 −6.3 | * | −0.1 | −0.2 | −0.2 | * |
| (R)-12 (S)-12 | 2.31 | 1.37 | 1.35 | <1.00 | −5.9 −5.8 | −6.2 −6.5 | −6.7 −6.5 | * | −0.1 | −0.3 | −0.2 | * |
| (R)-13 (S)-13 | <1.00 | 1.15 | <1.00 | <1.00 | * | ** | * | * | * | ** | * | * |
| (R)-14 (S)-14 | 1.55 | <1.00 | 2.24 | <1.00 | −6.5 −6.6 | * | −6.5 −6.6 | * | −0.1 | * | −0.1 | * |
| (R)-15 (S)-15 | 2.46 | <1.00 | 2.49 | <1.00 | −6.3 −6.3 | * | −7.3 −7.1 | * | 0.0 | * | −0.2 | * |
| (R)-16 (S)-16 | 1.92 | <1.00 | 2.08 | <1.00 | −6.0 −5.9 | * | −7.1 −7.4 | * | −0.2 | * | −0.3 | * |
| (R)-17 (S)-17 | 7.37 | 3.95 | 7.71 | 3.87 | ** | −7.3 −7.3 | ** | −4.9 −4.6 | ** | 0.0 | ** | −0.3 |
| (R)-18 (S)-18 | 1.43 | <1.00 | 1.89 | <1.00 | −5.6 −5.7 | * | −7.7 −7.9 | * | −0.1 | * | −0.2 | * |
| (R)-19 (S)-19 | 1.30 | 1.00 | 2.79 | <1.00 | −6.5 −6.4 | −7.2 −7.3 | −7.7 −7.5 | * | −0.1 | −0.1 | −0.2 | * |
| (R,R)-20 (S,S)-20 | 2.24 | 1.18 | 1.32 | <1.00 | −6.5 −6.5 | −7.4 −7.1 | −7.6 −7.8 | * | 0.0 | −0.3 | −0.2 | * |
| (S,R)-21 (R,S)-21 | <1.00 | <1.00 | <1.00 | <1.00 | * | * | * | * | * | * | * | * |
| (R)-22 (S)-22 | 2.45 | 2.81 | <1.00 | <1.00 | −6.5 −6.2 | −7.1 −7.0 | * | * | −0.3 | −0.1 | * | * |
| (R)-23 (S)-23 | 6.79 | 1.51 | 4.94 | <1.00 | ** | ** | −6.3 −6.7 | * | ** | ** | −0.4 | * |
| (R)-24 (S)-24 | 1.55 | <1.00 | 1.80 | <1.00 | −6.7 −7.2 | * | −8.0 −7.5 | * | −0.5 | * | −0.5 | * |
| (R)-25 (S)-25 | 1.08 | <1.00 | 1.66 | <1.00 | −6.1 −6.7 | * | −7.9 −8.3 | * | −0.6 | * | −0.4 | * |
| (R)-26 (S)-26 | 1.49 | <1.00 | 1.39 | <1.00 | −6.5 −7.2 | * | −7.6 −7.8 | * | −0.5 | * | −0.2 | * |
| (R)-27 (S)-27 | 1.97 | <1.00 | 1.91 | <1.00 | −7.0 −7.1 | * | −7.5 −7.6 | * | −0.1 | * | −0.1 | * |
| (R)-28 (S)-28 | 1.87 | 1.13 | 1.50 | <1.00 | −6.3 −6.8 | −7.5 −7.2 | −7.8 −7.5 | * | −0.5 | −0.3 | −0.3 | * |
| (R)-29 (S)-29 | 1.18 | 1.18 | 2.08 | <1.00 | −6.6 −7.1 | −7.6 −7.6 | −7.7 −7.5 | * | −0.5 | 0.0 | −0.2 | * |
| (R,R)-30 (S,S)-30 | 1.93 | <1.00 | <1.00 | 1.16 | −6.6 −6.8 | * | * | −5.4 −5.5 | −0.2 | * | * | −0.1 |
| (S,R)-31 (R,S)-31 | <1.00 | <1.00 | <1.00 | <1.00 | * | * | * | * | * | * | * | * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phyo, Y.Z.; Cravo, S.; Palmeira, A.; Tiritan, M.E.; Kijjoa, A.; Pinto, M.M.M.; Fernandes, C. Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns. Molecules 2018, 23, 142. https://doi.org/10.3390/molecules23010142
Phyo YZ, Cravo S, Palmeira A, Tiritan ME, Kijjoa A, Pinto MMM, Fernandes C. Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns. Molecules. 2018; 23(1):142. https://doi.org/10.3390/molecules23010142
Chicago/Turabian StylePhyo, Ye‛ Zaw, Sara Cravo, Andreia Palmeira, Maria Elizabeth Tiritan, Anake Kijjoa, Madalena M. M. Pinto, and Carla Fernandes. 2018. "Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns" Molecules 23, no. 1: 142. https://doi.org/10.3390/molecules23010142
APA StylePhyo, Y. Z., Cravo, S., Palmeira, A., Tiritan, M. E., Kijjoa, A., Pinto, M. M. M., & Fernandes, C. (2018). Enantiomeric Resolution and Docking Studies of Chiral Xanthonic Derivatives on Chirobiotic Columns. Molecules, 23(1), 142. https://doi.org/10.3390/molecules23010142