Anesthetic Agents of Plant Origin: A Review of Phytochemicals with Anesthetic Activity

Abstract
1. Introduction
2. Method
3. Plants That Contributed to the Development of Anesthesia
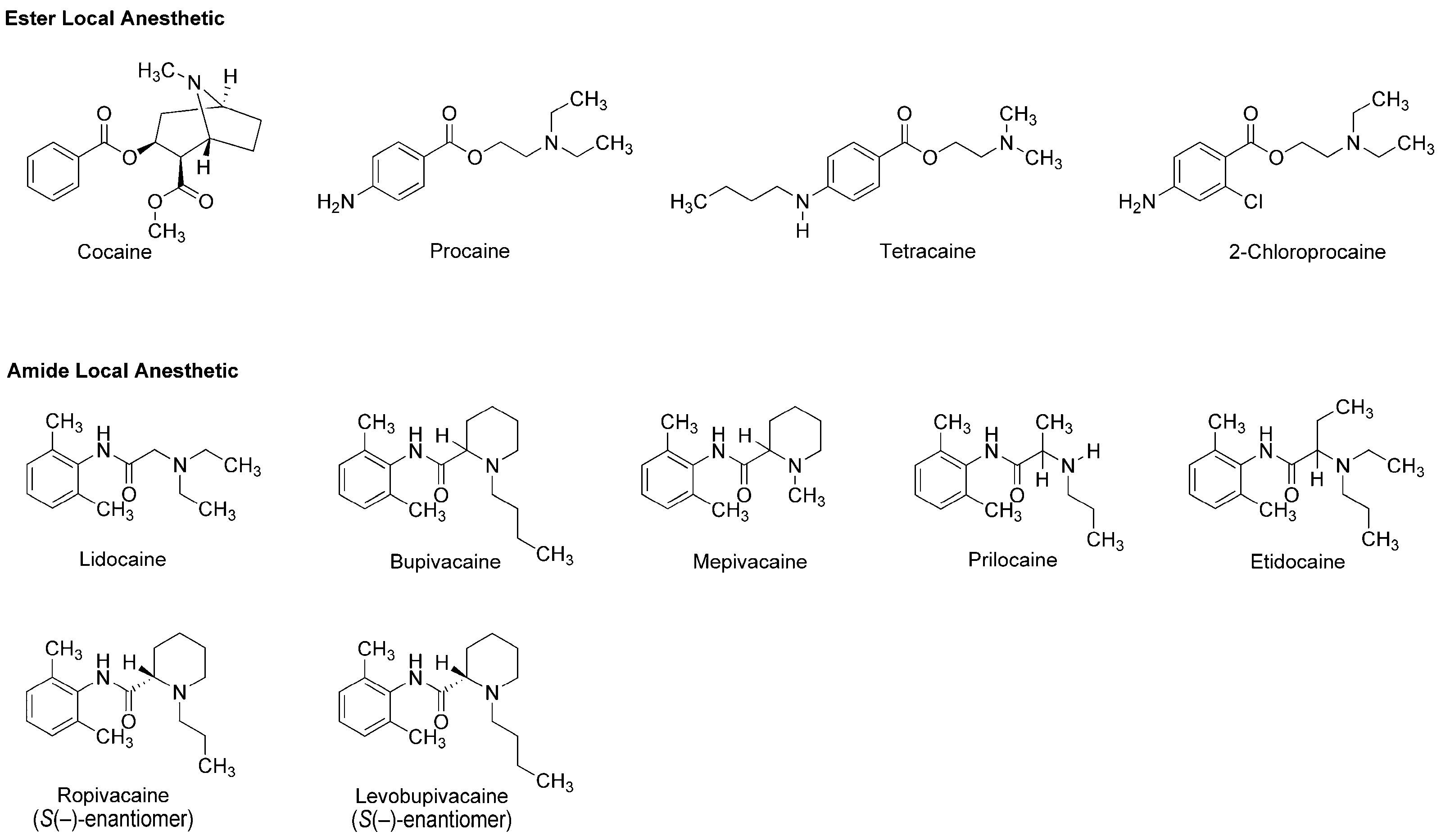
3.1. Local Anesthetics Derived from Plant Alkaloid
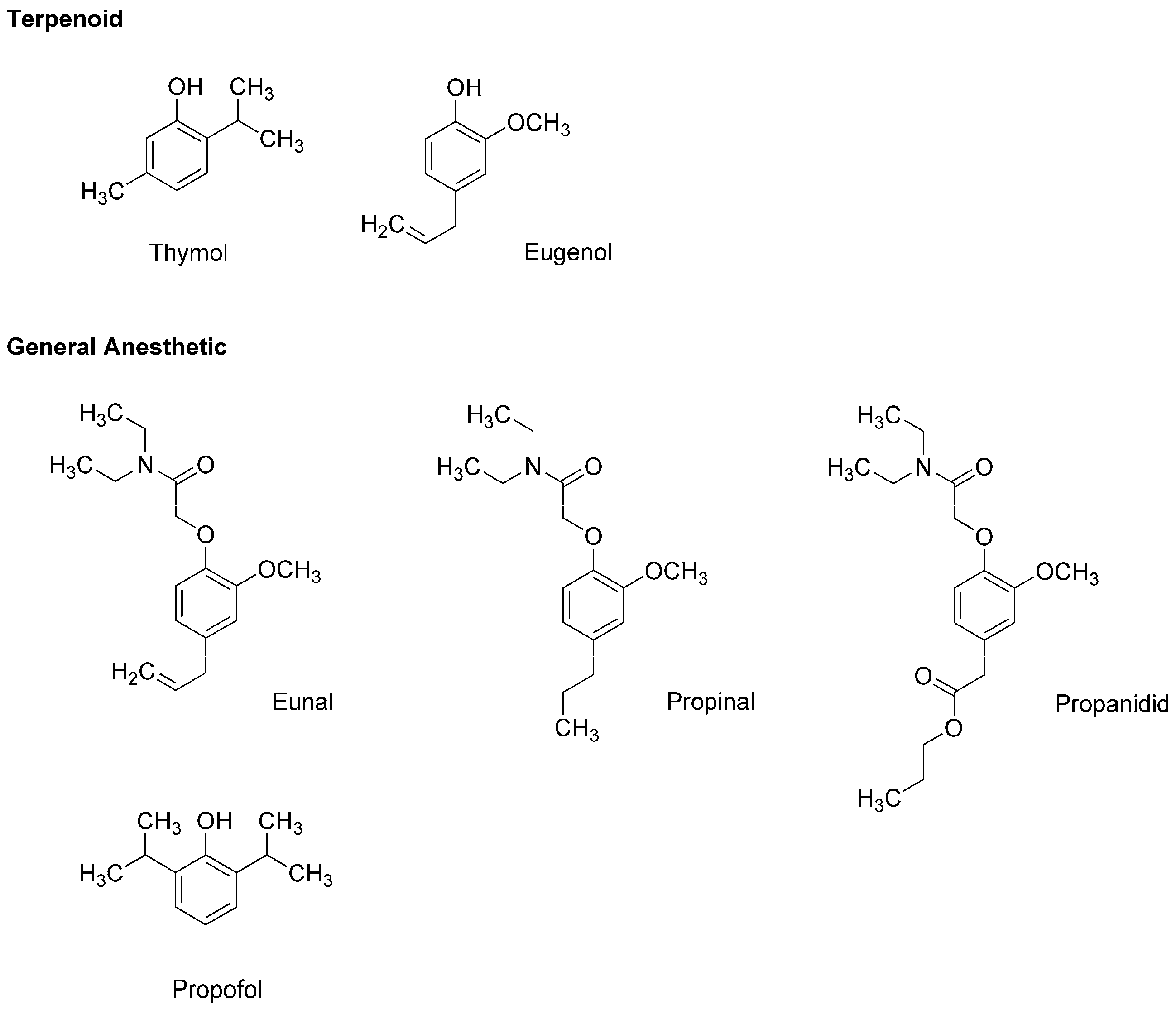
3.2. General Anesthetics Associated with Plant Terpenoids
4. Phytochemicals with the Local Anesthetic Activity
4.1. Plant Preparations
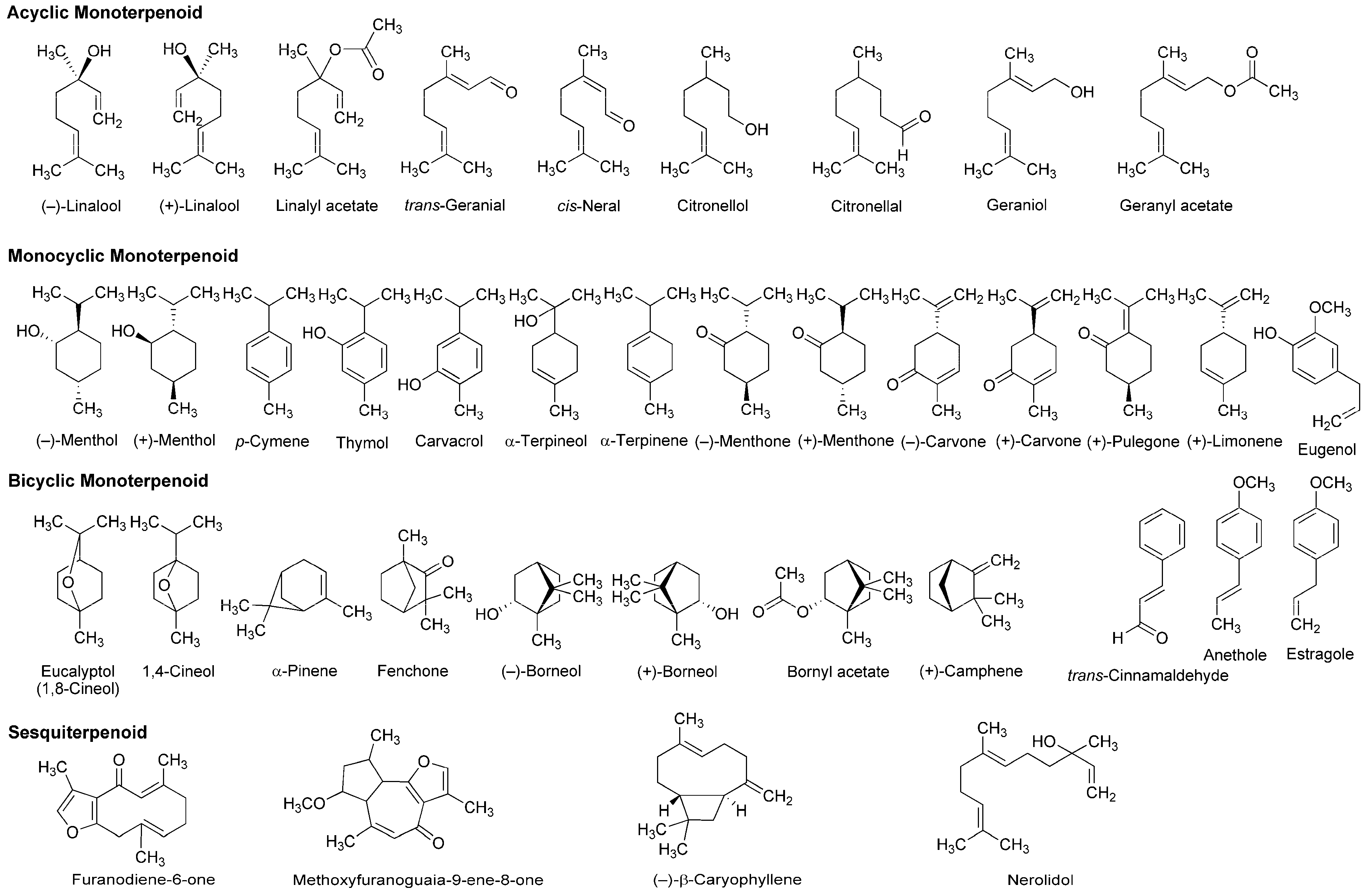
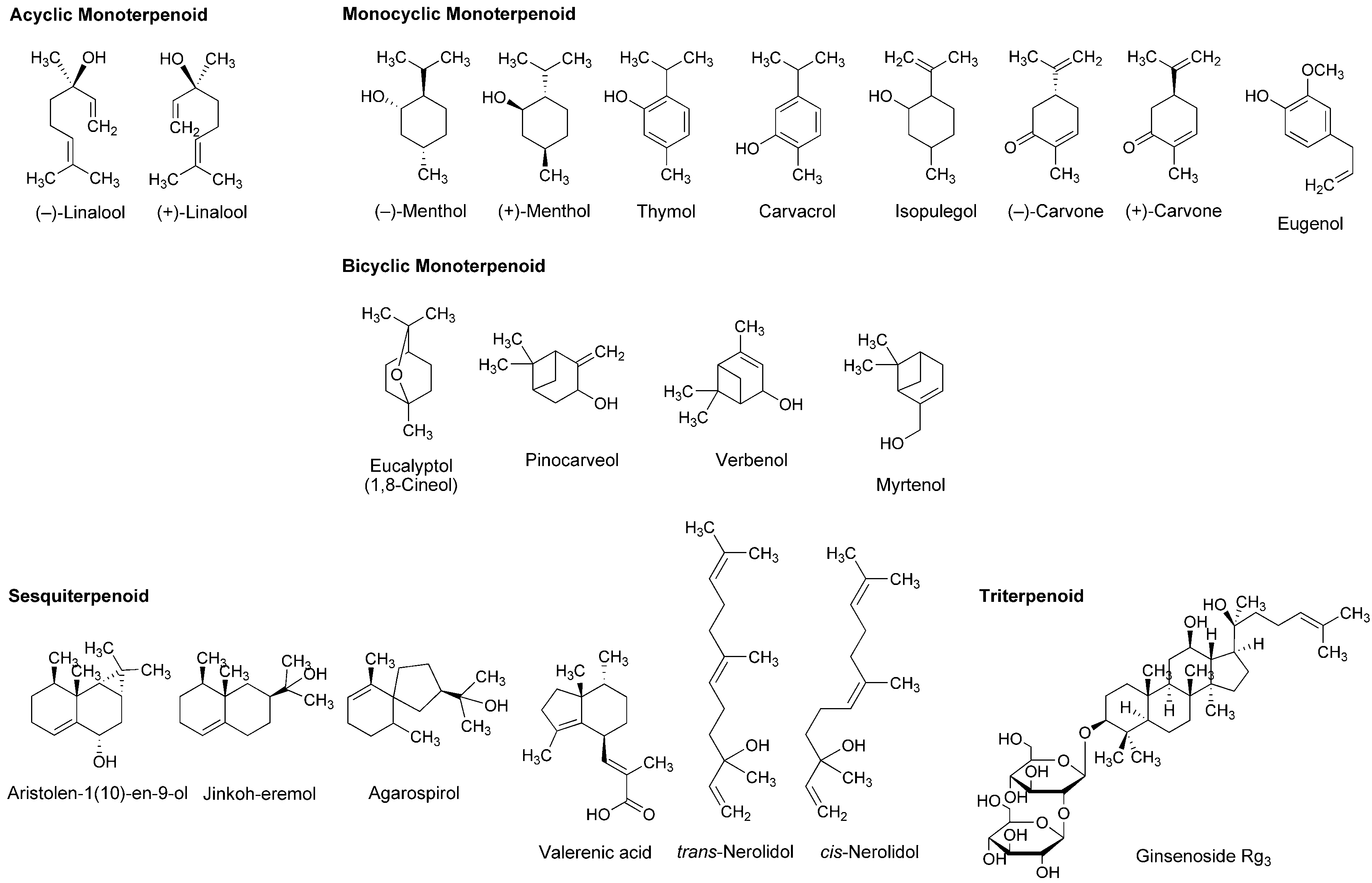
4.2. Essential Oils and Terpenoids
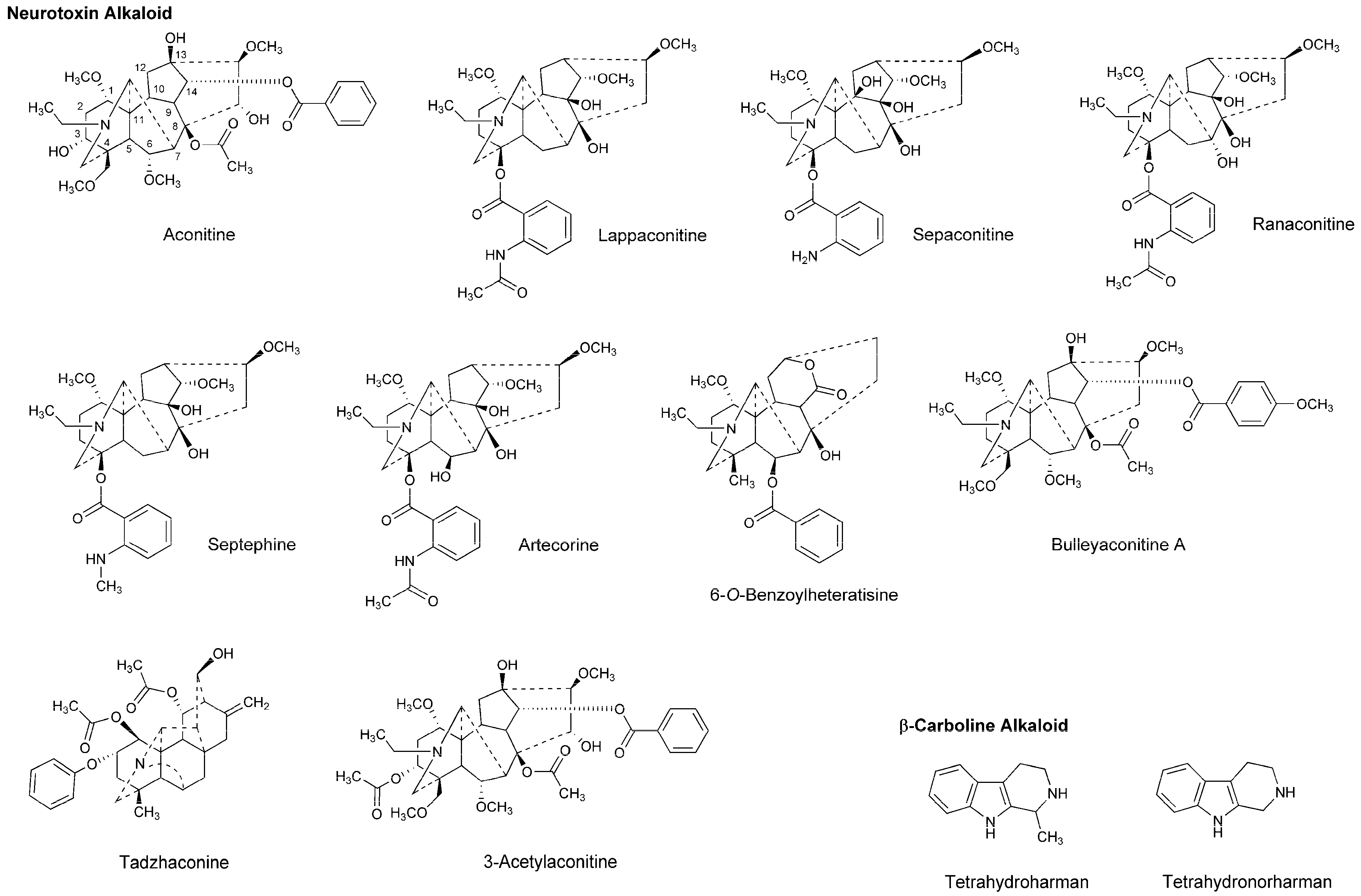
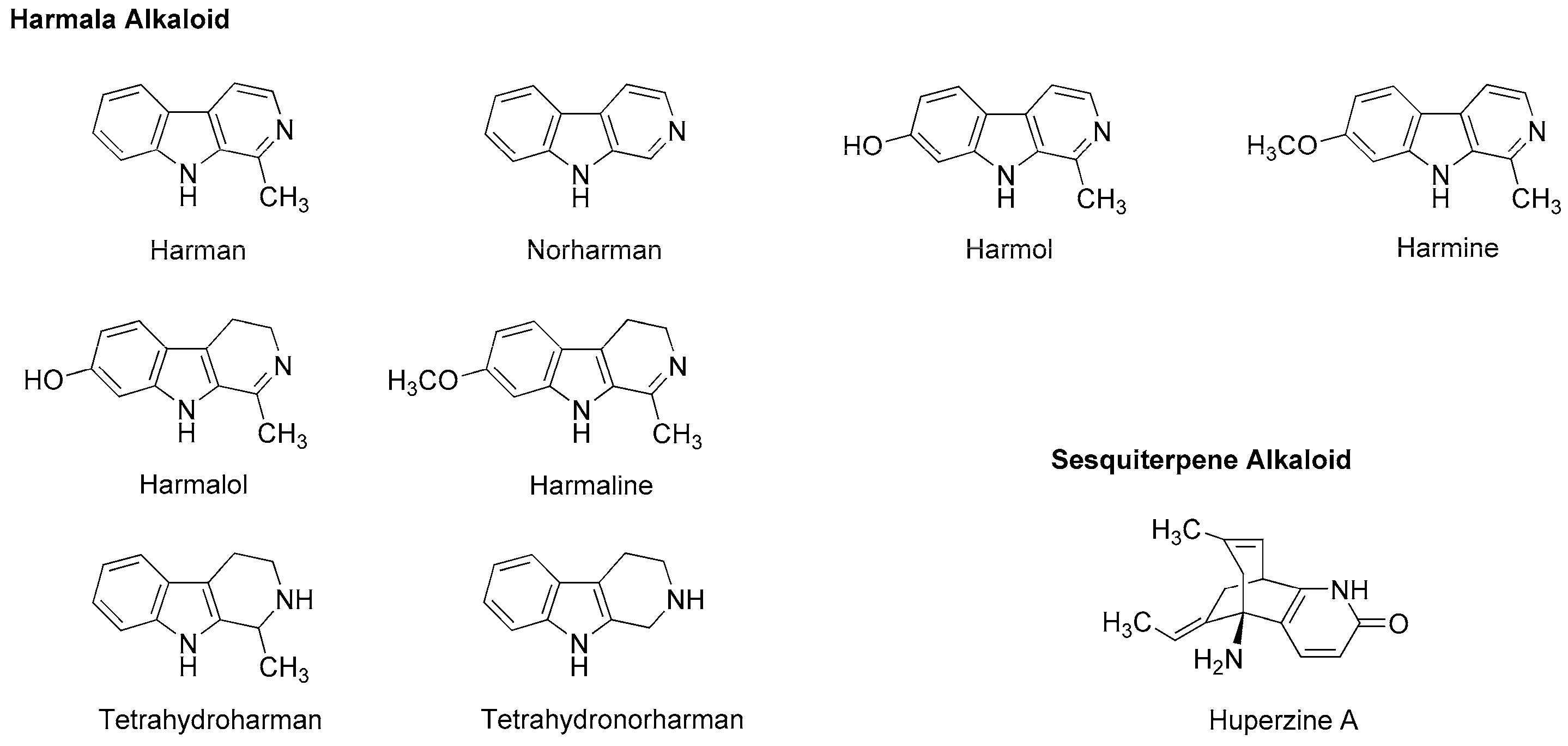
4.3. Alkaloids
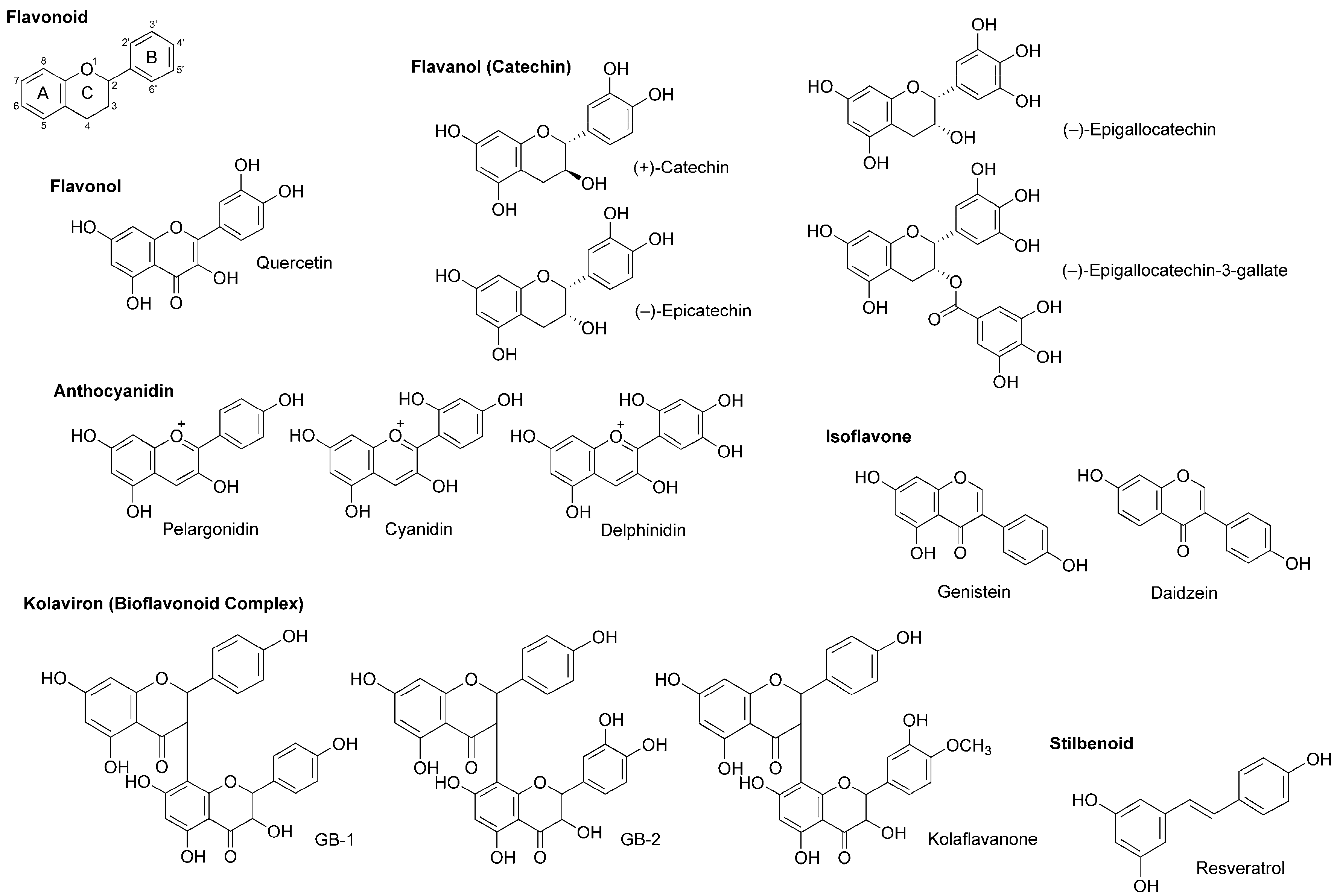
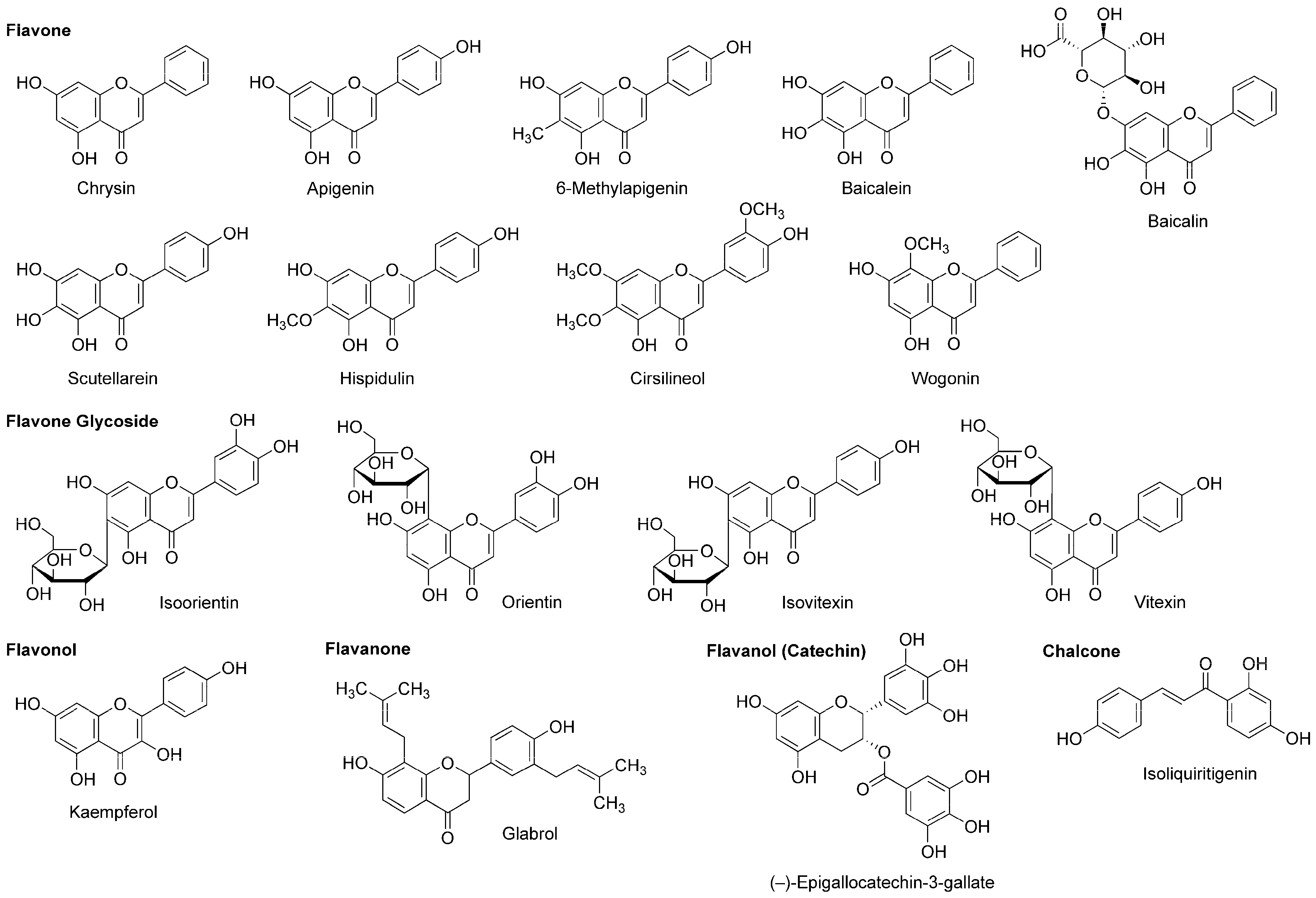
4.4. Flavonoids
4.5. Stilbenoids
5. Phytochemicals with the General Anesthetic Activity
5.1. Plant Preparations
5.2. Essential Oils and Terpenoids
5.3. Alkaloids
5.4. Flavonoids
6. Clinical Applicability of Phytochemicals
6.1. Local Anesthetic Phytochemicals
6.2. General Anesthetic Phytochemicals
6.3. Methodological Considerations in Discovering Phytochemical Lead Compounds
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CAP | Compound action potential |
| GABAA receptor | γ-aminobutyric acid type A receptor |
| Nav channel | Voltage-gated Na+ channel |
| NMDA receptor | N-methyl-d-aspartate receptor |
| TTX | Tetrodotoxin |
References
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs and over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Chidiac, E.J.; Kaddoum, R.N.; Fuleihan, S.F. Mandragora: Anesthetic of the ancients. Anesth. Analg. 2012, 115, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Fabricant, D.S.; Farnsworth, N.R. The value of plants used in traditional medicine for drug discovery. Environ. Health Perspect. 2001, 109, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Jachak, S.M.; Saklani, A. Challenges and opportunities in drug discovery from plants. Curr. Sci. 2007, 92, 1251–1257. [Google Scholar]
- Grémiaux, A.; Yokawa, K.; Mancuso, S.; Baluška, F. Plant anesthesia supports similarities between animals and plants: Claude Bernard’s forgotten studies. Plant Signal. Behav. 2014, 9, e27886. [Google Scholar] [CrossRef] [PubMed]
- Baluška, F.; Yokawa, K.; Mancuso, S.; Baverstock, K. Understanding of anesthesia—Why consciousness is essential for life and not based on genes. Commun. Integr. Biol. 2016, 9, e1238118. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, J.; González, Á. History of the development and evolution of local anesthesia since the coca leaf. Anesthesiology 2003, 98, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Koller, C. The sub-conjunctival application of cocaine in eye operations. Trans. Am. Ophthalmol. Soc. 1892, 6, 421–424. [Google Scholar] [PubMed]
- Fozzard, H.A.; Lee, P.J.; Lipkind, G.M. Mechanism of local anesthetic drug action on voltage-gated sodium channels. Curr. Pharm. Des. 2005, 11, 2671–2686. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Mizogami, M. Interaction of local anesthetics with biomembranes consisting of phospholipids and cholesterol: Mechanistic and clinical implications for anesthetic and cardiotoxic effects. Anesthesiol. Res. Pract. 2013, 2013, 297141. [Google Scholar] [CrossRef] [PubMed]
- James, R.; Glen, J.B. Synthesis, biological evaluation, and preliminary structure-activity considerations of a series of alkylphenols as intravenous anesthetic agent. J. Med. Chem. 1980, 23, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Kay, B.; Stephenson, D.K. ICI 35868 (Diprivan): A new intravenous anaesthetic. A comparison with Althesin. Anaesthesia 1980, 35, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- García, D.A.; Bujons, J.; Vale, C.; Suñol, C. Allosteric positive interaction of thymol with the GABAA receptor in primary cultures of mouse cortical neurons. Neuropharmacology 2006, 50, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Reiner, G.N.; Delgado-Marín, L.; Olguín, N.; Sánchez-Redondo, S.; Sánchez-Borzone, M.; Rodríguez-Farré, E.; Suñol, C.; García, D.A. Gabaergic pharmacological activity of propofol related compounds as possible enhancers of general anesthetics and interaction with membranes. Cell Biochem. Biophys. 2013, 67, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Structure-specific membrane-fluidizing effect of propofol. Clin. Exp. Pharmacol. Physiol. 2001, 28, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Reiner, G.N.; Fraceto, L.F.; de Paula, E.; Perillo, M.A.; García, D.A. Effects of gabaergic phenols on phospholipid bilayers as evaluated by 1H-NMR. J. Biomater. Nanobiotechnol. 2013, 4, 28–34. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, S.; Geetha, V.S.; Kuruvilla, A. Determination of local anesthetic action of Betel leaf extract alone and with Betel nut using infiltration and surface anesthesia. J. Nat. Rem. 2001, 1, 28–32. [Google Scholar] [CrossRef]
- Abdulrahman, F.I.; Onyeyili, P.A.; Sandabe, U.K.; Ogugbuaja, V.O. Evaluation of the effects of the aqueous extract of Vitex doniana root-bark on the peripheral and central nervous system of laboratory animals. J. Appl. Sci. 2007, 7, 1397–1403. [Google Scholar] [CrossRef]
- Tijjani, M.A.; Abdulrahman, F.I.; Khan, I.Z.; Sandabe, U.K. The effects of ethanolic extract of Vitex doniana stem bark on peripheral and central nervous system of laboratory animals. J. Appl. Pharm. Sci. 2012, 2, 74–79. [Google Scholar] [CrossRef]
- Chakraborty, A.; Devi, B.R.; Sanjebam, R.; Khumbong, S.; Thokchom, I.S. Preliminary studies on local anesthetic and antipyretic activities of Spilanthes acmella Murr. in experimental animal models. Indian J. Pharmacol. 2010, 42, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Udegbunam, R.I.; Asuzu, U.I.; Kene, R.O.; Udegbunam, S.O.; Nwaehujor, C.O. Local anesthetic and tissue effects of the leaf extract and fractions of Sterculia tragacantha Lindl. J. Pharmacol. Toxicol. 2012, 7, 192–198. [Google Scholar] [CrossRef]
- Udegbunam, R.I.; Asuzu, U.I.; Kene, R.O.; Udegbunam, S.O. Evaluation of local anesthetic efficacy of the crude extract of Sterculia tragacantha using West African Dwarf goats. Sokoto J. Vet. Sci. 2013, 11, 13–21. [Google Scholar] [CrossRef]
