Crystallization of Covalent Organic Frameworks for Gas Storage Applications


Abstract
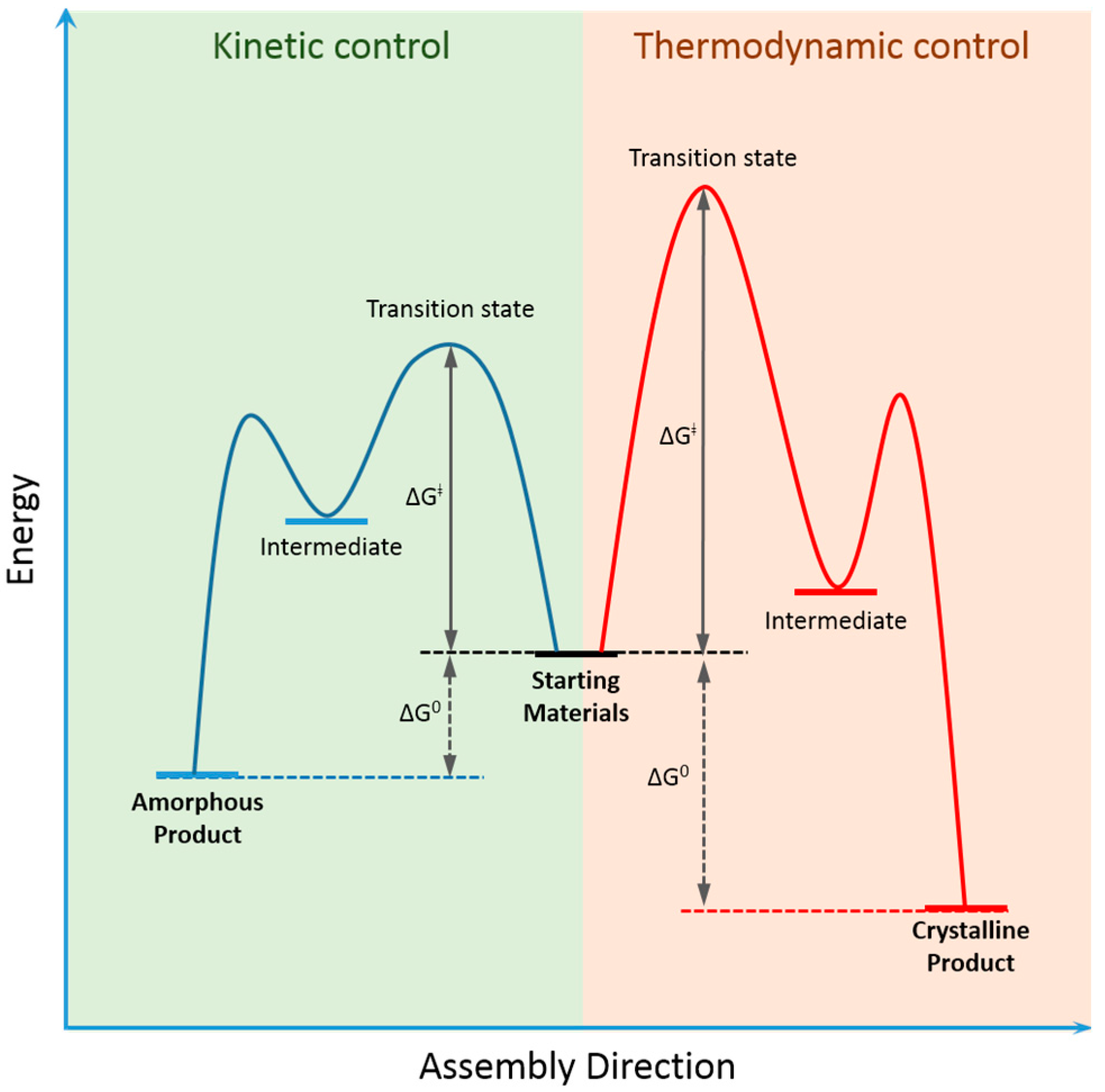
:1. Introduction
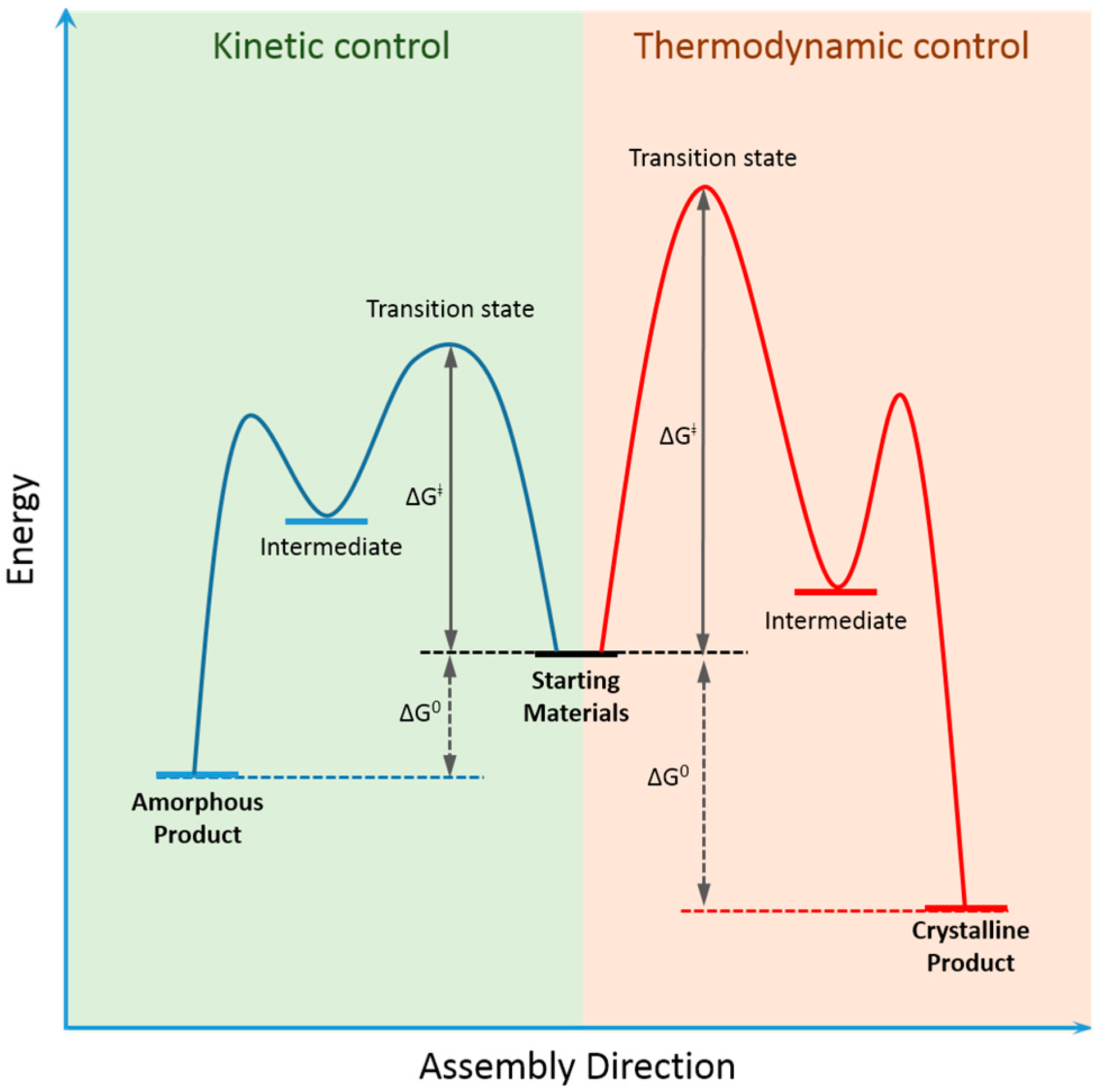
2. Dynamic Covalent Chemistry of COFs