- De Sousa, D.P. Analgesic-like activity of essential oils constituents. Molecules 2011, 16, 2233–2252. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, A.G.; Quintans, J.S.; Quintans, L.J., Jr. Monoterpenes with analgesic activity—A systematic review. Phytother. Res. 2013, 27, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Djilani, A.; Dicko, A. The therapeutic benefits of essential oils. In Nutrition, Well-being and Health; Bouayed, J., Bohn, T., Eds.; InTech: Shanghai, China, 2012; pp. 155–178. [Google Scholar] [CrossRef]
- Oz, M.; Lozon, Y.; Sultan, A.; Yang, K.H.; Galadari, S. Effects of monoterpenes on ion channels of excitable cells. Pharmacol. Ther. 2015, 152, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Zalachoras, I.; Kagiava, A.; Vokou, D.; Theophilidis, G. Assessing the local anesthetic effect of five essential oil constituents. Planta Med. 2010, 76, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, C.; Galeotti, N.; Salvatore, G.; Mazzanti, G. Local anaesthetic activity of the essential oil of Lavandula angustifolia. Planta Med. 1999, 65, 700–703. [Google Scholar] [CrossRef] [PubMed]
- Leal-Cardoso, J.H.; da Silva-Alves, K.S.; Ferreira-da-Silva, F.W.; dos Santos-Nascimento, T.; Joca, H.C.; de Macedo, F.H.; de Albuquerque-Neto, P.M.; Magalhães, P.J.; Lahlou, S.; Cruz, J.S.; et al. Linalool blocks excitability in peripheral nerves and voltage-dependent Na+ current in dissociated dorsal root ganglia neurons. Eur. J. Pharmacol. 2010, 645, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Peana, A.T.; D’Aquila, P.S.; Panin, F.; Serra, G.; Pippia, P.; Moretti, M.D. Anti-inflammatory activity of linalool and linalyl acetate constituents of essential oils. Phytomedicine 2002, 9, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, N.; Ghelardini, C.; Mannelli, L.; Mazzanti, G.; Baghiroli, L.; Bartolini, A. Local anaesthetic activity of (+)- and (−)-menthol. Planta Med. 2001, 67, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, C.; Hao, J.; Martin-Eauclaire, M.F.; Gabriac, M.; Delmas, P. Menthol pain relief through cumulative inactivation of voltage-gated sodium channels. Pain 2012, 153, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Tian, Y.; Gao, R.; Li, H.; Zhao, X.; Barrett, J.E.; Hu, H. Central mechanisms of menthol-induced analgesia. J. Pharmacol. Exp. Ther. 2012, 343, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Mizuta, K.; Fujita, T.; Kumamoto, E. Inhibition by menthol and its related chemicals of compound action potentials in frog sciatic nerves. Life Sci. 2013, 92, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Tomohiro, D.; Mizuta, K.; Fujita, T.; Nishikubo, Y.; Kumamoto, E. Inhibition by capsaicin and its related vanilloids of compound action potentials in frog sciatic nerves. Life Sci. 2013, 92, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, S.; Fujita, T.; Matsushita, A.; Kumamoto, E. Inhibition of the compound action potentials of frog sciatic nerves by aroma oil compounds having various chemical structures. Pharmacol. Res. Perspect. 2015, 3, e00127. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, R.; Fujita, T.; Koga, A.; Liu, T.; Nakatsuka, T.; Nakashima, M.; Kumamoto, E. Tramadol, but not its major metabolite (mono-O-demethyl tramadol) depresses compound action potentials in frog sciatic nerves. Br. J. Pharmacol. 2006, 149, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, K.; Fujita, T.; Nakatsuka, T.; Kumamoto, E. Inhibitory effects of opioids on compound action potentials in frog sciatic nerves and their chemical structures. Life Sci. 2008, 83, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Uemura, Y.; Fujita, T.; Ohtsubo, S.; Hirakawa, N.; Sakaguchi, Y.; Kumamoto, E. Effects of various antiepileptics used to alleviate neuropathic pain on compound action potential in frog sciatic nerves: Comparison with those of local anesthetics. Biomed. Res. Int. 2014, 2014, 540238. [Google Scholar] [CrossRef] [PubMed]
- Joca, H.C.; Cruz-Mendes, Y.; Oliveira-Abreu, K.; Maia-Joca, R.P.; Barbosa, R.; Lemos, T.L.; Lacerda Beirão, P.S.; Leal-Cardoso, J.H. Carvacrol decreases neuronal excitability by inhibition of voltage-gated sodium channels. J. Nat. Prod. 2012, 75, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante Melo, F.H.; Rios, E.R.; Rocha, N.F.; Citó Mdo, C.; Fernandes, M.L.; de Sousa, D.P.; de Vasconcelos, S.M.; de Sousa, F.C. Antinociceptive activity of carvacrol (5-isopropyl-2-methylphenol) in mice. J. Pharm. Pharmacol. 2012, 64, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.C.; de Sousa Oliveira, F.; Benedito, R.B.; de Sousa, D.P.; de Almeida, R.N.; de Araújo, D.A. Antinociceptive activity of (−)-carvone: Evidence of association with decreased peripheral nerve excitability. Biol. Pharm. Bull. 2008, 31, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Leal-Cardoso, J.H.; Matos-Brito, B.G.; Lopes-Junior, J.E.; Viana-Cardoso, K.V.; Sampaio-Freitas, A.B.; Brasil, R.O.; Coelho-De-Souza, A.N.; Albuquerque, A.A. Effects of estragole on the compound action potential of the rat sciatic nerve. Braz. J. Med. Biol. Res. 2004, 37, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Silva-Alves, K.S.; Ferreira-da-Silva, F.W.; Peixoto-Neves, D.; Viana-Cardoso, K.V.; Moreira-Júnior, L.; Oquendo, M.B.; Oliveira-Abreu, K.; Albuquerque, A.A.; Coelho-de-Souza, A.N.; Leal-Cardoso, J.H. Estragole blocks neuronal excitability by direct inhibition of Na+ channels. Braz. J. Med. Biol. Res. 2013, 46, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Alves, K.S.; Ferreira-da-Silva, F.W.; Coelho-de-Souza, A.N.; Albuquerque, A.A.; do Vale, O.C.; Leal-Cardoso, J.H. Essential oil of Croton zehntneri and its main constituent anethole block excitability of rat peripheral nerve. Planta Med. 2015, 81, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.G.; Sousa, S.D.; Silva, R.E.; Silva-Alves, K.S.; Ferreira-da-Silva, F.W.; Kerntopf, M.R.; Menezes, I.R.; Leal-Cardoso, J.H.; Barbosa, R. Essential oil of Lippia alba and its main constituent citral block the excitability of rat sciatic nerves. Braz. J. Med. Biol. Res. 2015, 48, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Lima-Accioly, P.M.; Lavor-Porto, P.R.; Cavalcante, F.S.; Magalhães, P.J.; Lahlou, S.; Morais, S.M.; Leal-Cardoso, J.H. Essential oil of Croton nepetaefolius and its main constituent, 1,8-cineole, block excitability of rat sciatic nerve in vitro. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-da-Silva, F.W.; Barbosa, R.; Moreira-Júnior, L.; dos Santos-Nascimento, T.; de Oliveira-Martins, M.D.; Coelho-de-Souza, A.N.; Cavalcante, F.S.; Ceccatto, V.M.; de Lemos, T.L.; Magalhães, P.J.; et al. Effects of 1,8-cineole on electrophysiological parameters of neurons of the rat superior cervical ganglion. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, C.; Galeotti, N.; Mazzanti, G. Local anaesthetic activity of monoterpenes and phenylpropanes of essential oils. Planta Med. 2001, 67, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Quintans-Júnior, L.; Moreira, J.C.; Pasquali, M.A.; Rabie, S.M.; Pires, A.S.; Schröder, R.; Rabelo, T.K.; Santos, J.P.; Lima, P.S.; Cavalcanti, S.C.; et al. Antinociceptive activity and redox profile of the monoterpenes (+)-camphene, p-cymene, and geranyl acetate in experimental models. ISRN Toxicol. 2013, 2013, 459530. [Google Scholar] [CrossRef] [PubMed]
- Dolara, P.; Corte, B.; Ghelardini, C.; Pugliese, A.M.; Cerbai, E.; Menichetti, S.; Lo Nostro, A. Local anaesthetic, antibacterial and antifungal properties of sesquiterpenes from myrrh. Planta Med. 2000, 66, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, C.; Galeotti, N.; Di Cesare Mannelli, L.; Mazzanti, G.; Bartolini, A. Local anaesthetic activity of β-caryophyllene. Farmaco 2001, 56, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Mendanha, S.A.; Moura, S.S.; Anjos, J.L.; Valadares, M.C.; Alonso, A. Toxicity of terpenes on fibroblast cells compared to their hemolytic potential and increase in erythrocyte membrane fluidity. Toxicol. In Vitro 2013, 27, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Shi, X.; Ding, H.; Dai, X.; Wan, G.; Qiao, Y. Interactions of borneol with DPPC phospholipid membranes: A molecular dynamics simulation study. Int. J. Mol. Sci. 2014, 15, 20365–20381. [Google Scholar] [CrossRef] [PubMed]
- Nowotarska, S.W.; Nowotarski, K.J.; Friedman, M.; Situ, C. Effect of structure on the interactions between five natural antimicrobial compounds and phospholipids of bacterial cell membrane on model monolayers. Molecules 2014, 19, 7497–7515. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Mizogami, M. Comparative interactions of anesthetic alkylphenols with lipid membranes. Open J. Anesthesiol. 2014, 4, 308–317. [Google Scholar] [CrossRef]
- Sarmento-Neto, J.F.; do Nascimento, L.G.; Felipe, C.F.; de Sousa, D.P. Analgesic potential of essential oils. Molecules 2016, 21, 20. [Google Scholar] [CrossRef] [PubMed]
- Vadhanan, C.P.; Narendren, G. Future local anesthetics—Neurotoxins? Int. J. Anesthesiol. Res. 2014, 2, 11–15. [Google Scholar] [CrossRef]
- Dzhakhangirov, F.N.; Kasymova, K.R.; Sultankhodzhaev, M.N.; Salimov, B.T.; Usmanova, S.K.; Shakirov, R.S. Toxicity and local anesthetic activity of diterpenoid alkaloids. Chem. Nat. Compd. 2007, 43, 581–589. [Google Scholar] [CrossRef]
- Wright, S.N. Irreversible block of human heart (hH1) sodium channels by the plant alkaloid lappaconitine. Mol. Pharmacol. 2001, 59, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Gutser, U.T.; Friese, J.; Heubach, J.F.; Matthiesen, T.; Selve, N.; Wilffert, B.; Gleitz, J. Mode of antinociceptive and toxic action of alkaloids of Aconitum spec. Naunyn Schmiedebergs Arch. Pharmacol. 1998, 357, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Z.; Xiao, Y.Q.; Zhang, C.; Sun, X.M. Study of analgesic and anti-inflammatory effects of lappaconitine gelata. J. Tradit. Chin. Med. 2009, 29, 141–145. [Google Scholar] [CrossRef]
- Wang, C.F.; Gerner, P.; Wang, S.Y.; Wang, G.K. Bulleyaconitine A isolated from Aconitum plant displays long-acting local anesthetic properties in vitro and in vivo. Anesthesiology 2007, 107, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.F.; Gerner, P.; Schmidt, B.; Xu, Z.Z.; Nau, C.; Wang, S.Y.; Ji, R.R.; Wang, G.K. Use of bulleyaconitine A as an adjuvant for prolonged cutaneous analgesia in the rat. Anesth. Analg. 2008, 107, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Turabekova, M.A.; Rasulev, B.F.; Dzhakhangirov, F.N.; Toropov, A.A.; Leszczynska, D.; Leszczynski, J. Aconitum and Delphinium diterpenoid alkaloids of local anesthetic activity: Comparative QSAR analysis based on GA-MLRA/PLS and optimal descriptors approach. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2014, 32, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Biphasic effects of acetaldehyde-biogenic amine condensation products on membrane fluidity. J. Pharm. Pharmacol. 2001, 53, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Mizogami, M. Drinking-related tetrahydroharmans counteract the membrane effects of local anesthetic lidocaine. J. Drug Alcohol Res. 2014, 3, 235887. [Google Scholar] [CrossRef]
- Wu, Y.L.; Ohsaga, A.; Oshiro, T.; Iinuma, K.; Kondo, Y.; Ebihara, S.; Sasaki, H.; Maruyama, Y. Suppressive effects of red wine polyphenols on voltage-gated ion channels in dorsal root ganglionic neuronal cells. Tohoku J. Exp. Med. 2005, 206, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Lim, J.M.; Kim, S.S.; Kim, J.; Park, M.; Song, J.H. Effects of (−)epigallocatechin-3-gallate on Na+ currents in rat dorsal root ganglion neurons. Eur. J. Pharmacol. 2009, 604, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Paillart, C.; Carlier, E.; Guedin, D.; Dargent, B.; Couraud, F. Direct block of voltage-sensitive sodium channels by genistein, a tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther. 1997, 280, 521–526. [Google Scholar] [PubMed]
- Adaramoye, O.A. Protective effect of kolaviron, a bioflavonoid from Garcinia kola seeds, in brain of Wistar albino rats exposed to gamma-radiation. Biol. Pharm. Bull. 2010, 33, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Tchimene, M.K.; Aanaga, A.O.; Ugwoke, C.E.; Ezugu, C.O.; Okunji, C.; Iwu, M.M. Bio-flavonoids and garcinoic acid from Garcinia kola seeds with promising local anesthetic potentials. Int. J. Pharmacogn. Phytochem. Res. 2015, 7, 764–767. [Google Scholar]