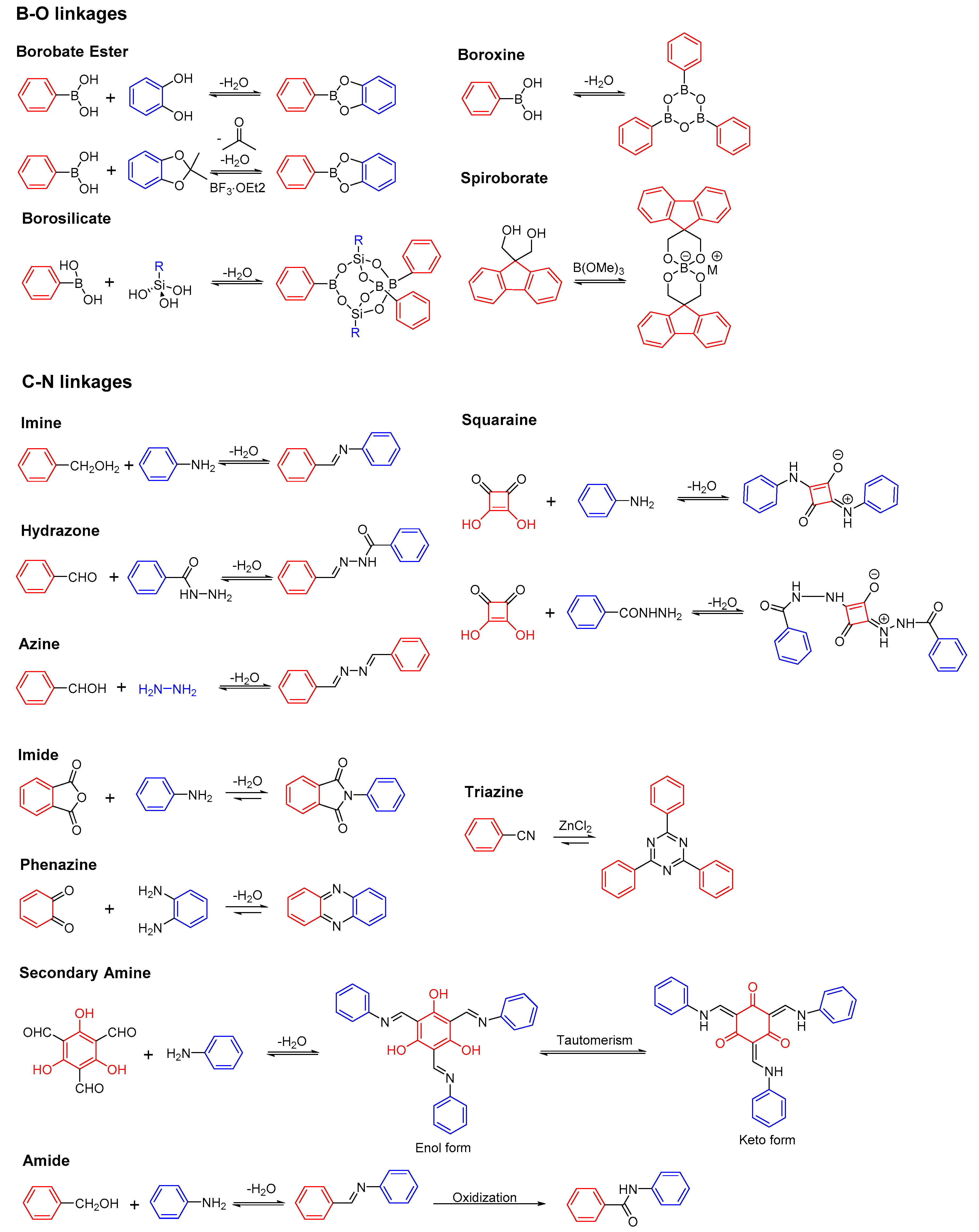
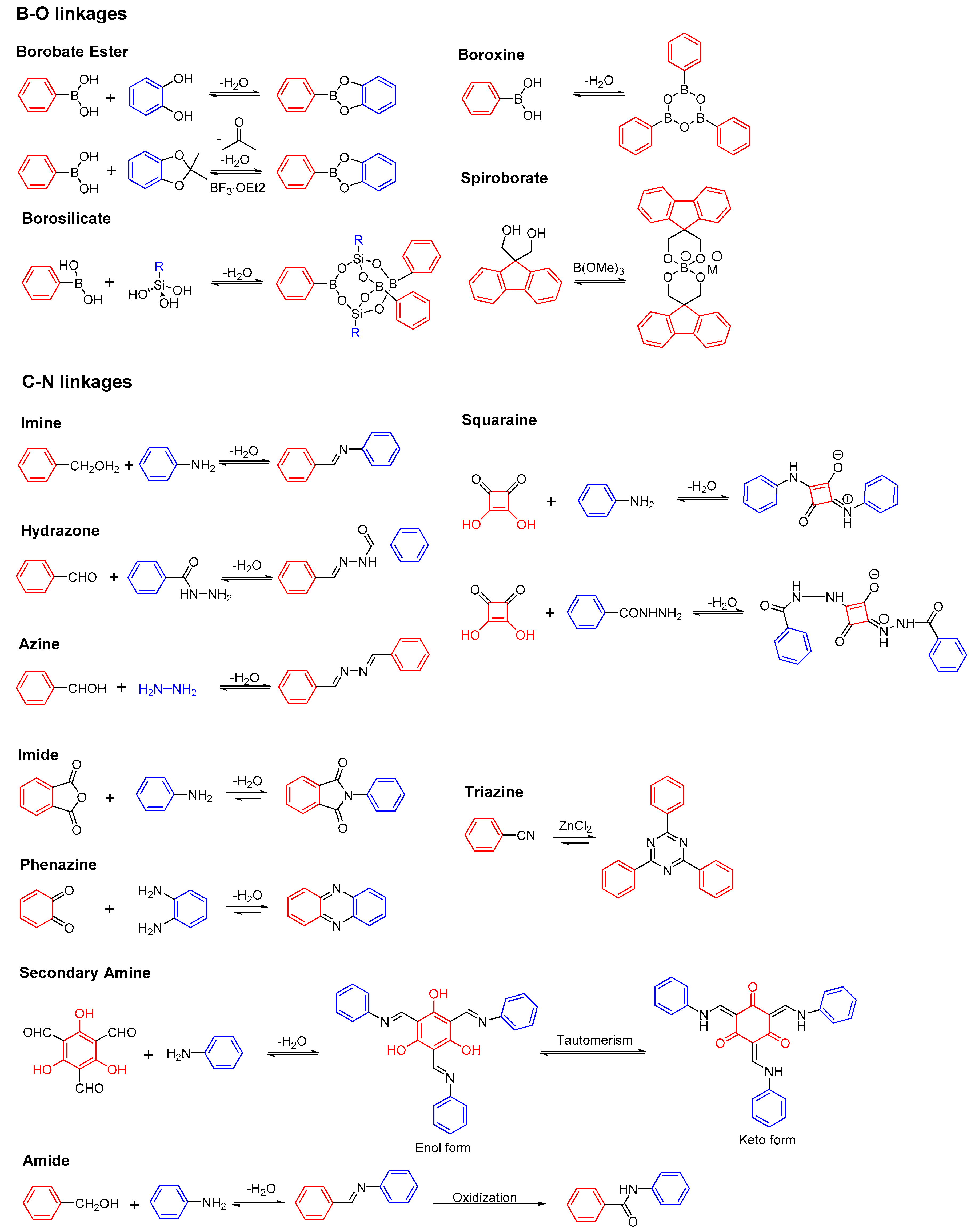
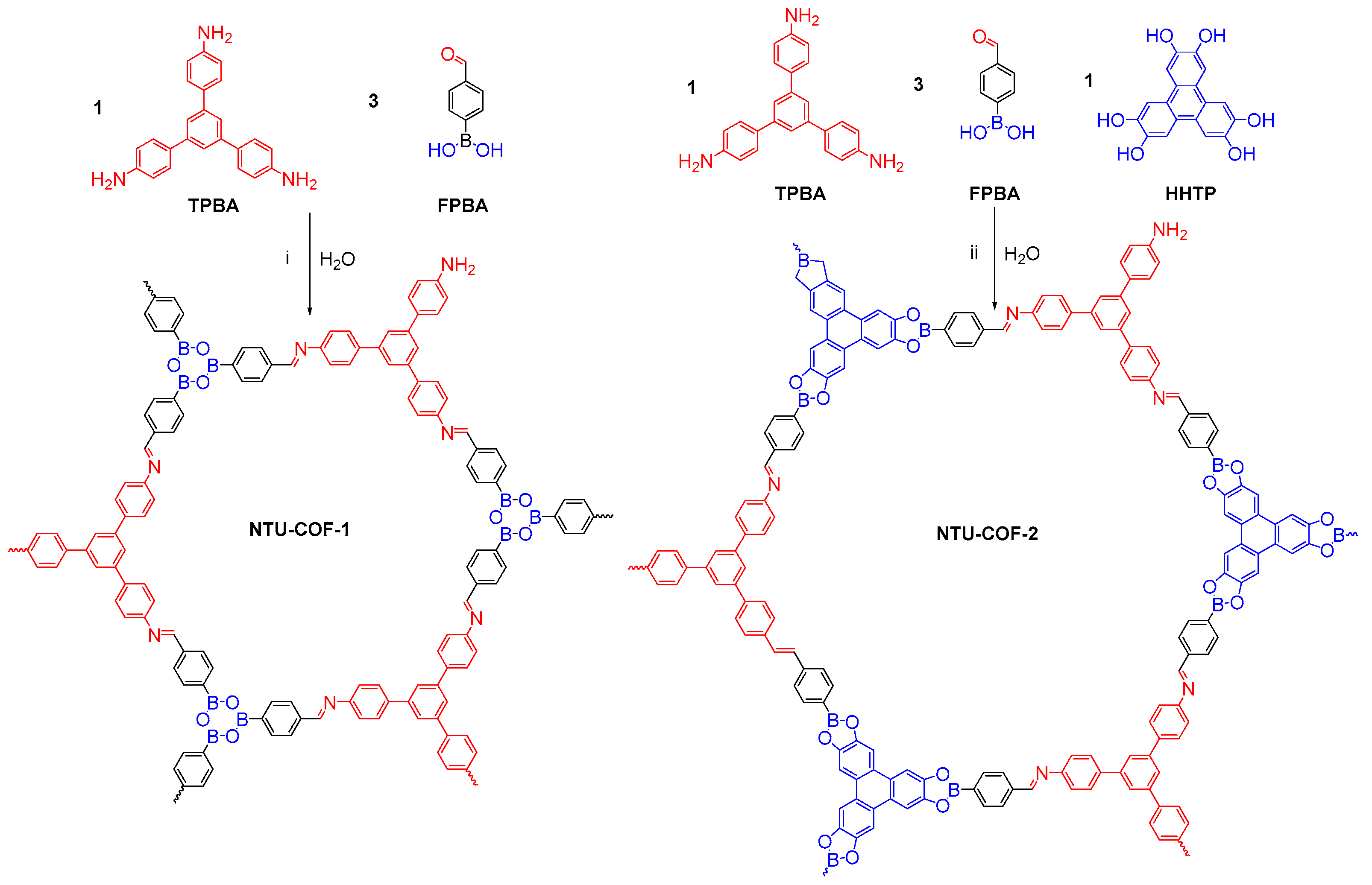
2.1. B-O Linkages
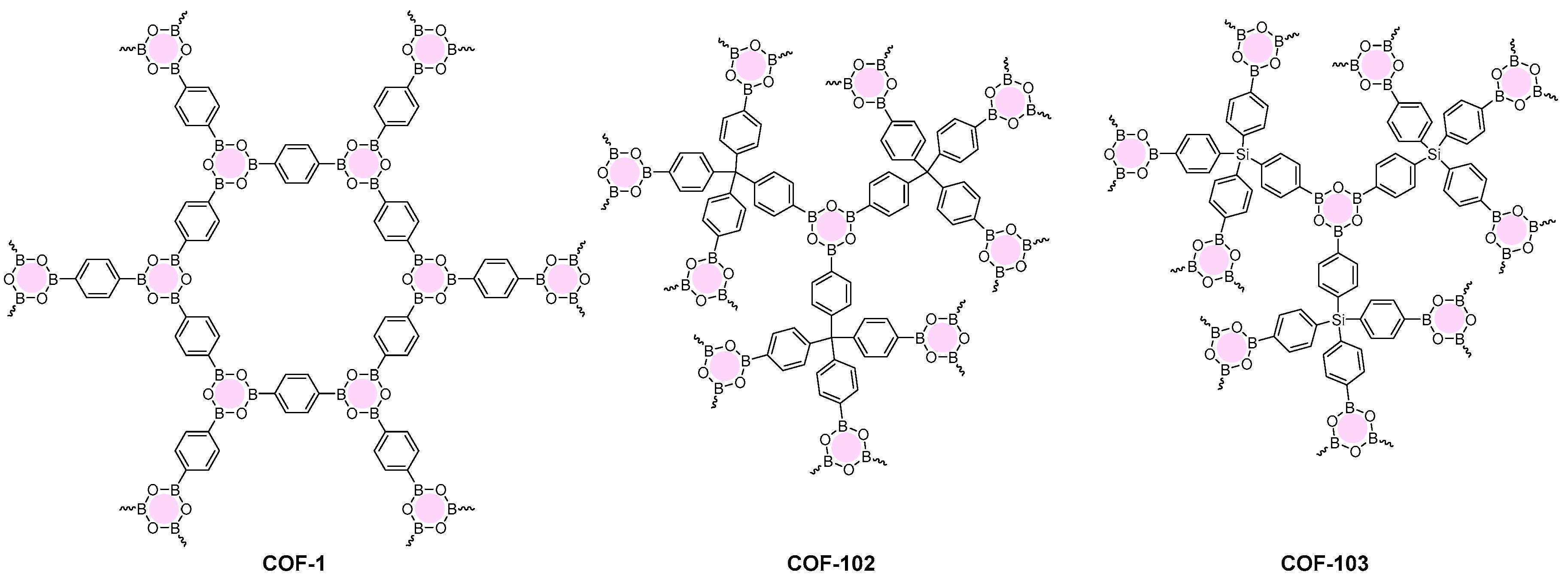
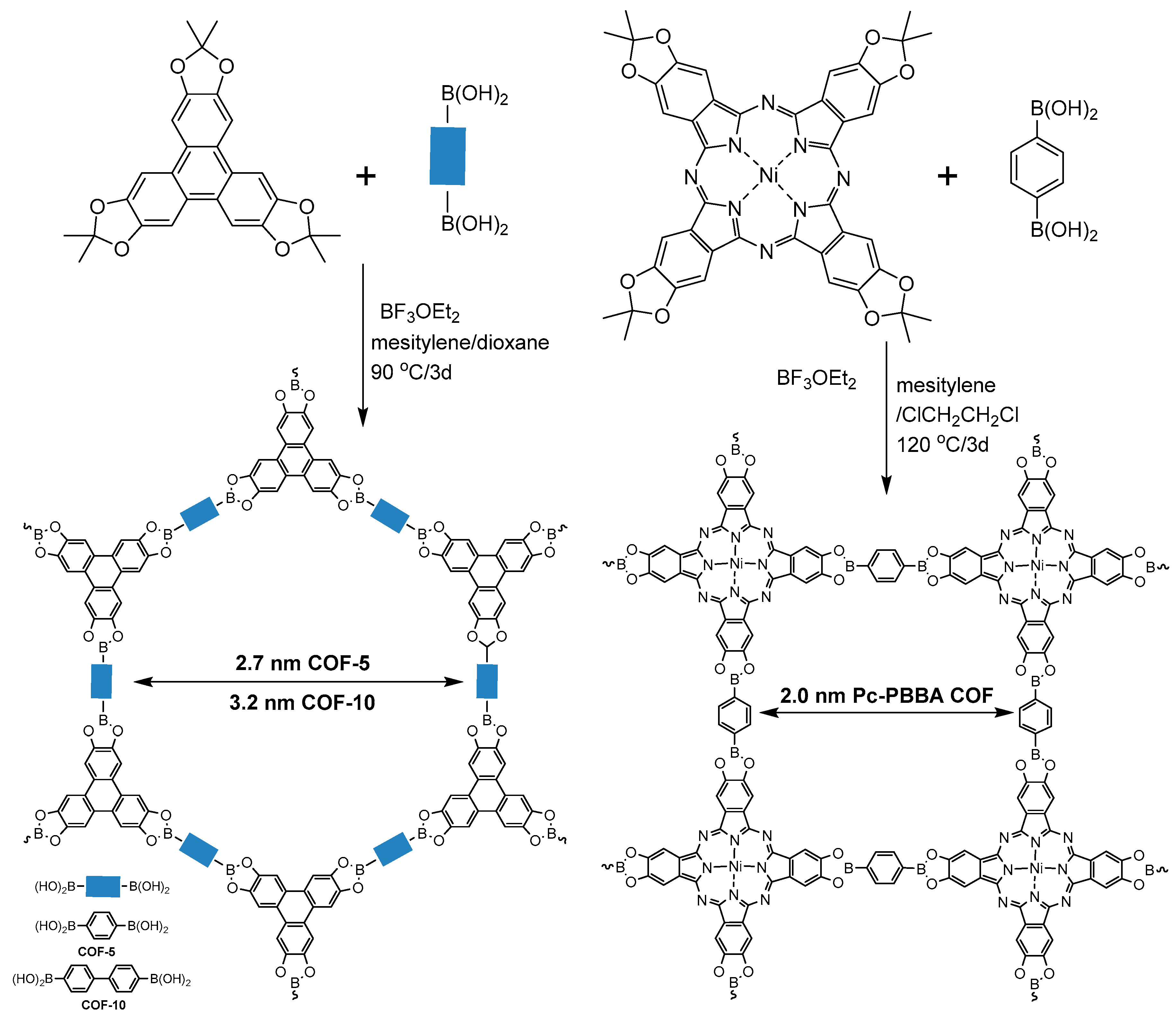