- Kim, H.I.; Kim, T.H.; Song, J.H. Resveratrol inhibits Na+ currents in rat dorsal root ganglion neurons. Brain Res. 2005, 1045, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Chau, P.L. New insights into the molecular mechanisms of general anaesthetics. Br. J. Pharmacol. 2010, 161, 288–307. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; Li, G.D. GABAA receptors as molecular targets of general anesthetics: Identification of binding sites provides clues to allosteric modulation. Can. J. Anaesth. 2011, 58, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Membrane interactions of phytochemicals as their molecular mechanism applicable to the discovery of drug leads from plants. Molecules 2015, 20, 18923–18966. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.Y.; Xue, H. Development of effective therapeutics targeting the GABAA receptor: Naturally occurring alternatives. Curr. Pharm. Des. 2004, 10, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Zaku, S.G.; Abdulrahaman, F.A.; Onyeyili, P.A.; Aguzue, O.C.; Thomas, S.A. Phytochemical constituents and effects of aqueous root-bark extract of Ficus sycomorus L. (Moraceae) on muscular relaxation, anaesthetic and sleeping time on laboratory animals. Afr. J. Biotechnol. 2009, 8, 6004–6006. [Google Scholar]
- Grundmann, O.; Wang, J.; McGregor, G.P.; Butterweck, V. Anxiolytic activity of a phytochemically characterized Passiflora incarnata extract is mediated via the GABAergic system. Planta Med. 2008, 74, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Appel, K.; Rose, T.; Fiebich, B.; Kammler, T.; Hoffmann, C.; Weiss, G. Modulation of the γ-aminobutyric acid (GABA) system by Passiflora incarnata L. Phytother. Res. 2011, 25, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Lolli, L.F.; Sato, C.M.; Romanini, C.V.; de Villas-Boas, L.B.; Santos, C.A.; de Oliveira, R.M. Possible involvement of GABAA-benzodiazepine receptor in the anxiolytic-like effect induced by Passiflora actinia extracts in mice. J. Ethnopharmacol. 2007, 111, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, C.G.; Silva, L.L.; Reckziegel, P.; Barros, F.M.; Bürger, M.E.; Baldisserotto, B.; Mallmann, C.A.; Schmidt, D.; Caron, B.O.; Heinzmann, B.M. Participation of the GABAergic system in the anesthetic effect of Lippia alba (Mill.) N.E. Brown essential oil. Braz. J. Med. Biol. Res. 2012, 45, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Parodi, T.V.; Cunha, M.A.; Heldwein, C.G.; de Souza, D.M.; Martins, Á.C.; de Garcia, L.O.; Wasielesky, W., Jr.; Monserrat, J.M.; Schmidt, D.; Caron, B.O.; et al. The anesthetic efficacy of eugenol and the essential oils of Lippia alba and Aloysia triphylla in post-larvae and sub-adults of Litopenaeus vannamei (Crustacea, Penaeidae). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2012, 155, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.L.; Garlet, Q.I.; Benovit, S.C.; Dolci, G.; Mallmann, C.A.; Bürger, M.E.; Baldisserotto, B.; Longhi, S.J.; Heinzmann, B.M. Sedative and anesthetic activities of the essential oils of Hyptis mutabilis (Rich.) Briq. and their isolated components in silver catfish (Rhamdia quelen). Braz. J. Med. Biol. Res. 2013, 46, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Toni, C.; Becker, A.G.; Simões, L.N.; Pinheiro, C.G.; de Lima Silva, L.; Heinzmann, B.M.; Caron, B.O.; Baldisserotto, B. Fish anesthesia: Effects of the essential oils of Hesperozygis ringens and Lippia alba on the biochemistry and physiology of silver catfish (Rhamdia quelen). Fish Physiol. Biochem. 2014, 40, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, H.; Ito, M.; Shiraki, T.; Yagura, T.; Honda, G. Sedative effects of vapor inhalation of agarwood oil and spikenard extract and identification of their active components. J. Nat. Med. 2008, 62, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, H.; Ito, M.; Asada, Y.; Kobayashi, Y. Inhalation administration of the sesquiterpenoid aristolen-1(10)-en-9-ol from Nardostachys chinensis has a sedative effect via the GABAergic system. Planta Med. 2015, 81, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, H.; Ueda, R.; Matsumoto, K.; Kawanishi, K.; Kato, A. Effect of jinkoh-eremol and agarospirol from agarwood on the central nervous system in mice. Planta Med. 1996, 62, 2–6. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, D.P.; de Almeida Soares Hocayen, P.; Andrade, L.N.; Andreatini, R. A systematic review of the anxiolytic-like effects of essential oils in animal models. Molecules 2015, 20, 18620–18660. [Google Scholar] [CrossRef] [PubMed]
- Kessler, A.; Sahin-Nadeem, H.; Lummis, S.C.; Weigel, I.; Pischetsrieder, M.; Buettner, A.; Villmann, C. GABAA receptor modulation by terpenoids from Sideritis extracts. Mol. Nutr. Food Res. 2014, 58, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.S.; Mehendale, S.; Xiao, Y.; Aung, H.H.; Xie, J.T.; Ang-Lee, M.K. The gamma-aminobutyric acidergic effects of valerian and valerenic acid on rat brainstem neuronal activity. Anesth. Analg. 2004, 98, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Khom, S.; Baburin, I.; Timin, E.; Hohaus, A.; Trauner, G.; Kopp, B.; Hering, S. Valerenic acid potentiates and inhibits GABAA receptors: Molecular mechanism and subunit specificity. Neuropharmacology 2007, 53, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Trauner, G.; Khom, S.; Baburin, I.; Benedek, B.; Hering, S.; Kopp, B. Modulation of GABAA receptors by valerian extracts is related to the content of valerenic acid. Planta Med. 2008, 74, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Benke, D.; Barberis, A.; Kopp, S.; Altmann, K.H.; Schubiger, M.; Vogt, K.E.; Rudolph, U.; Möhler, H. GABAA receptors as in vivo substrate for the anxiolytic action of valerenic acid, a major constituent of valerian root extracts. Neuropharmacology 2009, 56, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Peana, A.T.; De Montis, M.G.; Sechi, S.; Sircana, G.; D’Aquila, P.S.; Pippia, P. Effects of (−)-linalool in the acute hyperalgesia induced by carrageenan, l-glutamate and prostaglandin E2. Eur. J. Pharmacol. 2004, 497, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.; Taylor, J.E.; Flores, J.; Bracken, S.; McCall, S.; Ceremuga, T.E. Investigation of the anxiolytic effects of linalool, a lavender extract, in the male Sprague-Dawley rat. AANA J. 2008, 76, 47–52. [Google Scholar] [PubMed]
- Linck, V.M.; da Silva, A.L.; Figueiró, M.; Piato, A.L.; Herrmann, A.P.; Dupont Birck, F.; Caramão, E.B.; Nunes, D.S.; Moreno, P.R.; Elisabetsky, E. Inhaled linalool-induced sedation in mice. Phytomedicine 2009, 16, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, Y.; Hara, C.; Tamura, K.; Fujii, T.; Nakamura, K.; Masujima, T.; Aoki, T. Sedative effect on humans of inhalation of essential oil of linalool: Sensory evaluation and physiological measurements using optically active linalools. Anal. Chim. Acta 1998, 365, 293–299. [Google Scholar] [CrossRef]
- Heldwein, C.G.; de Silva, L.L.; Gai, E.Z.; Roman, C.; Parodi, T.V.; Bürger, M.E.; Baldisserotto, B.; Flores, É.M.; Heinzmann, B.M. S-(+)-Linalool from Lippia alba: Sedative and anesthetic for silver catfish (Rhamdia quelen). Vet. Anaesth. Analg. 2014, 41, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Watt, E.E.; Betts, B.A.; Kotey, F.O.; Humbert, D.J.; Griffith, T.N.; Kelly, E.W.; Veneskey, K.C.; Gill, N.; Rowan, K.C.; Jenkins, A.; et al. Menthol shares general anesthetic activity and sites of action on the GABAA receptor with the intravenous agent, propofol. Eur. J. Pharmacol. 2008, 590, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.K.; Karim, S.; Goodchild, A.K.; Vaughan, C.W.; Drew, G.M. Menthol enhances phasic and tonic GABAA receptor-mediated currents in midbrain periaqueductal grey neurons. Br. J. Pharmacol. 2014, 171, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Priestley, C.M.; Williamson, E.M.; Wafford, K.A.; Sattelle, D.B. Thymol, a constituent of thyme essential oil, is a positive allosteric modulator of human GABAA receptors and a homo-oligomeric GABA receptor from Drosophila melanogaster. Br. J. Pharmacol. 2003, 140, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, B.; Haeseler, G.; Leuwer, M.; Dengler, R.; Krampfl, K.; Bufler, J. Structural requirements of phenol derivatives for direct activation of chloride currents via GABAA receptors. Eur. J. Pharmacol. 2001, 421, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ahn, K.; Oh, T.H.; Nah, S.Y.; Rhim, H. Inhibitory effect of ginsenosides on NMDA receptor-mediated signals in rat hippocampal neurons. Biochem. Biophys. Res. Commun. 2002, 296, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Lundbæk, J.A. Lipid bilayer-mediated regulation of ion channel function by amphiphilic drugs. J. Gen. Physiol. 2008, 131, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Mizogami, M. Analgesic agents share the membrane interactivity possibly associated with the diversity of their pharmacological properties. Br. J. Pharm. Res. 2015, 7, 110–121. [Google Scholar] [CrossRef]
- DeFeudis, F.V.; Drieu, K. Ginkgo biloba extract (EGb 761) and CNS functions: Basic studies and clinical applications. Curr. Drug Targets 2000, 1, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Aricioglu, F.; Altunbas, H. Harmane induces anxiolysis and antidepressant-like effects in rats. Ann. N. Y. Acad. Sci. 2003, 1009, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Farzin, D.; Mansouri, N. Antidepressant-like effect of harmane and other β-carbolines in the mouse forced swim test. Eur. Neuropsychopharmacol. 2006, 16, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Inhibition of membrane effects of general anesthetic propofol by benzodiazepine inverse agonist tetrahydro-β-carboline. Int. J. Pharmacol. 2012, 8, 542–548. [Google Scholar] [CrossRef]
- Zhang, J.M.; Hu, G.Y. Huperzine A, a nootropic alkaloid, inhibits N-methyl-d-aspartate-induced current in rat dissociated hippocampal neurons. Neuroscience 2001, 105, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A. Flavonoid nutraceuticals and ionotropic receptors for the inhibitory neurotransmitter GABA. Neurochem. Int. 2015, 89, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.K.; Krydsfeldt, K.; Rasmussen, H.B. Bioassay-guided isolation of apigenin with GABA-benzodiazepine activity from Tanacetum parthenium. Phytother. Res. 2009, 23, 1642–1644. [Google Scholar] [CrossRef] [PubMed]
- Wasowski, C.; Marder, M.; Viola, H.; Medina, J.H.; Paladini, A.C. Isolation and identification of 6-methylapigenin, a competitive ligand for the brain GABAA receptors, from Valeriana wallichii. Planta Med. 2002, 68, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Han, J.Y.; Moon, D.C.; Hong, J.T.; Oh, K.W. (−)-Epigallocatechin-3-O-gallate augments pentobarbital-induced sleeping behaviors through Cl− channel activation. J. Med. Food 2011, 14, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.L.; Chebib, M.; Johnston, G.A. The dietary flavonoids apigenin and (−)-epigallocatechin gallate enhance the positive modulation by diazepam of the activation by GABA of recombinant GABAA receptors. Biochem. Pharmacol. 2004, 68, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Salah, S.M.; Jäger, A.K. Two flavonoids from Artemisia herba-alba Asso with in vitro GABAA-benzodiazepine receptor activity. J. Ethnopharmacol. 2005, 99, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Gazola, A.C.; Costa, G.M.; Castellanos, L.; Ramosb, F.A.; Reginattoa, F.H.; de Limac, T.C.; Schenkel, E.P. Involvement of GABAergic pathway in the sedative activity of apigenin, the main flavonoid from Passiflora quadrangularis pericarp. Rev. Bras. Farm. 2015, 25, 158–163. [Google Scholar] [CrossRef]
- Brown, E.; Hurd, N.S.; McCall, S.; Ceremuga, T.E. Evaluation of the anxiolytic effects of chrysin, a Passiflora incarnata extract, in the laboratory rat. AANA J. 2007, 75, 333–337. [Google Scholar] [PubMed]
- Awad, R.; Arnason, J.T.; Trudeau, V.; Bergeron, C.; Budzinski, J.W.; Foster, B.C.; Merali, Z. Phytochemical and biological analysis of skullcap (Scutellaria lateriflora L.): A medicinal plant with anxiolytic properties. Phytomedicine 2003, 10, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.F.; Hung, W.Y.; Chen, C.F. Anxiolytic-like effects of baicalein and baicalin in the Vogel conflict test in mice. Eur. J. Pharmacol. 2003, 464, 141–146. [Google Scholar] [CrossRef]