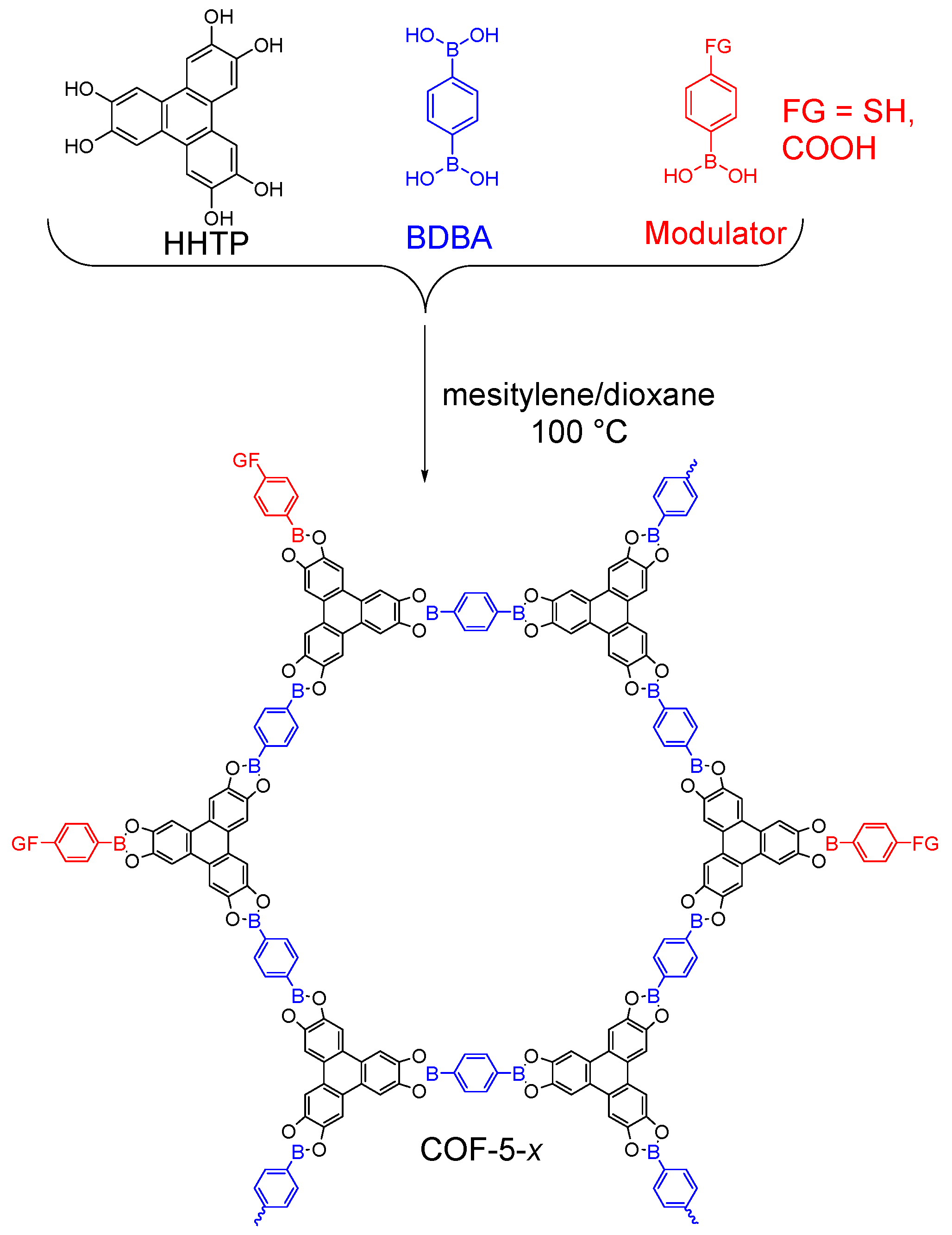
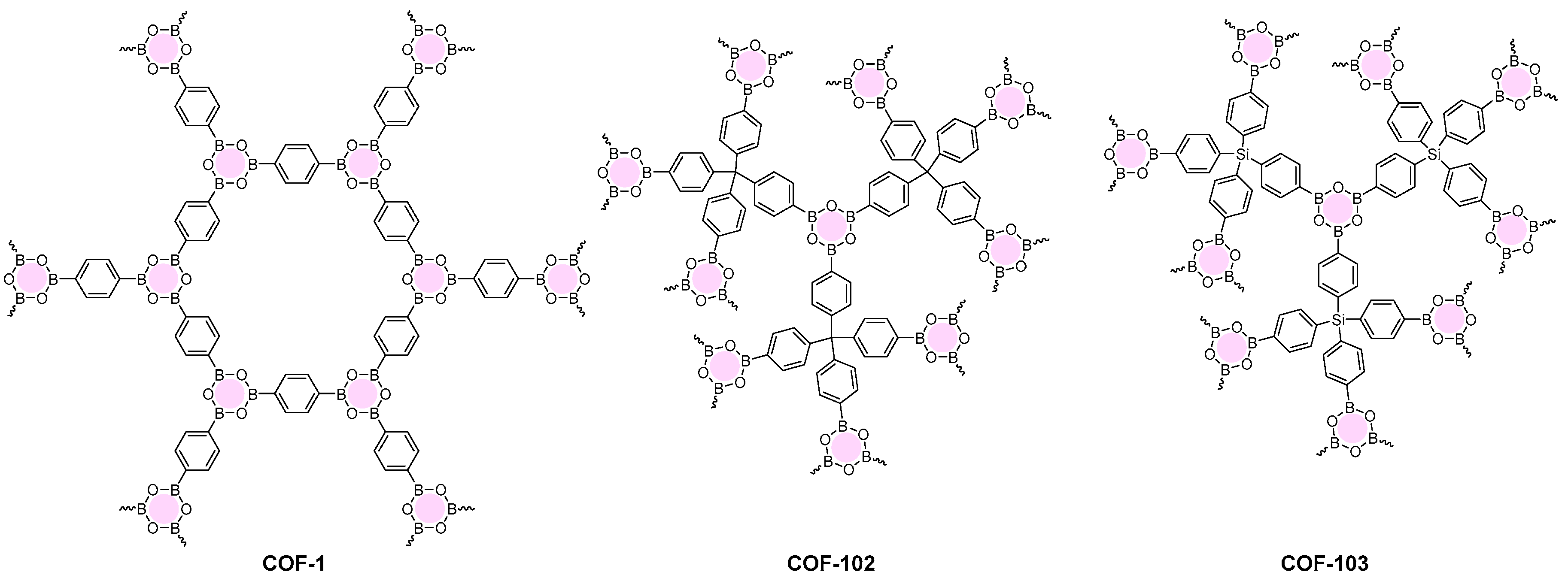
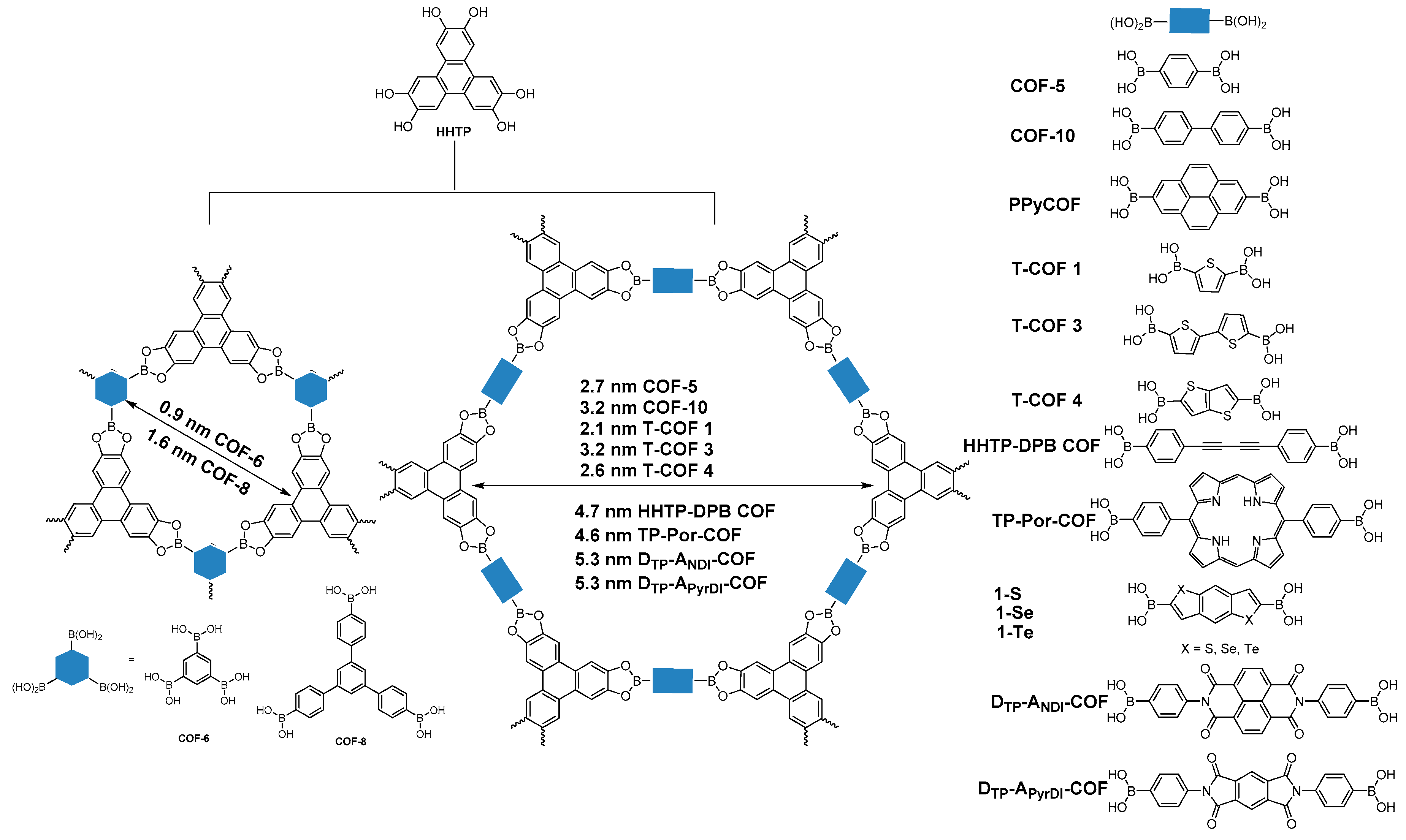
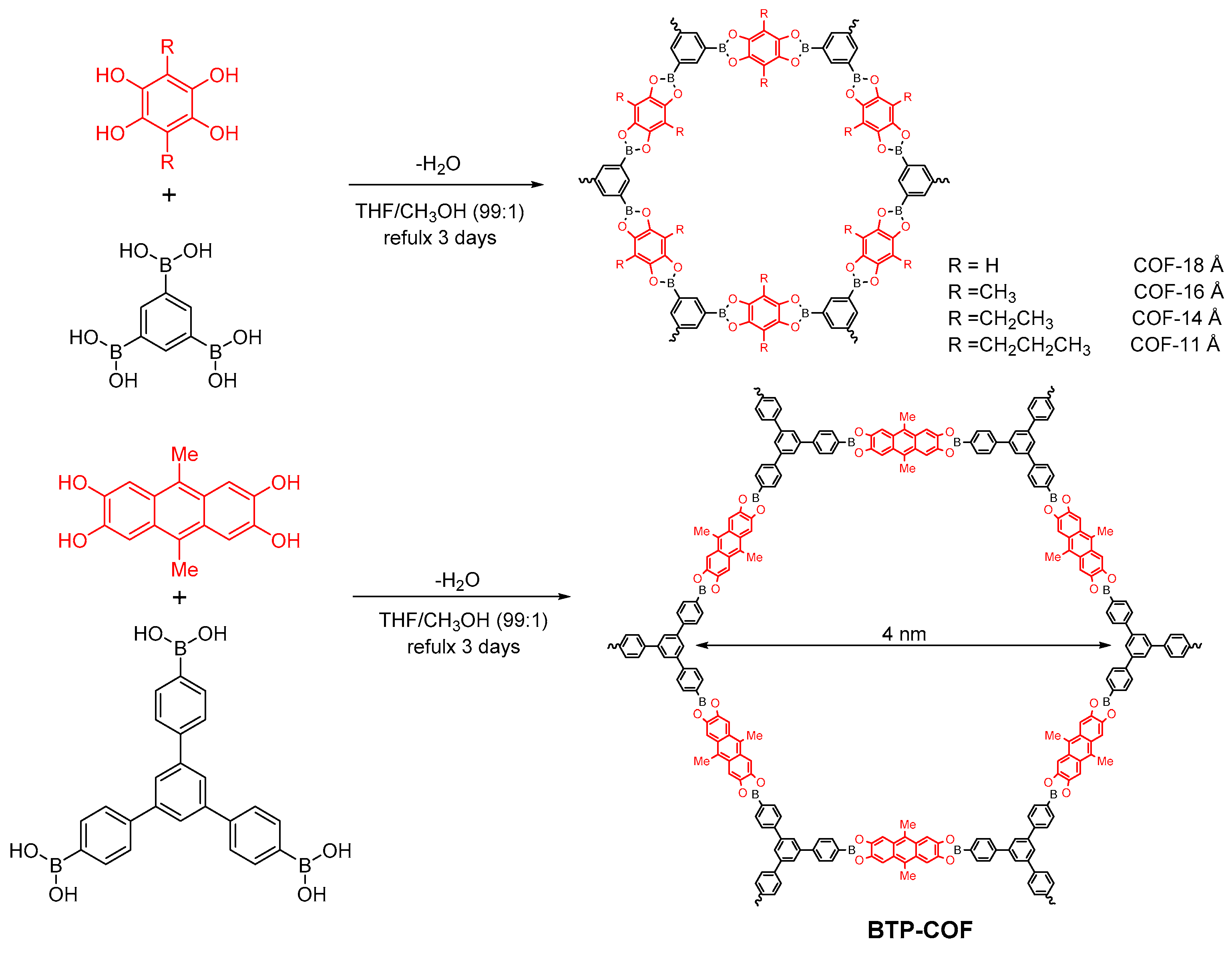
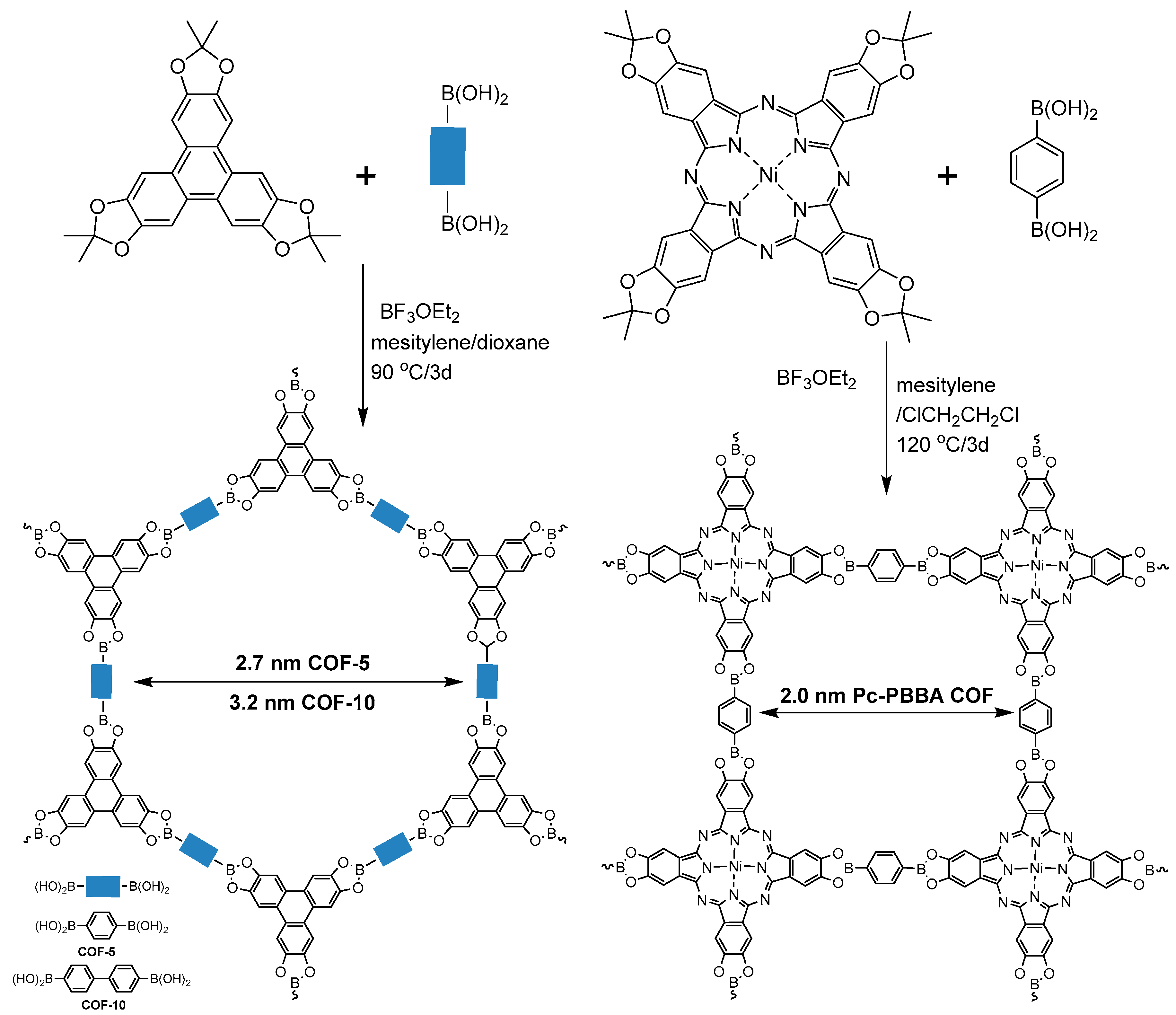