- Grundmann, O.; Nakajima, J.; Kamata, K.; Seo, S.; Butterweck, V. Kaempferol from the leaves of Apocynum venetum possesses anxiolytic activities in the elevated plus maze test in mice. Phytomedicine 2009, 16, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.M.; Huen, M.S.; Wang, H.Y.; Zheng, H.; Sigel, E.; Baur, R.; Ren, H.; Li, Z.W.; Wong, J.T.; Xue, H. Anxiolytic effect of wogonin, a benzodiazepine receptor ligand isolated from Scutellaria baicalensis Georgi. Biochem. Pharmacol. 2002, 64, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.M.; Wang, X.H.; Xue, H. Interaction of flavones from the roots of Scutellaria baicalensis with the benzodiazepine site. Planta Med. 2000, 66, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Jamal, H.; Ansari, W.H.; Rizvi, S.J. Evaluation of chalcones—A flavonoid subclass, for, their anxiolytic effects in rats using elevated plus maze and open field behaviour tests. Fundam. Clin. Pharmacol. 2008, 22, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Kim, S.; Jin, Z.; Yang, H.; Han, D.; Baek, N.I.; Jo, J.; Cho, C.W.; Park, J.H.; Shimizu, M.; et al. Isoliquiritigenin, a chalcone compound, is a positive allosteric modulator of GABAA receptors and shows hypnotic effects. Biochem. Biophys. Res. Commun. 2011, 413, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Cho, S.; Lee, C.J. Isoliquiritigenin, a chalcone compound, enhances spontaneous inhibitory postsynaptic response. Exp. Neurobiol. 2014, 23, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Z.; Ikarashi, Y.; Kase, Y. Isoliquiritigenin is a novel NMDA receptor antagonist in kampo medicine yokukansan. Cell. Mol. Neurobiol. 2011, 31, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Park, J.H.; Pae, A.N.; Han, D.; Kim, D.; Cho, N.C.; No, K.T.; Yang, H.; Yoon, M.; Lee, C.; et al. Hypnotic effects and GABAergic mechanism of licorice (Glycyrrhiza glabra) ethanol extract and its major flavonoid constituent glabrol. Bioorg. Med. Chem. 2012, 20, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, J.R.; Chebib, M.; Johnston, G.A. Interactions of flavonoids with ionotropic GABA receptors. Adv. Pharmacol. 2015, 72, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.K.; Saaby, L. Flavonoids and the CNS. Molecules 2011, 16, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H. Structure-dependent membrane interaction of flavonoids associated with their bioactivity. Food Chem. 2010, 120, 1089–1096. [Google Scholar] [CrossRef]
- Margina, D.; Llie, M.; Manda, G.; Neagoe, I.; Mocanu, M.; Ionescu, D.; Gradinaru, D.; Ganea, C. Quercetin and epigallocatechin gallate effects on the cell membranes biophysical properties correlate with their antioxidant potential. Gen. Physiol. Biophys. 2012, 31, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; He, M.; Zang, X.; Zhou, Y.; Qiu, T.; Pan, S.; Xu, X. A structure-activity relationship study of flavonoids as inhibitors of E. coli by membrane interaction effect. Biochim. Biophys. Acta 2013, 1828, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, H.; Mizogami, M. Plant components exhibit pharmacological activities and drug interactions by acting on lipid membranes. Pharmacog. Commun. 2012, 2, 58–71. [Google Scholar] [CrossRef]
- Wang, C.F.; Gerner, P.; Schmidt, B.; Wang, S.Y.; Wang, G.K. Prolonged cutaneous analgesia in rats induced by 3-acetylaconitine as an additive. Drug Dev. Res. 2008, 69, 83–88. [Google Scholar] [CrossRef]
- Xu, H.R.; Ji, G.X.; Wang, Q.; Li, X.N.; Gong, Y.J.; Zhang, Y.; Li, W.; Shen, T. Dose-dependent pharmacokinetics of bulleyaconitine A in rats. Pharmazie 2013, 68, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.Y.; Xu, H.N.; Huang, J.M.; Wang, G.Q.; Shen, T.; Zhang, J.F. A pharmacokinetic study of intramuscular administration of bulleyaconitine A in healthy volunteers. Biol. Pharm. Bull. 2005, 28, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.G.; Wang, Q.H.; Lin, Y.B.; Chen, K.L.; Lin, H.Y.; Lin, W.; Lin, Q.; Liu, F. Clinical study on epidural injected lappaconitine for post-operative analgesia. Chin. J. Integr. Med. 1996, 2, 26–29. [Google Scholar] [CrossRef]
- Chen, M.G.; Wang, Q.H.; Lin, W.; Lin, Y.B.; Chen, K.L.; Liu, F.; Lin, Q.; Lin, H.Y.; Cai, H.D. A clinical study in epidural injection with lappaconitine compound for post-operative analgesia. Chin. J. Integr. Med. 1997, 3, 257–260. [Google Scholar] [CrossRef]
- Friese, J.; Gleitz, J.; Gutser, U.T.; Heubach, J.F.; Matthiesen, T.; Wilffert, B.; Selve, N. Aconitum sp. alkaloids: The modulation of voltage-dependent Na+ channels, toxicity and antinociceptive properties. Eur. J. Pharmacol. 1997, 337, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Movafegh, A.; Alizadeh, R.; Hajimohamadi, F.; Esfehani, F.; Nejatfar, M. Preoperative oral Passiflora incarnata reduces anxiety in ambulatory surgery patients: A double-blind, placebo-controlled study. Anesth. Analg. 2008, 106, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Aslanargun, P.; Cuvas, O.; Dikmen, B.; Aslan, E.; Yuksel, M.U. Passiflora incarnata Linneaus as an anxiolytic before spinal anesthesia. J. Anesth. 2012, 26, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S.; Naghavi, H.R.; Vazirian, M.; Shayeganpour, A.; Rashidi, H.; Khani, M. Passionflower in the treatment of generalized anxiety: A pilot double-blind randomized controlled trial with oxazepam. J. Clin. Pharm. Ther. 2001, 26, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Modabbernia, A.; Akhondzadeh, S. Saffron, passionflower, valerian and sage for mental health. Psychiatr. Clin. N. Am. 2013, 36, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.J.; Li, Q.S.; Soeller, I.; Rockwell, K.; Xie, S.X.; Amsterdam, J.D. Long-term chamomile therapy of generalized anxiety disorder: A study protocol for a randomized, double-blind, placebo-controlled trial. J. Clin. Trials 2014, 4, 188. [Google Scholar] [CrossRef]
- Amsterdam, J.D.; Li, Y.; Soeller, I.; Rockwell, K.; Mao, J.J.; Shults, J. A randomized, double-blind, placebo-controlled trial of oral Matricaria recutita (chamomile) extract therapy for generalized anxiety disorder. J. Clin. Psychopharmacol. 2009, 29, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, P.; Hoffmann, D.L. An investigation into the efficacy of Scutellaria lateriflora in healthy volunteers. Altern. Ther. Health Med. 2003, 9, 74–78. [Google Scholar] [PubMed]
- Gao, J.; Sanchez-Medina, A.; Pendry, B.A.; Hughes, M.J.; Webb, G.P.; Corcoran, O. Validation of a HPLC method for flavonoid biomarkers in skullcap (Scutellaria) and its use to illustrate wide variability in the quality of commercial tinctures. J. Pharm. Pharm. Sci. 2008, 11, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Yun, I.; Cho, E.S.; Jang, H.O.; Kim, U.K.; Choi, C.H.; Chung, I.K.; Kim, I.S.; Wood, W.G. Amphiphilic effects of local anesthetics on rotational mobility in neuronal and model membranes. Biochim. Biophys. Acta 2002, 1564, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kopeć, W.; Telenius, J.; Khandelia, H. Molecular dynamics simulations of the interactions of medicinal plant extracts and drugs with lipid bilayer membranes. FEBS J. 2013, 280, 2785–2805. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, F.; Fabre, G.; Berka, K.; Ossman, T.; Chantemargue, B.; Paloncýová, M.; Marquet, P.; Otyepka, M.; Trouillas, P. In silico pharmacology: Drug membrane partitioning and crossing. Pharmacol. Res. 2016, 111, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.M. Synergy and other interactions in phytomedicines. Phytomedicine 2001, 8, 401–409. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytochemicals | Activities | Experiments | Results | References |
|---|---|---|---|---|
| Terpenoids | ||||
| Linalool | LA | In vitro: Monitoring of compound action potentials in frog sciatic nerves | Exerted local anesthetic effects at 7.5–30 mM as well as 3.5–30 mM lidocaine | [29] |
| Linalool
Linalyl acetate | LA | In vivo: Administration in rabbit conjunctival sac | Depressed conjunctival reflexes by 0.03–2.5 mg/mL administration | [30] |
| Linalool | LA | In vitro: Patch-clamp recording of rat sciatic nerves and rat dorsal root ganglion neurons | Reversely blocked nerve excitability and inhibited voltage-gated Na+ currents at sub-micromolar concentrations | [31] |
| (−)-Menthol
(+)-Menthol | LA | In vitro and in vivo: Evaluation of the activity using rat phrenic nerve hemidiaphragm and by rabbit conjunctival reflex test | Reduced the electrically evoked contractions at 0.1–100 ng/mL and increased the number of stimuli to provoke the conjunctival reflex at 30–300 μg/mL | [33] |
| Menthol | LA | In vitro: Patch-clamp recording of rat dorsal root ganglion neurons | Inhibited tetrodotoxin-resistant Nav1.8 and Nav1.9 and tetrodotoxin-sensitive Na+ channels depending on micromolar concentration, voltage and frequency | [34] |
| Carvacrol
Thymol Citronellol Bornyl acetate Citral | LA | In vitro: Recording of compound action potentials in frog sciatic nerves | Reduced compound action potential peak amplitudes (IC50 = 0.34–7.2 mM) and inhibited nerve conduction by blocking tetrodotoxin-sensitive voltage-gated Na+ channels | [36,37,38] |
| Carvacrol | LA | In vitro: Monitoring of rat sciatic nerve compound action potentials and patch-clamp recording of rat dorsal root ganglion neurons | Reversely blocked the excitability of sciatic nerves (IC50 = 0.5 mM) and reduced voltage-gated Na+ currents (IC50 = 0.37 mM) | [42] |
| Carvacrol | AA LA | In vivo: 50–100 mg/kg (p.o.) administration to mice, followed by acetic acid-induced abdominal constriction, formalin injection and hot plate tests | Inhibited nociception induced by different methods | [43] |
| (−)-Carvone | AA LA | In vivo: 100–200 mg/kg (i.p.) administration to mice | Inhibited acetic acid-induced writhing and formalin-induced hind paw nociception | [44] |
| Estragole | LA | In vitro: Patch-clamp recording of rat dorsal root ganglion neurons | Inhibited total Na+ currents (IC50 = 3.2 mM) and tetrodotoxin-resistant Na+ currents (IC50 = 3.6 mM) | [46] |
| Citral | LA | In vitro: Monitoring of compound action potentials in rat sciatic nerves | Inhibited compound action potentials (IC50 = 0.23 mM) | [48] |
| α-Terpineol
Anethole | LA | In vitro and in vivo: Evaluation of the activity using rat phrenic nerve-hemidiaphragm and by rabbit conjunctival reflex test | Reduced the electrically evoked contractions at 0.01–1 μg/mL and increased the number of stimuli to evoke the conjunctival reflex at 10–100 μg/mL | [51] |
| p-Cymene | AA LA | In vivo: 50–200 mg/kg (i.p.) administration to mice, followed by acetic acid-induced writhing and formalin-induced hind paw licking tests | Showed significant antinociceptive effects in both tests | [52] |
| β-Caryophyllene | LA | In vitro and in vivo: Evaluation of the activity using rat phrenic nerve-hemidiaphragm and by rabbit conjunctival reflex test | Reduced the electrically evoked contractions at 0.1 ng/mL to 1.0 μg/mL and increased the number of stimuli to evoke the conjunctival reflex at 10 μg/mL to 1.0 mg/mL | [54] |
| Alkaloids | ||||
| Lappaconitine other neurotoxins | LA | In vivo: Rabbit corneal reflex test to drop test solutions (0.01–1%) into the conjunctival sac and cat neck trunk anesthesia to inject test solutions (0.1 mL of 0.1–0.5%, i.c. and s.c.) | Showed greater potency and longer duration of anesthesia than lidocaine and procaine | [61] |
| Aconitine 3-Acetylaconitine | AA | In vivo: Intravenous administration to mice, followed by formaldehyde injection (s.c.) to induce hyperalgesia | Showed antinociceptive effects in the early phase (ED50 = 0.027–0.028 mg/kg) and the late phase (ED50 = 0.077–0.097 mg/kg) | [63] |
| Bulleyaconitine A | LA | In vivo: Injection of 0.2 mL test solution into rat sciatic notch | Blocked sensory and motor functions of the sciatic nerves at 0.375 mM Co-injection with 2% lidocaine or epinephrine (1:100,000) prolonged the nerve-blocking duration | [65] |
| Bulleyaconitine A | LA AA | In vitro and in vivo: Recording of Na+ currents of human embryonic kidney cells expressing Nav isoforms and measurement of the cutaneous trunci muscle reflex after injection (s.c.) of 0.6 mL test solution | Blocked Nav1.7 and Nav1.8 Na+ currents at 10 μM and induced the complete nociceptive blockade lasting for ~3 h at 0.125 mM Co-injection with 0.5% lidocaine/epinephrine (1:200,000) increased the duration of analgesia | [66] |
| 3-Acetylaconitine | AA LA | In vivo: 50–125 μM (s.c.) co-injection with 0.5% lidocaine and epinephrine (1:200,000) to rats | Produced the complete analgesia lasting for 3–12 h | [138] |
| Lappaconitine | AA LA | Pre-clinical: Patients received epidural injections of 0–12 mg lappaconitine | Produced satisfactory analgesia depending on dosages Suggested the clinical utility for postoperative analgesia | [141] |
| Lappaconitine | AA LA | Pre-clinical: Patients received epidural injections of test solutions consisting of 12 mg lappaconitine, 12 mg lappaconitine plus 22.5 mg bupivacaine or 22.5 mg bupivacaine | Epidural co-injection of lappaconitine with bupivacaine induced analgesia with greater potency, earlier initiation and longer maintenance than lappaconitine alone and bupivacaine alone | [142] |
| Flavonoids | ||||
| (−)-Epigallo-catechin-3-gallate | LA | In vitro: Measurement of Na+ currents in rat dorsal root ganglion neurons | Inhibited tetrodotoxin-sensitive and tetrodotoxin-resistant Na+ currents | [71] |
| Genistein Daidzein | LA | In vitro: Measurement of Na+ currents in cultured rat brain neurons | Blocked voltage-sensitive Na+ channels (IC50 = 60 μM for genistein and 195 μM for daidzein) | [72] |