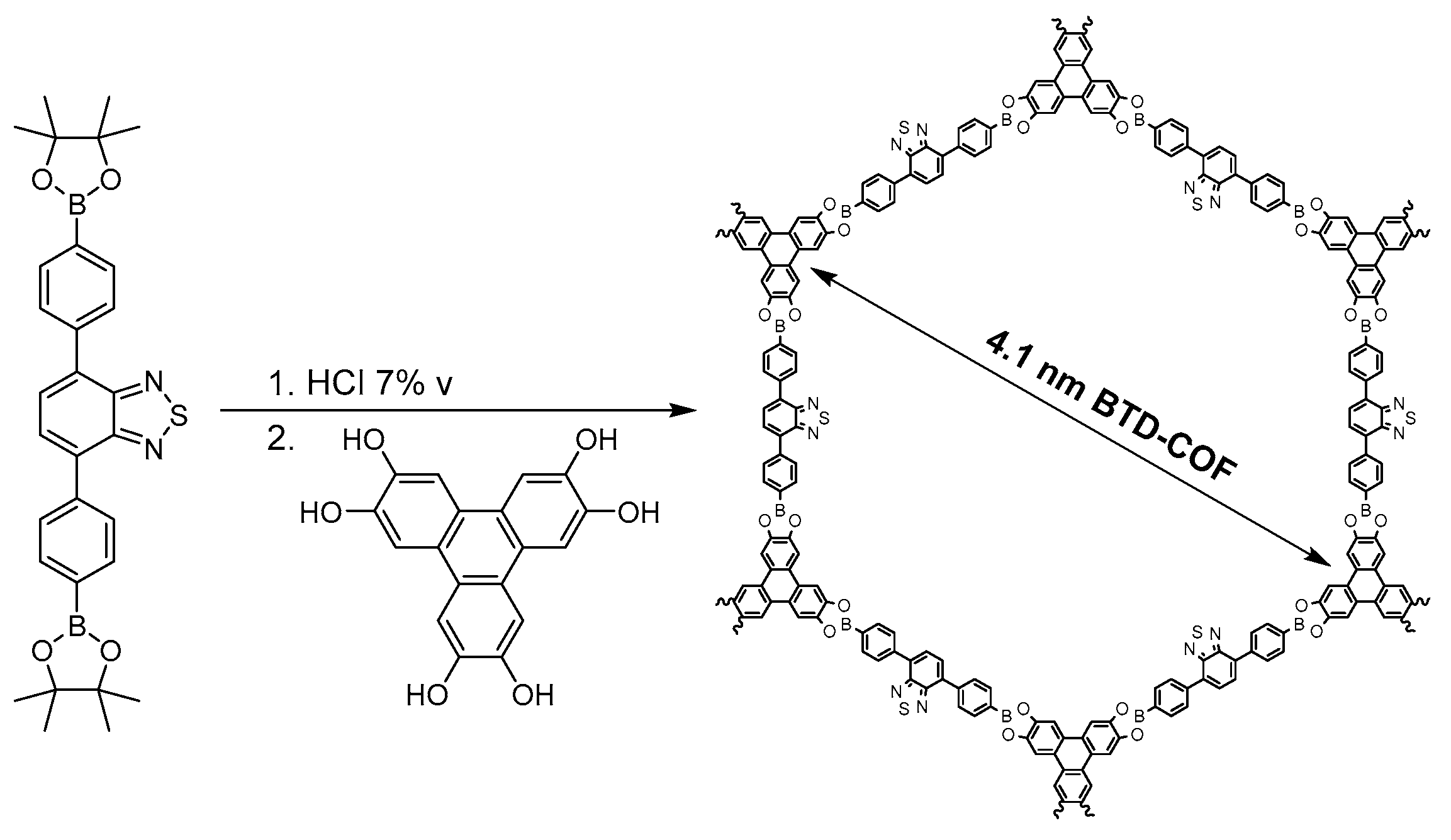
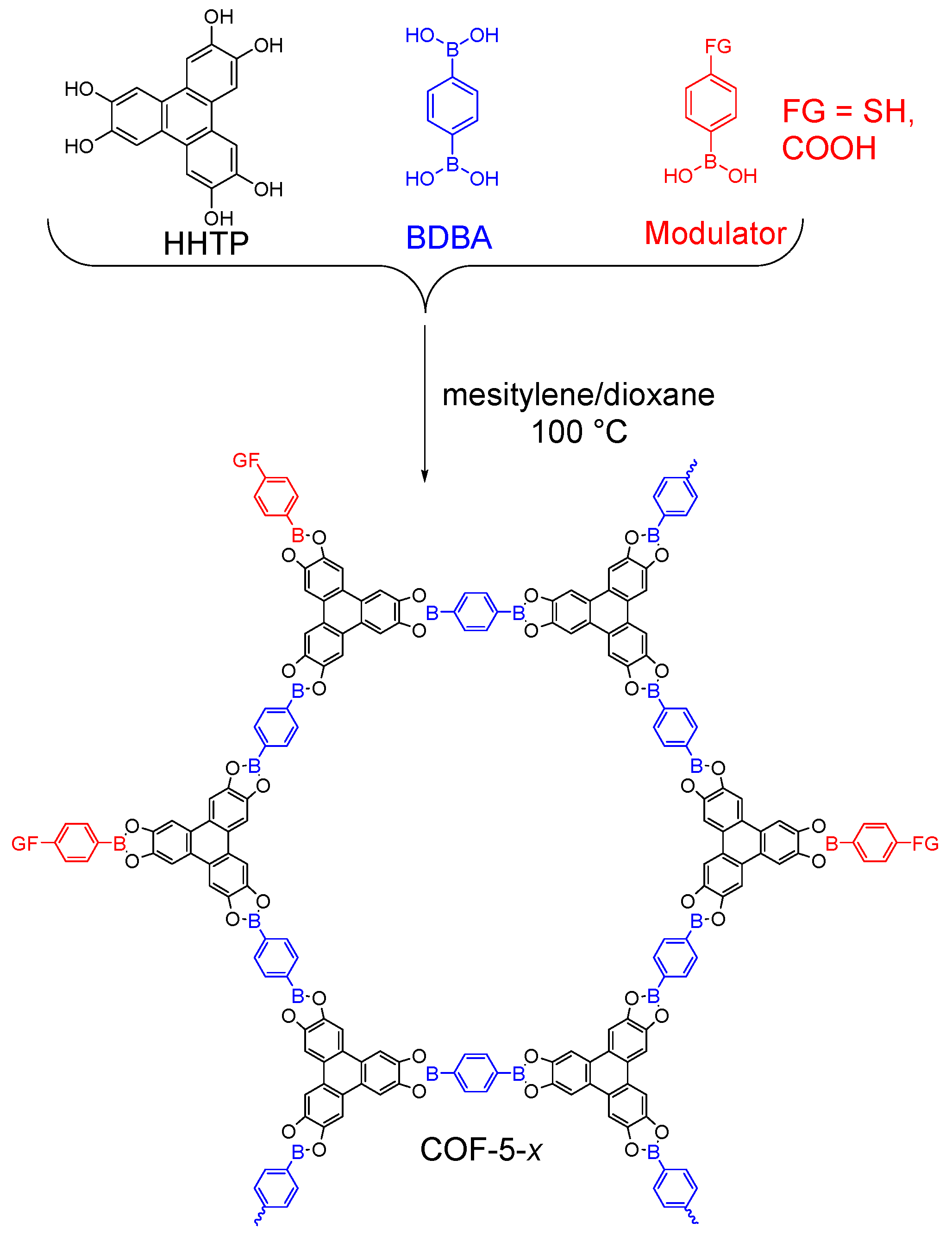
2.1.1. Boroxine-Linked COFs
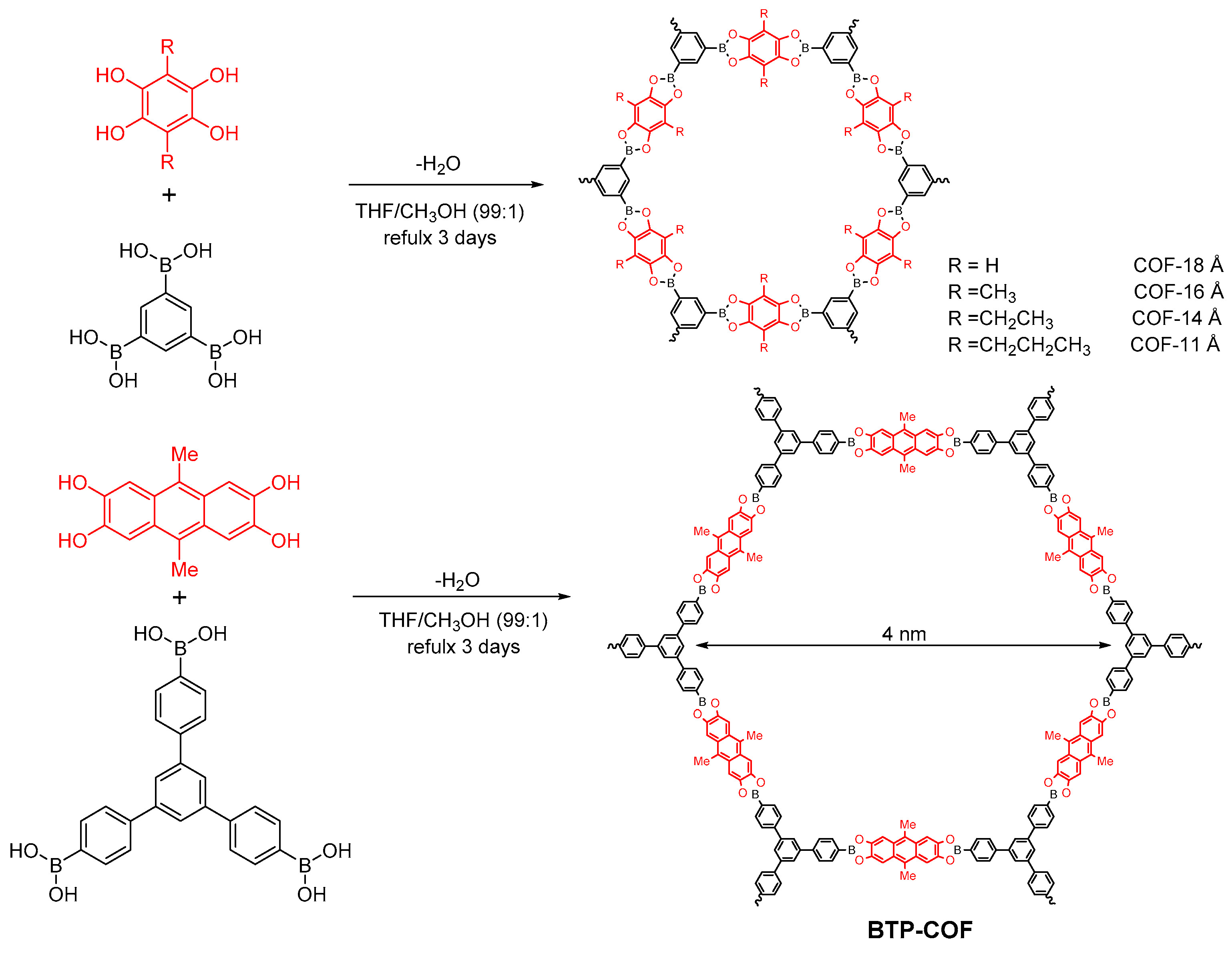
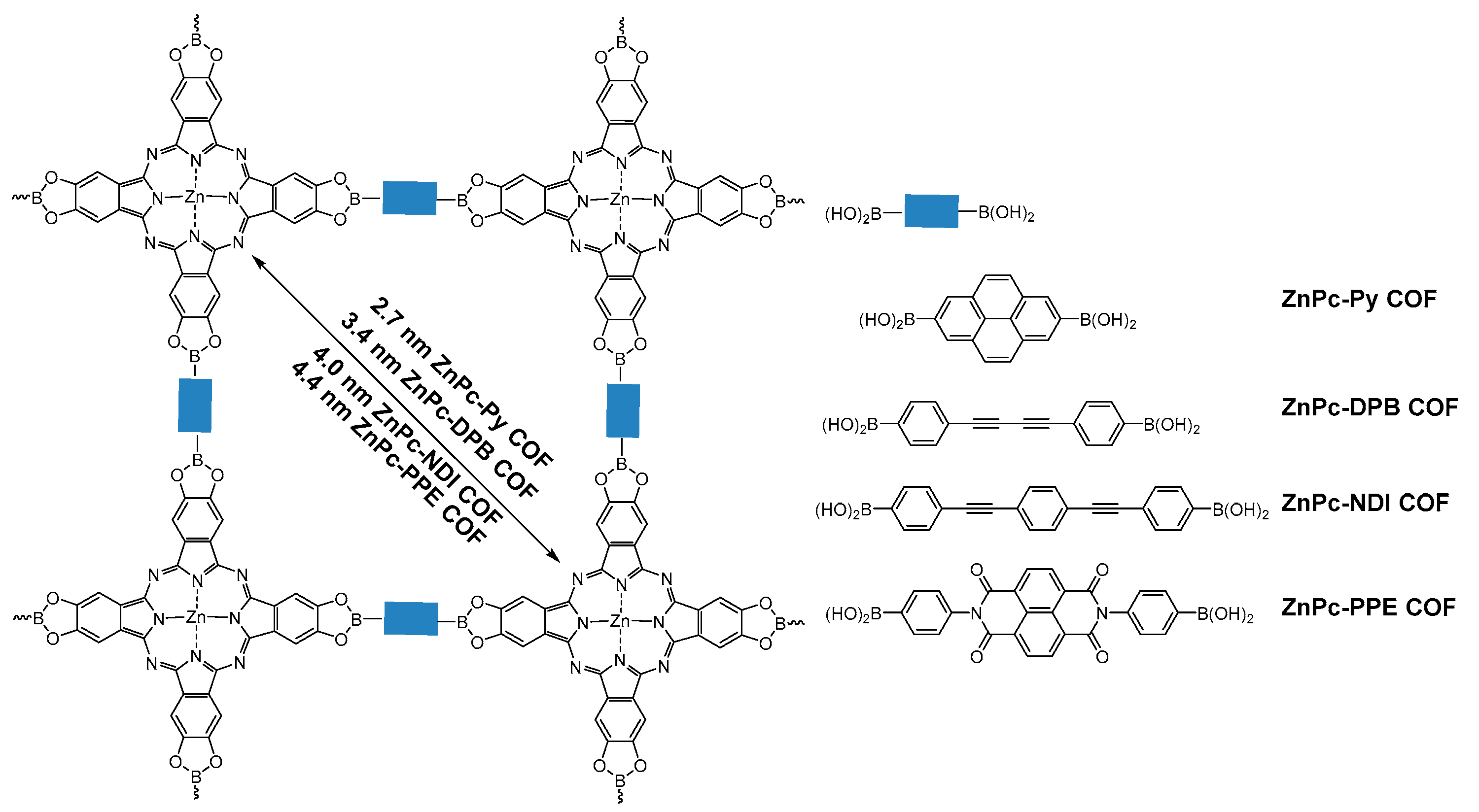
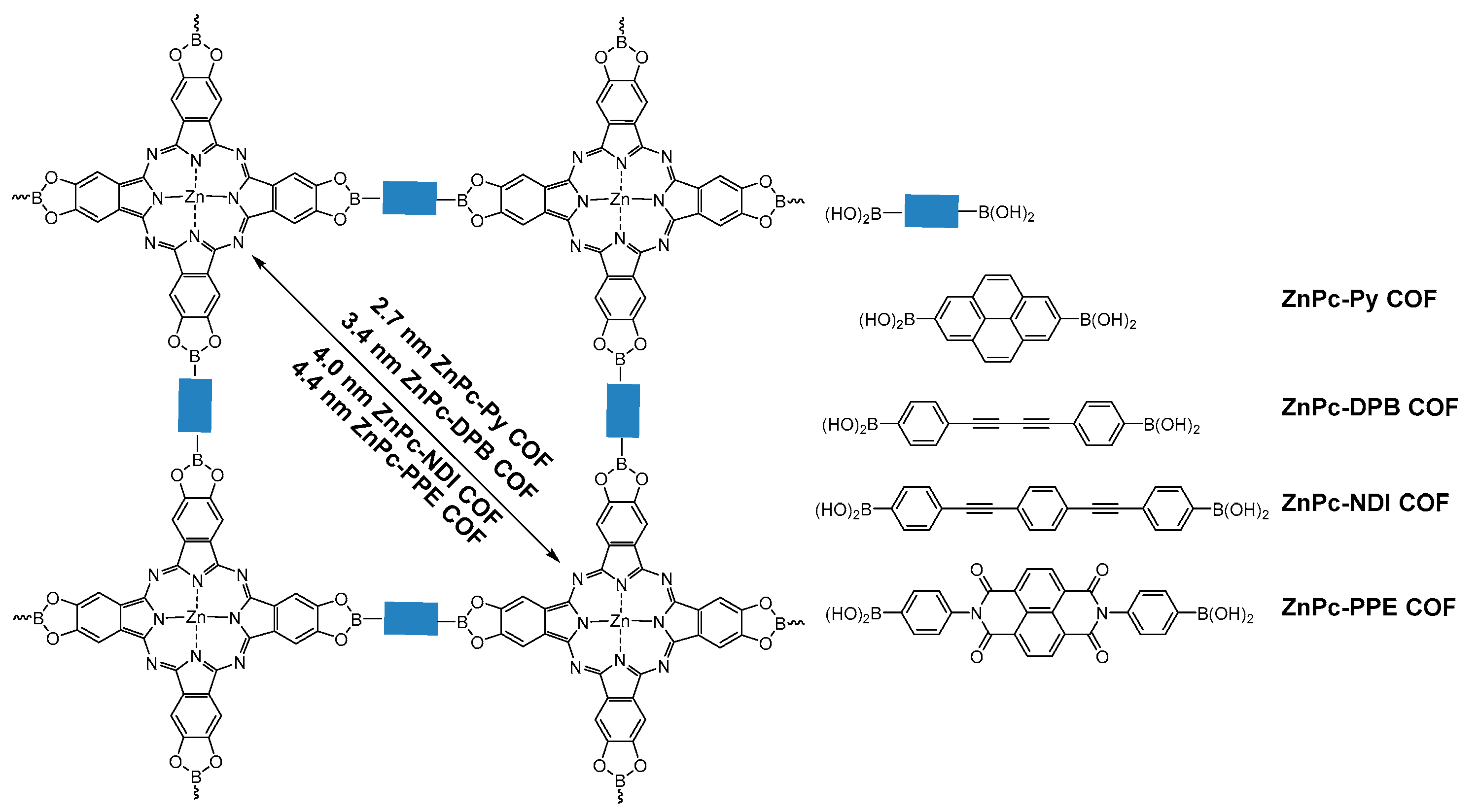
2.1.2. Dioxaborole-Linked COFs
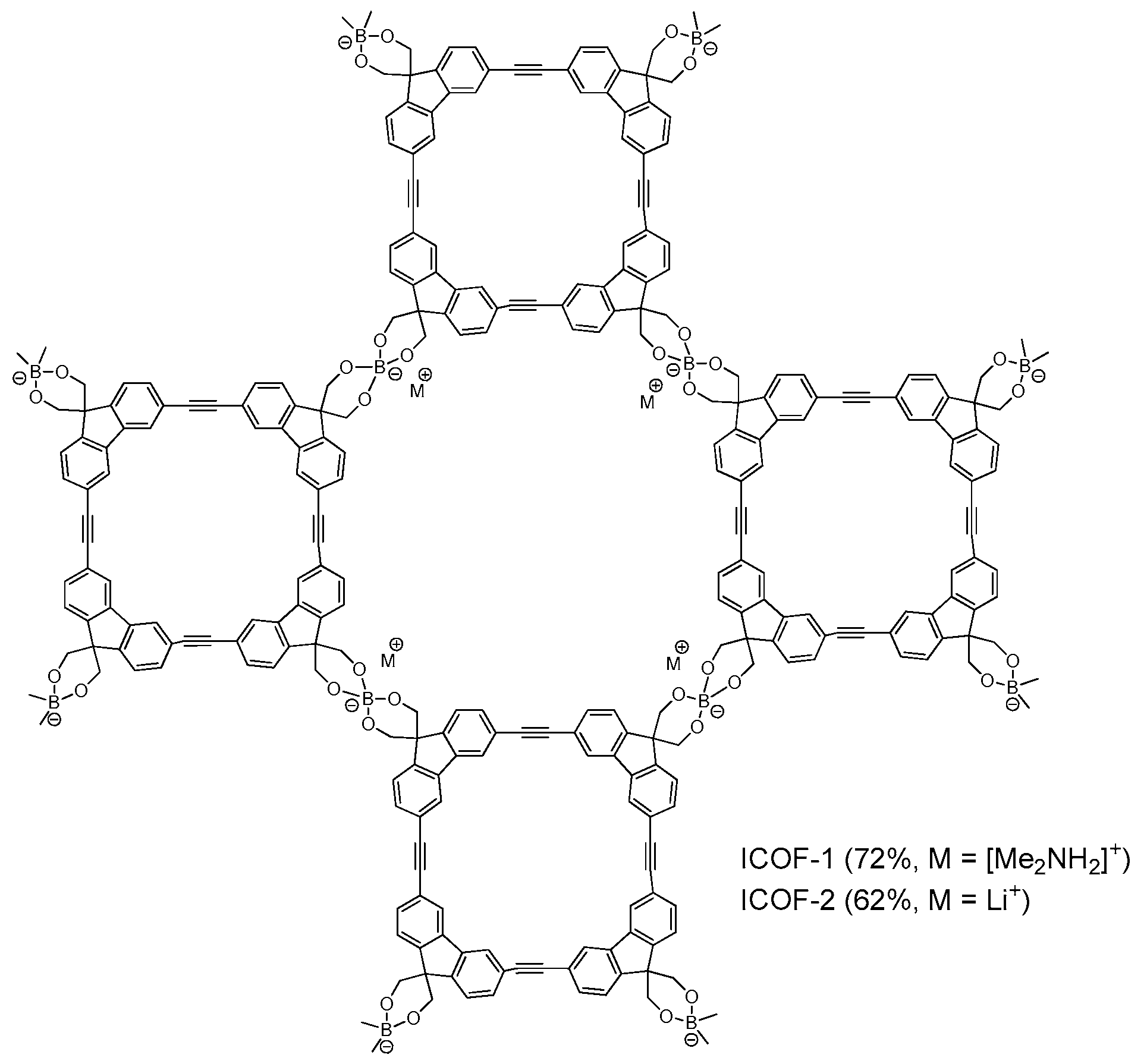
2.1.3. Spiroborate-Linked COFs
2.2. C-N Linkages
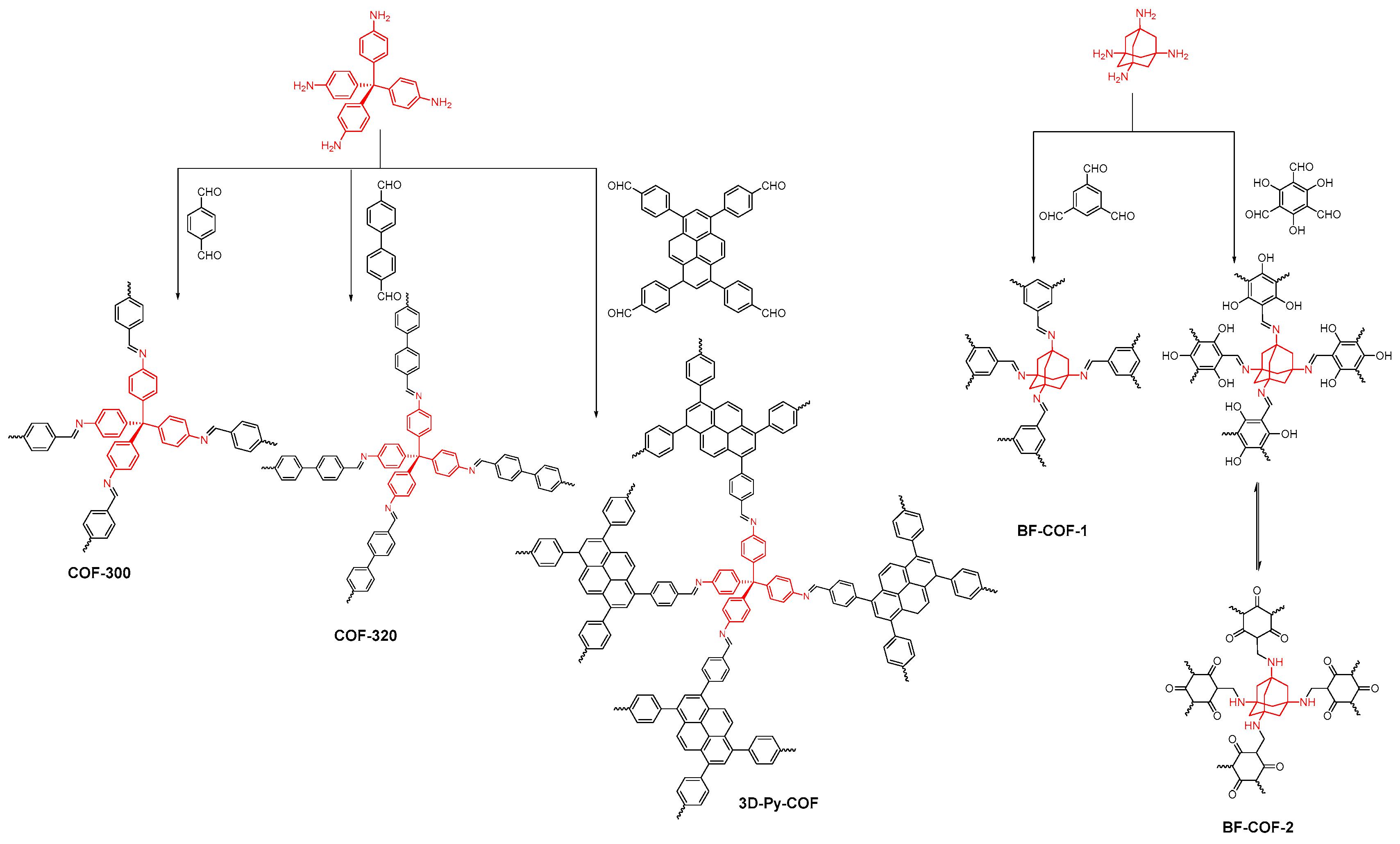
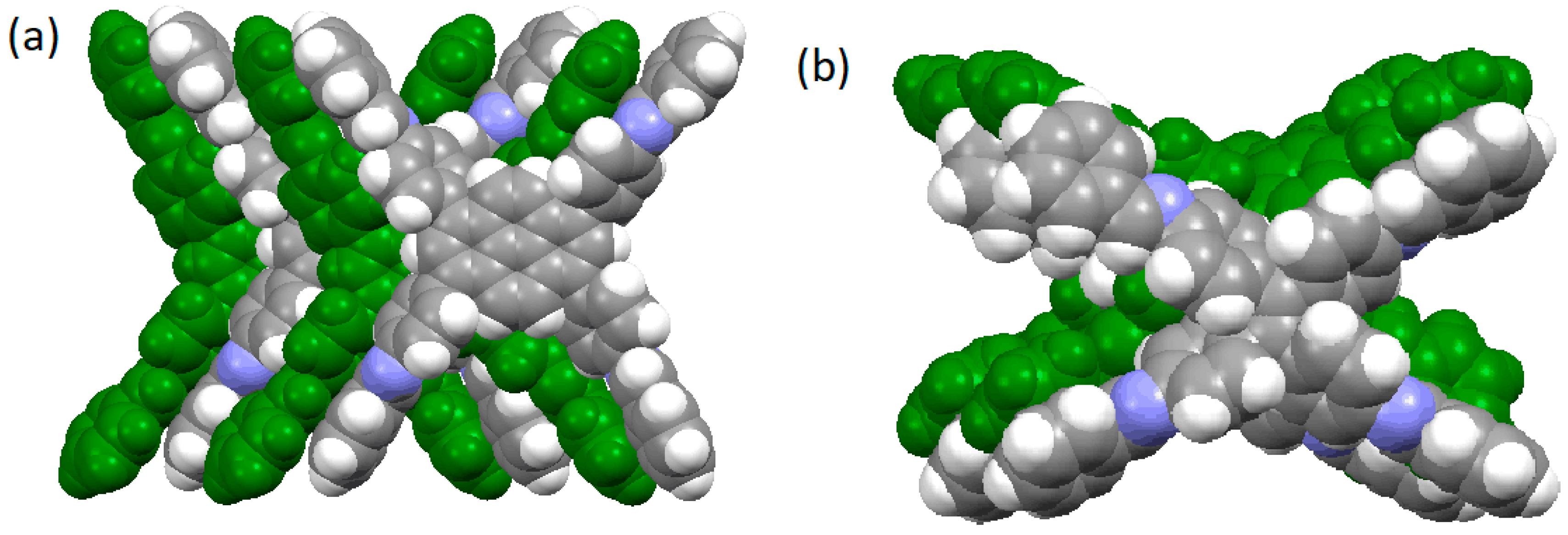