| Stilbenoid | ||||
| Resveratrol | LA | In vitro: Measurement of Na+ currents in rat dorsal root ganglion neurons | Suppressed tetrodotoxin-sensitive and tetrodotoxin-resistant Na+ channels | [75] |
| Terpenoids | ||||
| Aristolen-1(10)-en-9-ol | S H GA-like | In vivo: Spontaneous vapor administration to mice | Inhibited the locomotion of caffeine-treated mice at 300 μg/cage and prolonged the time of pentobarbital-induced sleep The effects were inhibited by flumazenil | [89] |
| Isopulegol
Pinocarveol Verbenol Myrtenol | GA-like | In vitro: Determination of the effects on GABAA receptor α1β2 or α1β2γ2 subunits expressed in Xenopus laevis oocytes or human embryonic kidney cells | Modulated GABAA receptor functions at 3–300 μM independently of the γ2 subunit | [92] |
| Valerenic acid | GA-like | In vitro: Determination of the effect on GABAA receptors in neonatal rat brainstem preparations | Inhibited muscimol-sensitive neurons (IC50 = 23 μM) The effect was antagonized by bicuculline | [93] |
| Valerenic acid | GA-like A | In vitro and in vivo: Radioligand binding assay with crude rat brain membranes and 1–6 mg/kg (i.p.) and 10 mg/kg (p.o.) administration to mice | Interacted allosterically with benzodiazepine and GABA binding site of GABAA receptors Showed anxiolytic effects on mice | [96] |
| Linalool | S H GA-like | In vivo: Placed mice in a chamber of an atomosphere saturated 1% or 3% vapor | Produced sedation and increased the time of pentobarbital-induced sleep | [99] |
| Menthol | GA-like | In vitro: Determination of the effect on recombinant human GABAA receptors expressed in Xenopus laevis oocytes | Enhanced sub-maximal GABA currents at 3–300 μM | [102] |
| Menthol | GA-like | In vitro: Whole-cell voltage clamp recording of rat midbrain periaqueductal grey neurons | Prolonged at 150–750 μM the duration of spontaneous inhibitory postsynaptic potentials mediated by GABAA receptors | [103] |
| Thymol
Carvacrol Eugenol | GA-like | In vitro: Determination of the effects on primary cultures of mouse cortical neurons at micromolar concentrations | Enhanced [3H]flunitrazepam binding to GABAA receptors and increased 2 μM GABA-evoked Cl− influx | [15] |
| Thymol | GA-like | In vitro: Determination of the effects on human GABAA receptor subunit combinations expressed in Xenopus laevis oocytes | Potentiated EC20 GABA response at 1–100 μM | [104] |
| Thymol | GA-like | In vitro: Determination of the positive allosteric modulatory effects on GABAA receptors in primary cultures of mouse cortical neurons | Enhanced 5 μM GABA-induced Cl− influx (EC50 = 12 μM) and directly enhanced Cl− influx (EC50 = 135 μM) | [14] |
| Alkaloids | ||||
| Harman | A S | In vivo: 2.5–10 mg/kg (i.p.) administration to rats | Decreased the time of immobility in a forced swim test and increased the time spent in open arms in an elevated plus maze | [110] |
| Harman
Norharman Harmine | S GA-like | In vivo: 2.5–15 mg/kg (i.p.) administration to mice | Decreased the time of immobility The effects were inhibited by flumazenil | [111] |
| Flavonoids | ||||
| (−)-Epigallo-catechin-3-gallate | H | In vivo: 5–20 mg/kg (p.o.) administration to mice | Prolonged the time of pentobarbital-induced sleep | [117] |
| Apigenin | S GA-like | In vivo: 0.6 mg/kg (p.o.) administration to mice | Induced sedation The effect was blocked by flumazenil | [120] |
| Chrysin | A GA-like | In vivo: 2 mg/kg (i.p.) administration to rats | Induced anxiolysis The effect was affected by flumazenil | [121] |
| Baicalein Baicalin | A GA-like | In vivo: 10–20 mg/kg (i.p.) administration to mice | Induced anxiolysis The effects were antagonized by flumazenil | [123] |
| Kaempferol | A GA-like | In vivo: 0.02–1.0 mg/kg (p.o.) administration to mice | Induced anxiolysis The effect was antagonized by flumazenil | [124] |
| Wogonin | GA-like A | In vitro and in vivo: Radioreceptor binding assay with rat forebrain synaptosomal membranes, electrophysiological experiment with rat dorsal root ganglion neurons and 7.5–30 mg/kg (p.o.) administration to mice | Showed the affinity for the benzodiazepine site of GABAA receptors (Ki = 0.92 μM), enhanced GABA-activated currents and induced anxiolysis | [125] |
| Isoliquiritigenin | GA-like H | In vitro and in vivo: Radioreceptor binding assay with rat cerebral cortex membranes, electrical measurement of rat dorsal raphe neurons and 25–50 mg/kg (p.o.) administration to mice | Showed the affinity for GABAA-benzodiazepine receptors (Ki = 0.45 μM), increased GABA-evoked currents and potentiated pentobarbital-induced sleep | [128] |
| Isoliquiritigenin | GA-like | In vitro: Measurement of the GABAergic synaptic renponses by the whole-cell patch clamp technique | Prolonged at 1 μM the decay of spontaneous inhibitory postsynaptic currents mediated by GABAA receptors The effect was inhibited by flumazenil | [129] |
| Glabrol | H GA-like | In vivo: 25–50 mg/kg (p.o.) administration to mice | Increased the sleep duration and decreased the sleep latency in mice treated with pentobarbital The effects were blocked by flumazenil | [131] |
| Apigenin | A | Pre-clinical: Randomized, double-blind, placebo-controlled trial for outpatients with 220 mg chamomile extracts standardized to a content of 1.2% apigenin | Showed clinically meaningful difference from controls in anxiety rating scores | [149] |
| Wogonin
Baicalein Baicalin Scutellarein | A | Pre-clinical: Double-blind and placebo-controlled study for human subjects with 100–200 mg skullcap extracts containing flavones | Exhibited anxiolytic effects | [150,151] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsuchiya, H. Anesthetic Agents of Plant Origin: A Review of Phytochemicals with Anesthetic Activity. Molecules 2017, 22, 1369. https://doi.org/10.3390/molecules22081369
Tsuchiya H. Anesthetic Agents of Plant Origin: A Review of Phytochemicals with Anesthetic Activity. Molecules. 2017; 22(8):1369. https://doi.org/10.3390/molecules22081369
Chicago/Turabian StyleTsuchiya, Hironori. 2017. "Anesthetic Agents of Plant Origin: A Review of Phytochemicals with Anesthetic Activity" Molecules 22, no. 8: 1369. https://doi.org/10.3390/molecules22081369
APA StyleTsuchiya, H. (2017). Anesthetic Agents of Plant Origin: A Review of Phytochemicals with Anesthetic Activity. Molecules, 22(8), 1369. https://doi.org/10.3390/molecules22081369