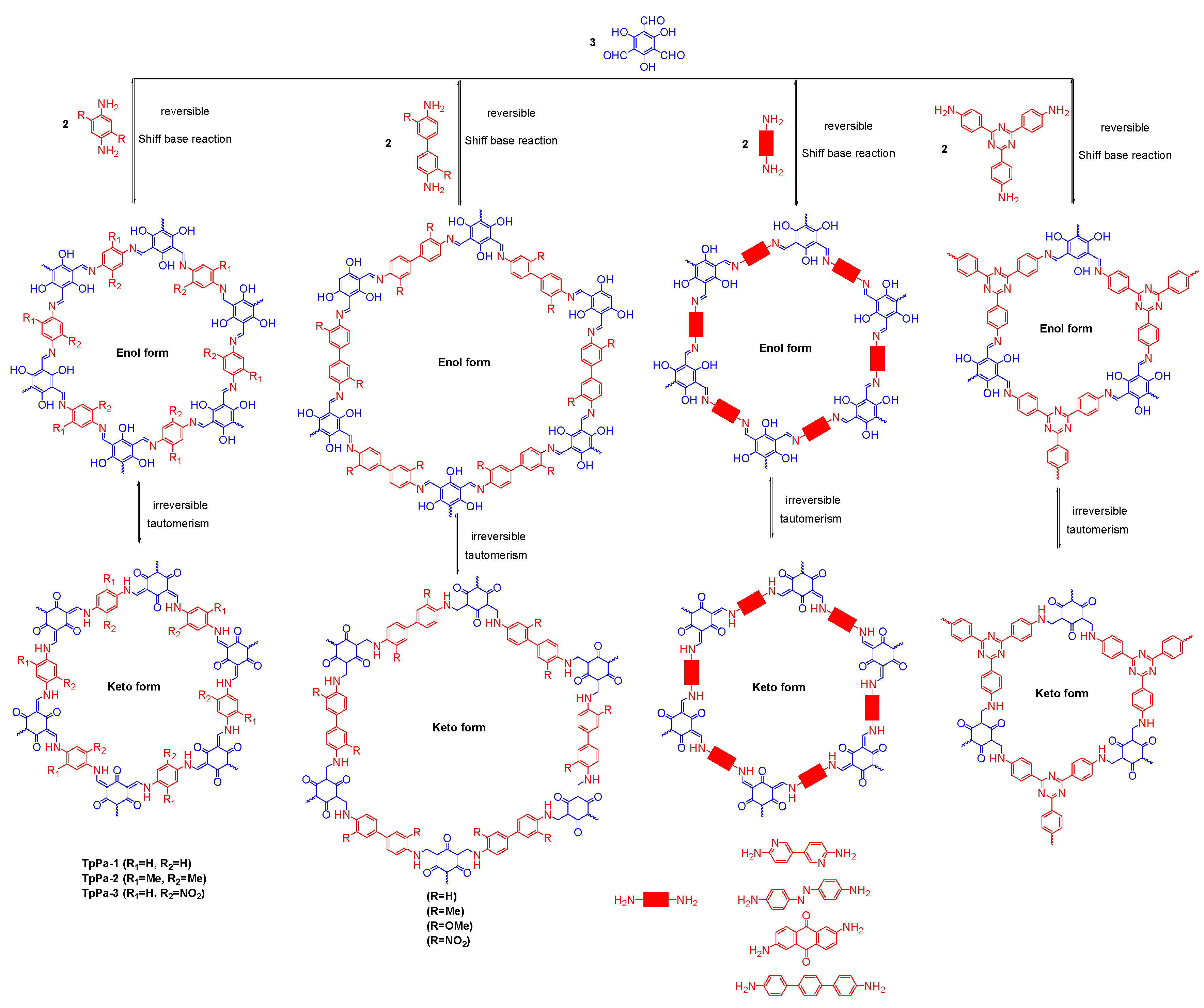
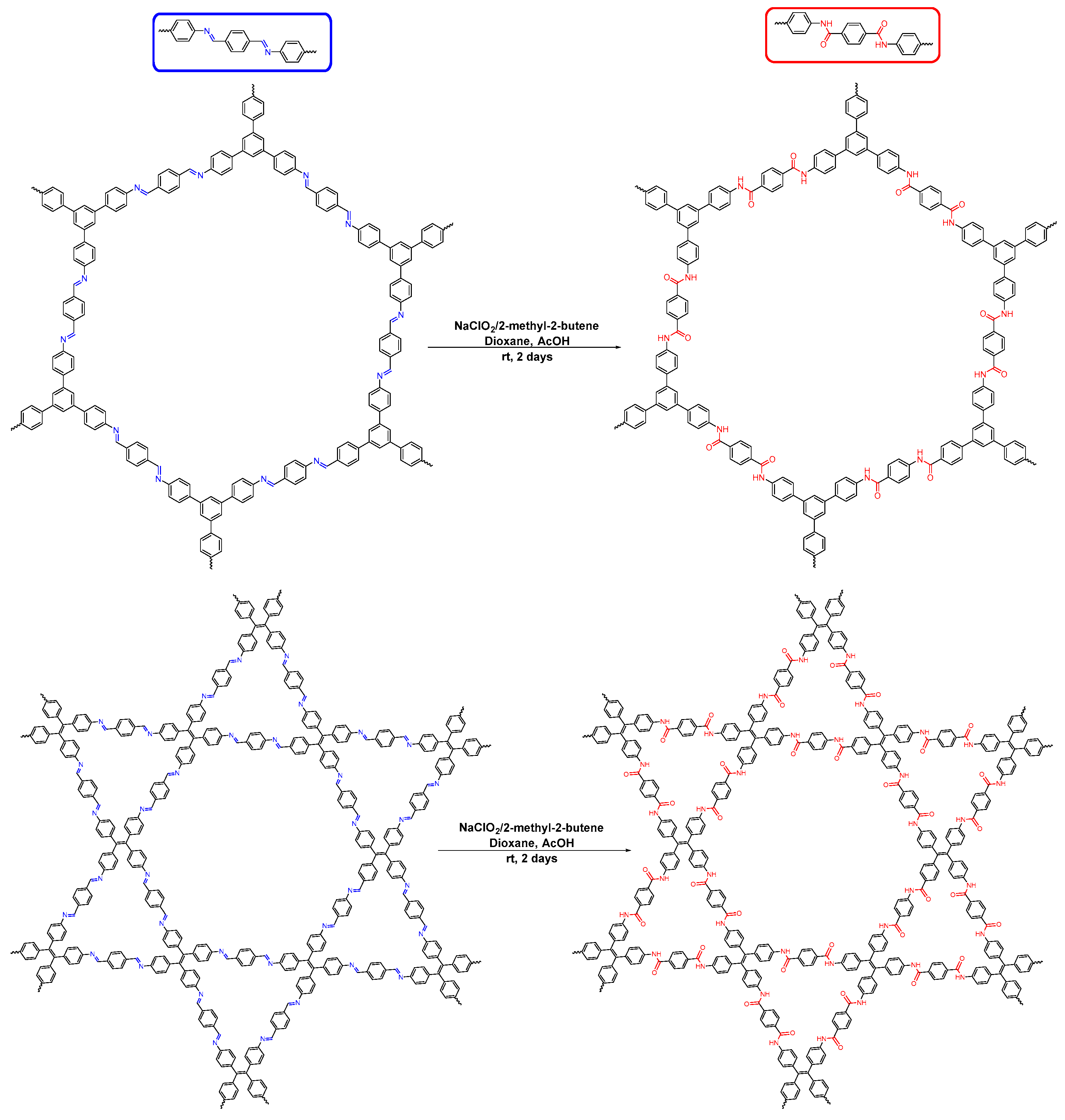
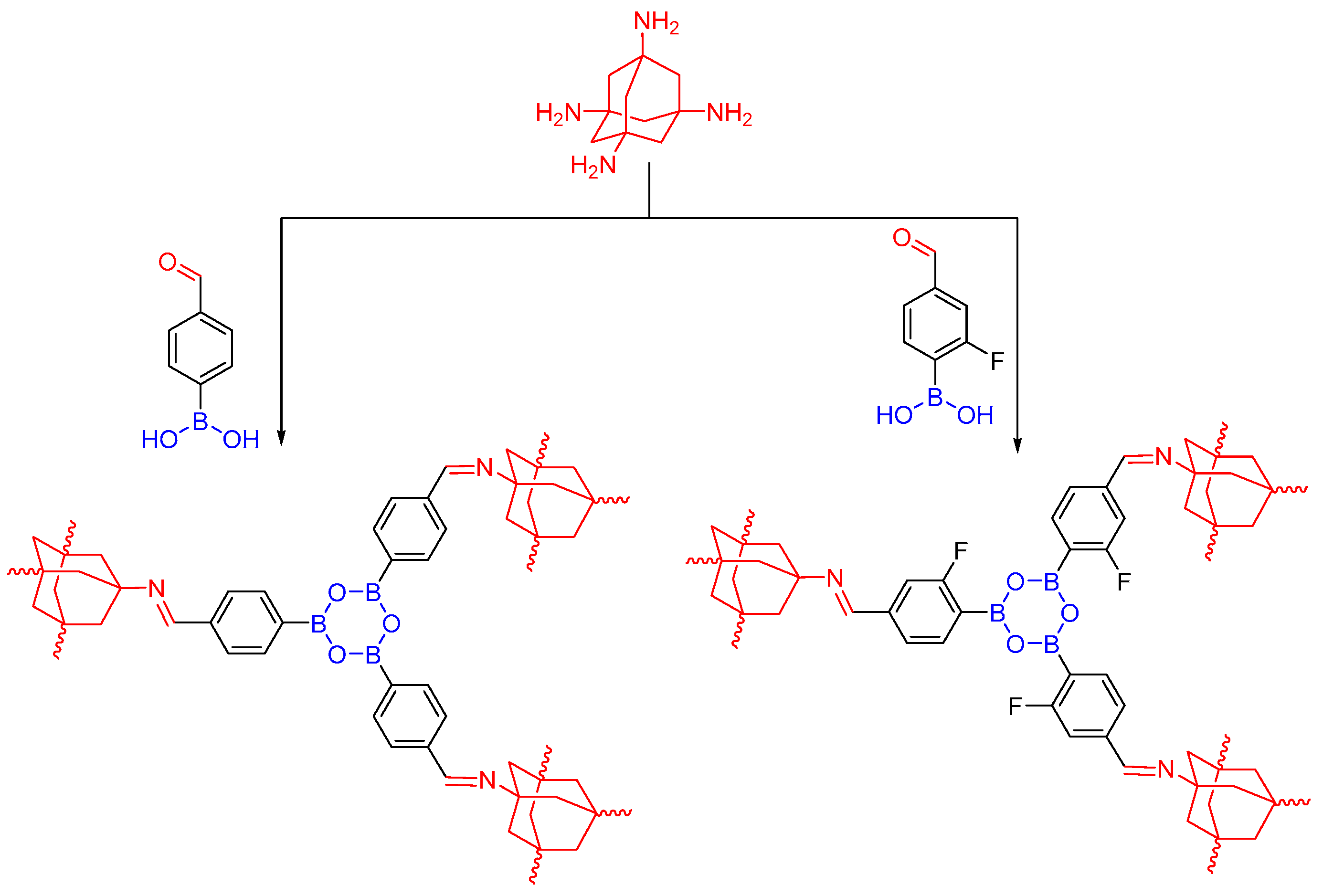
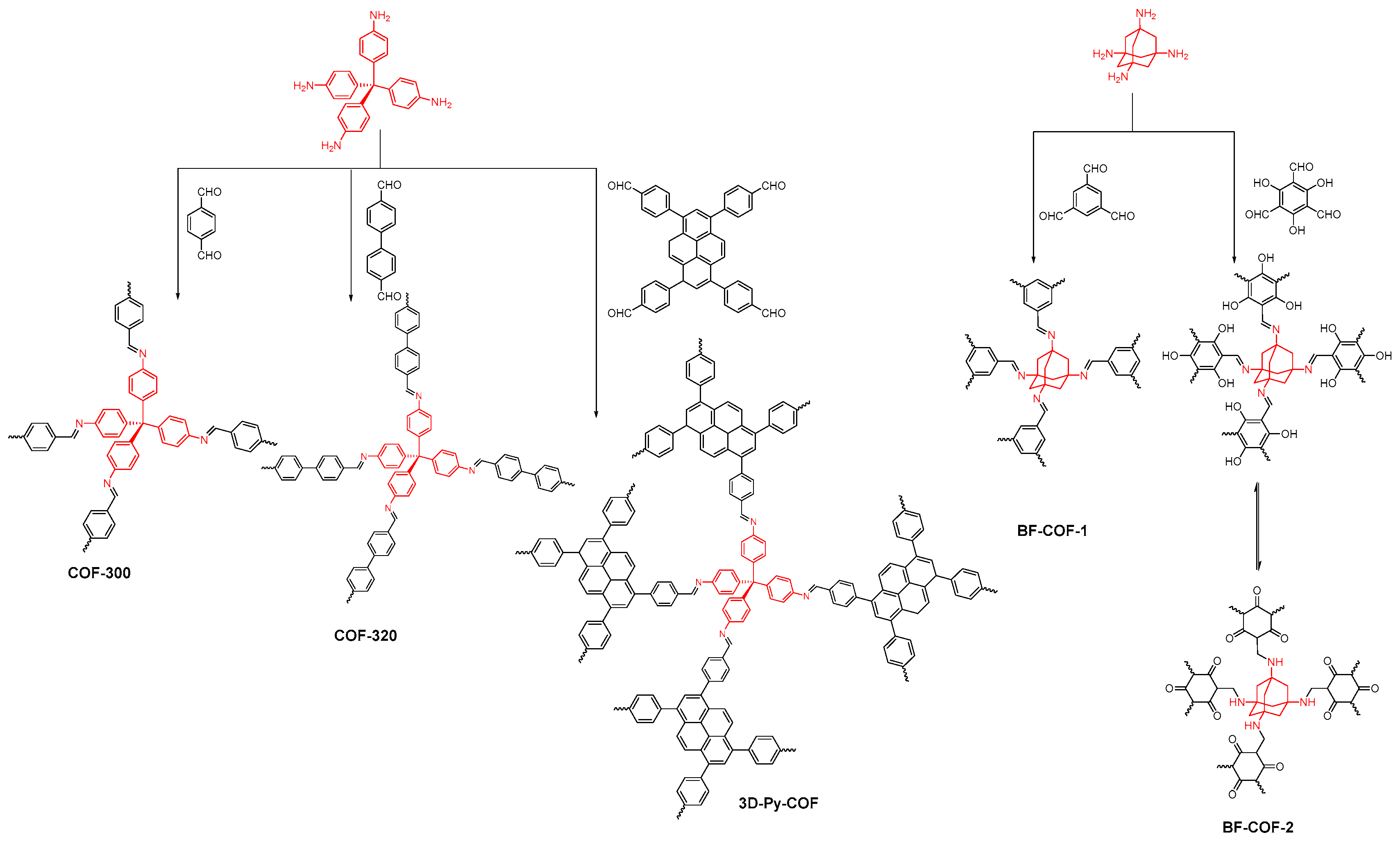
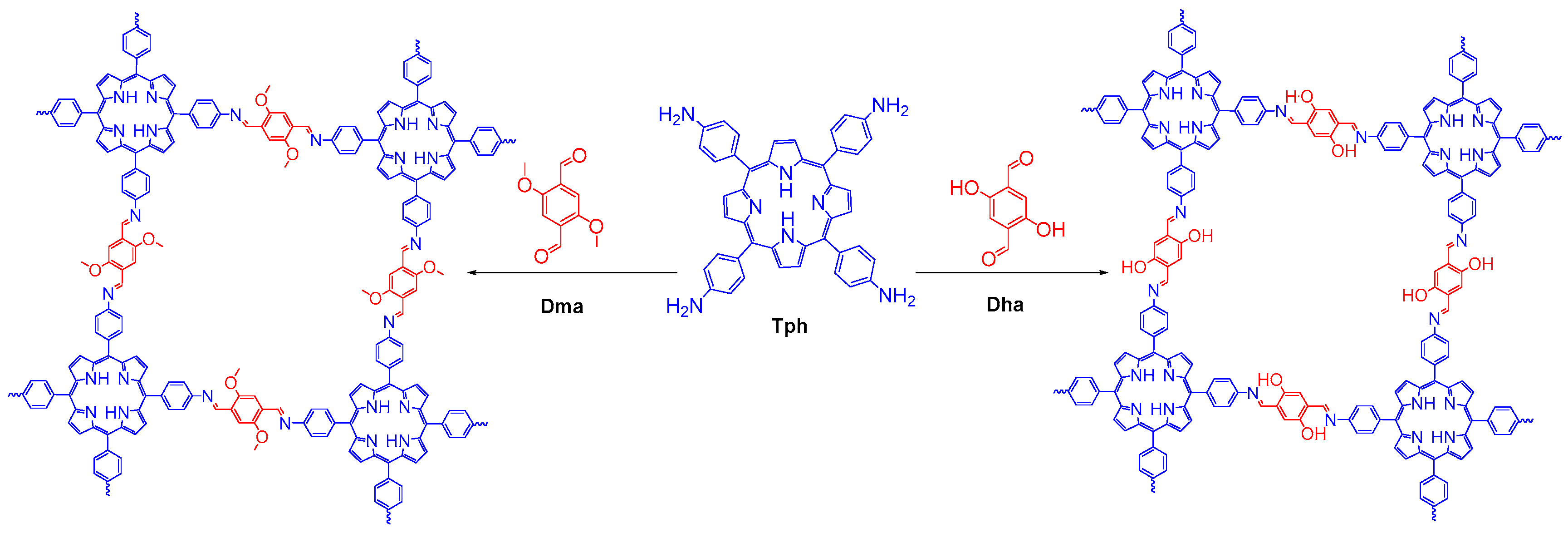
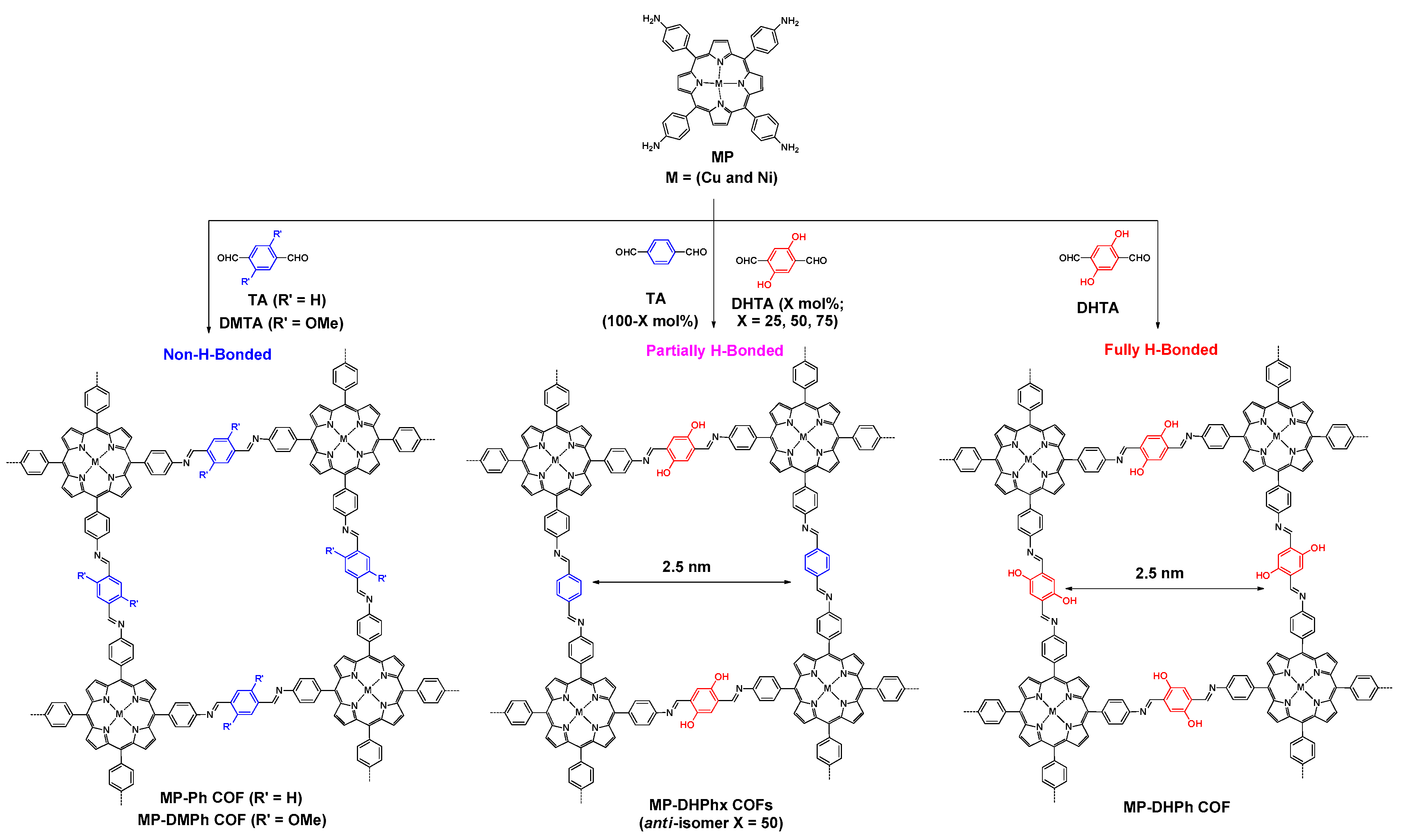
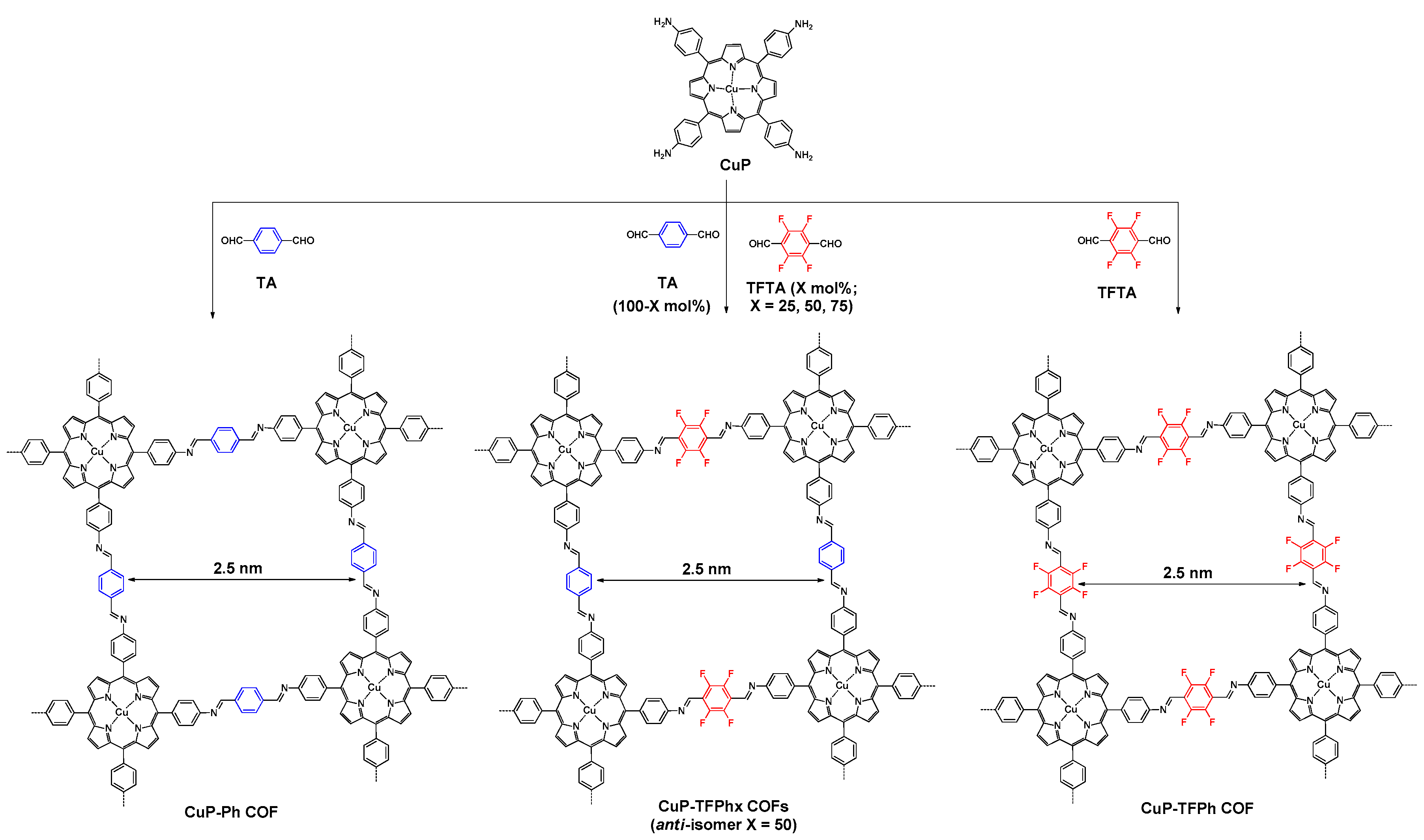
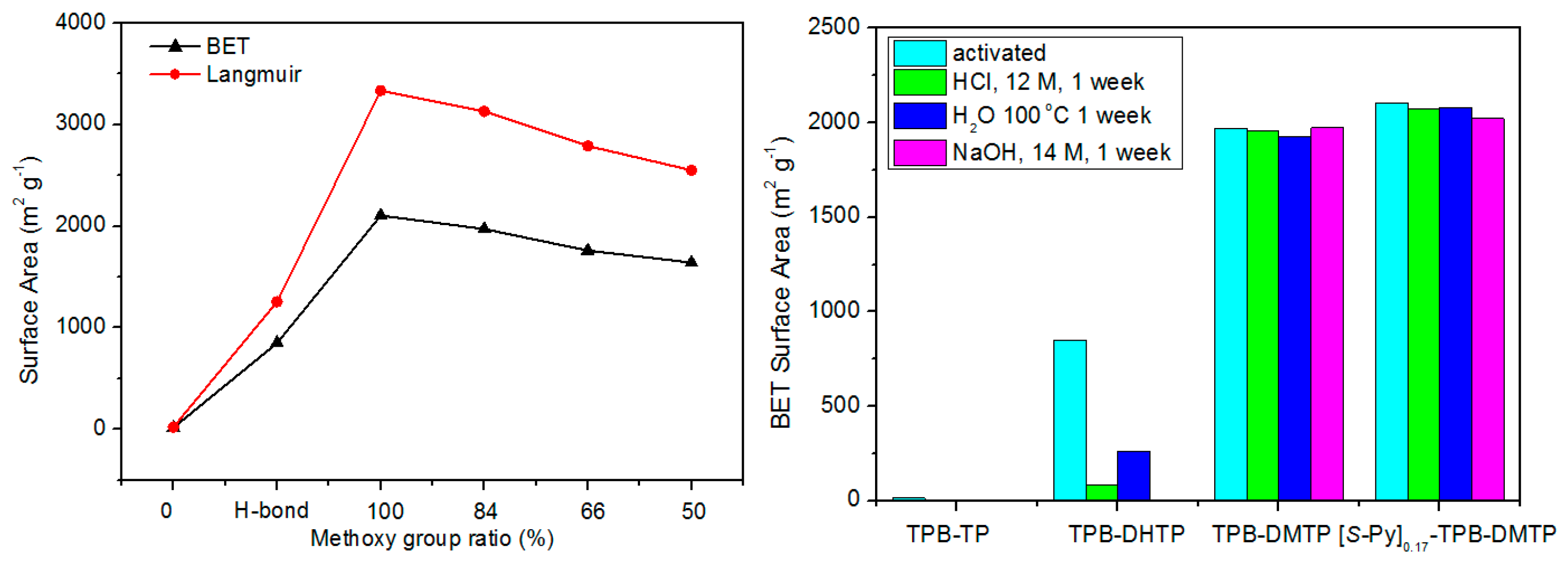
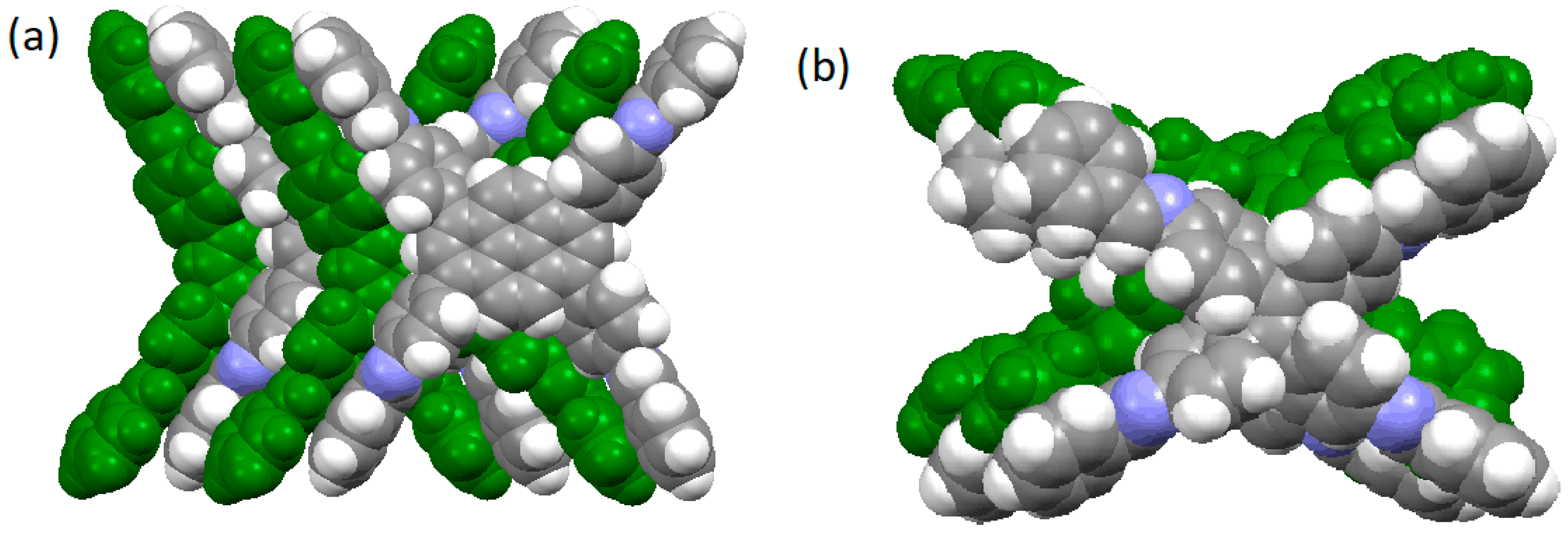
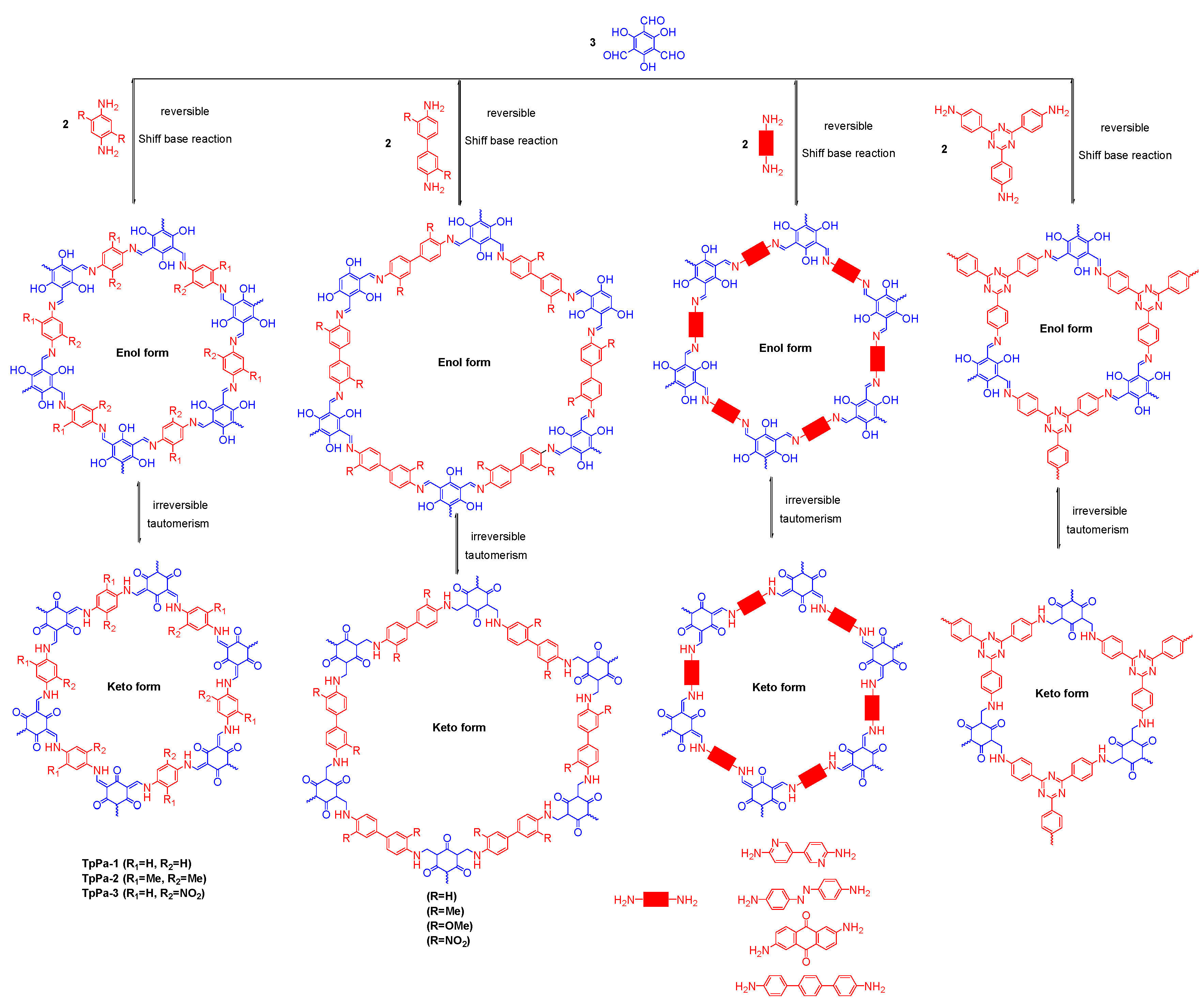
2.2.1. Imine-Linked COFs
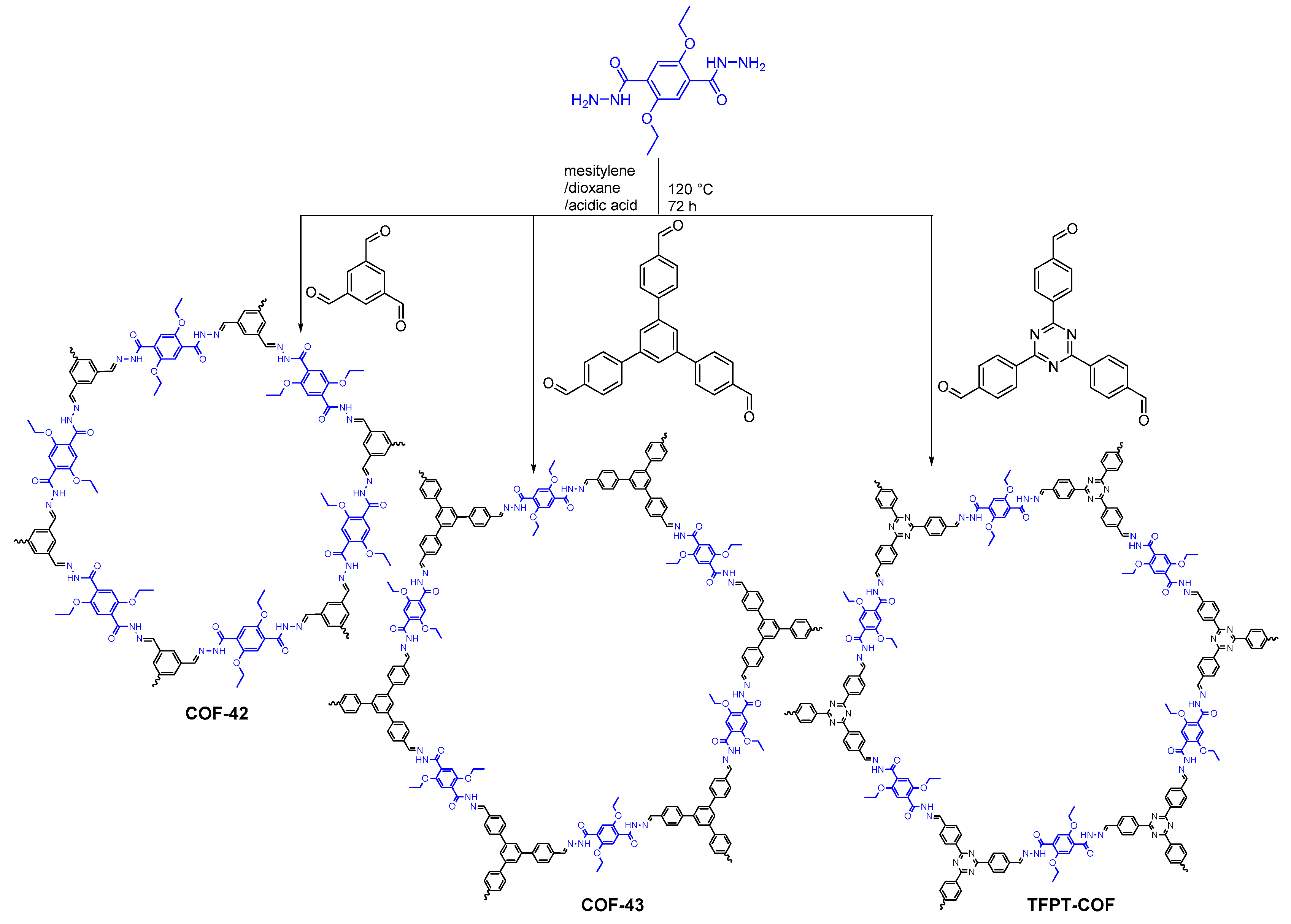
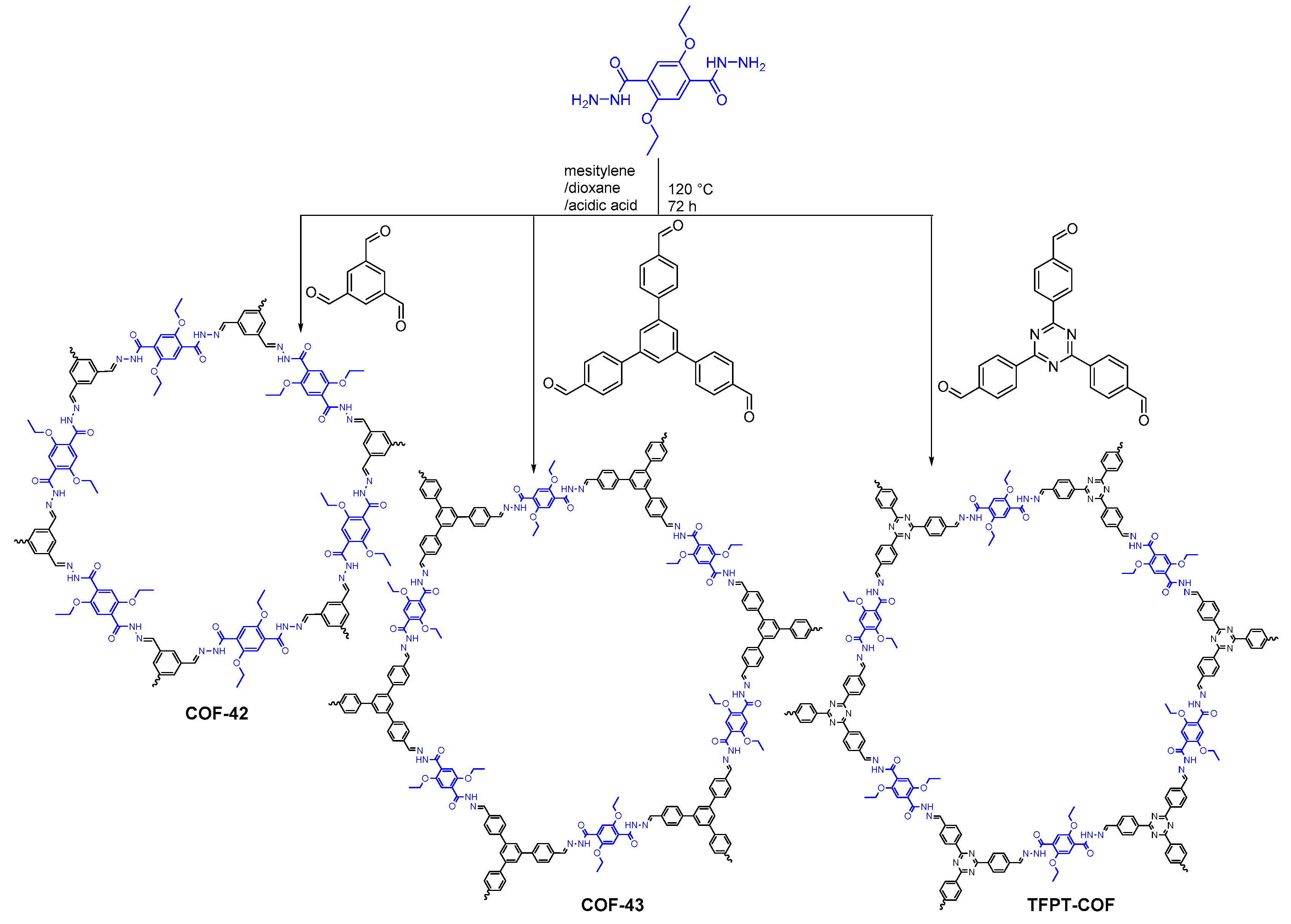
2.2.2. Hydrazone-Linked COFs
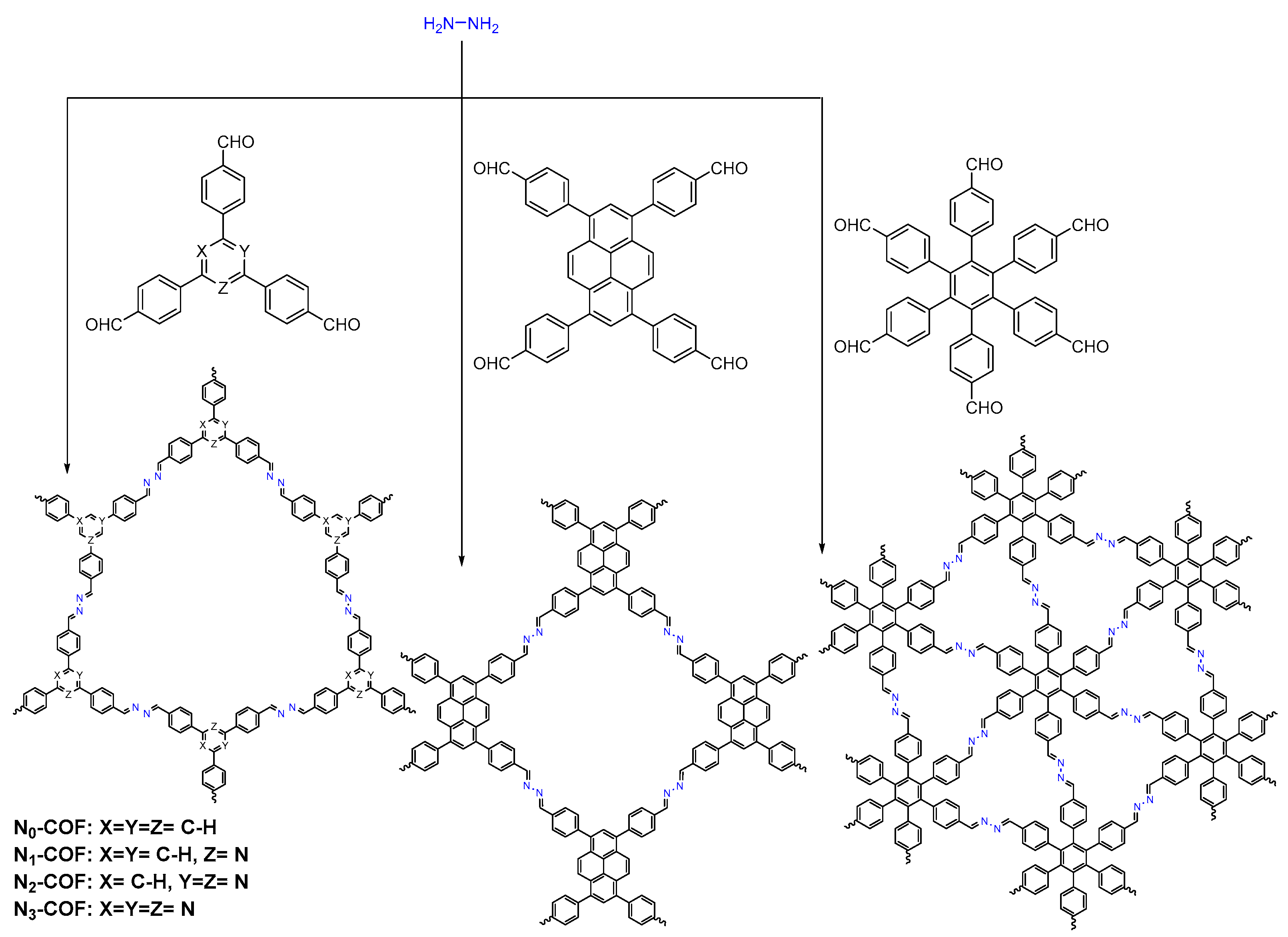
2.2.3. Azine-Linked COFs
2.2.4. Squaraine-Linked COF
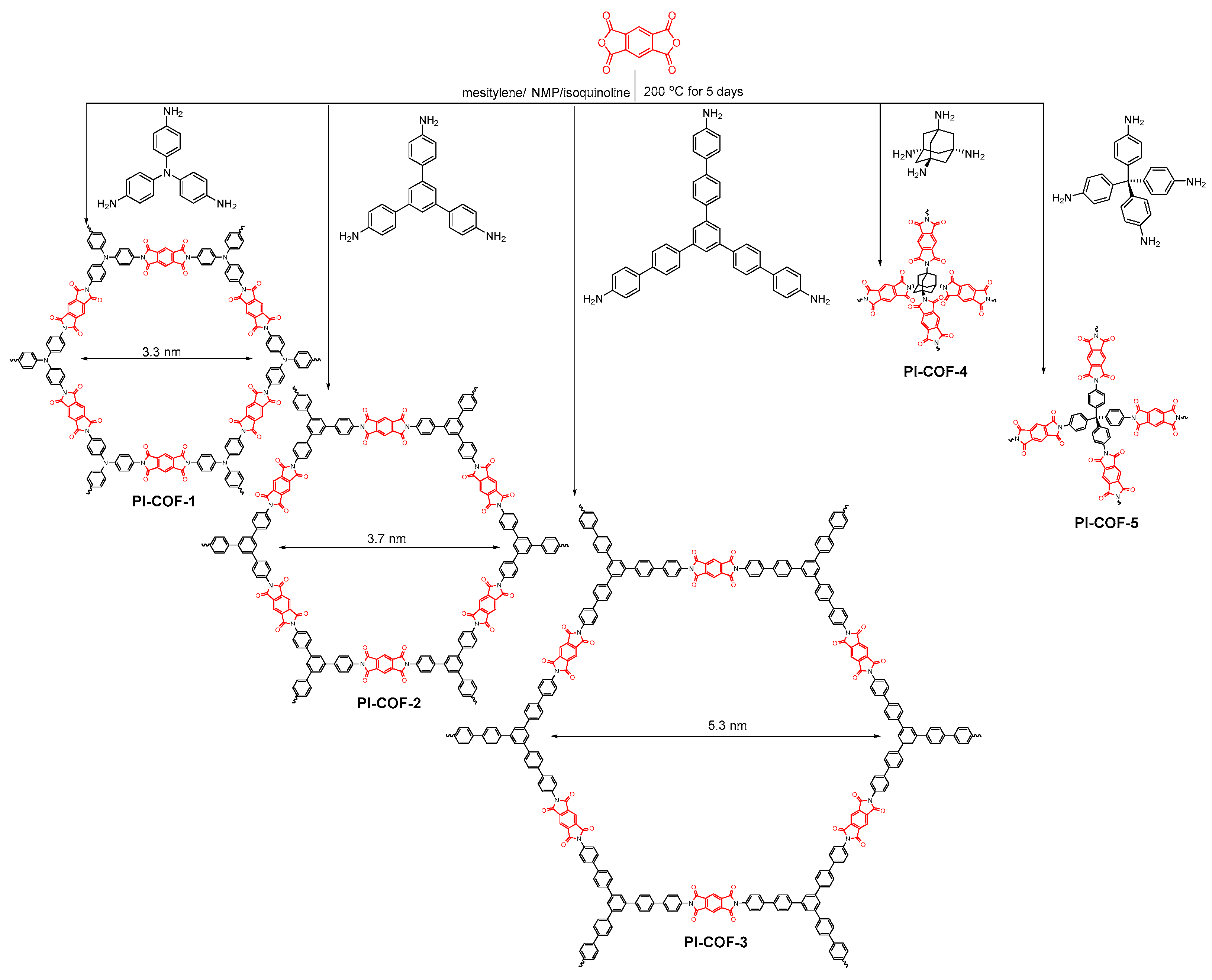
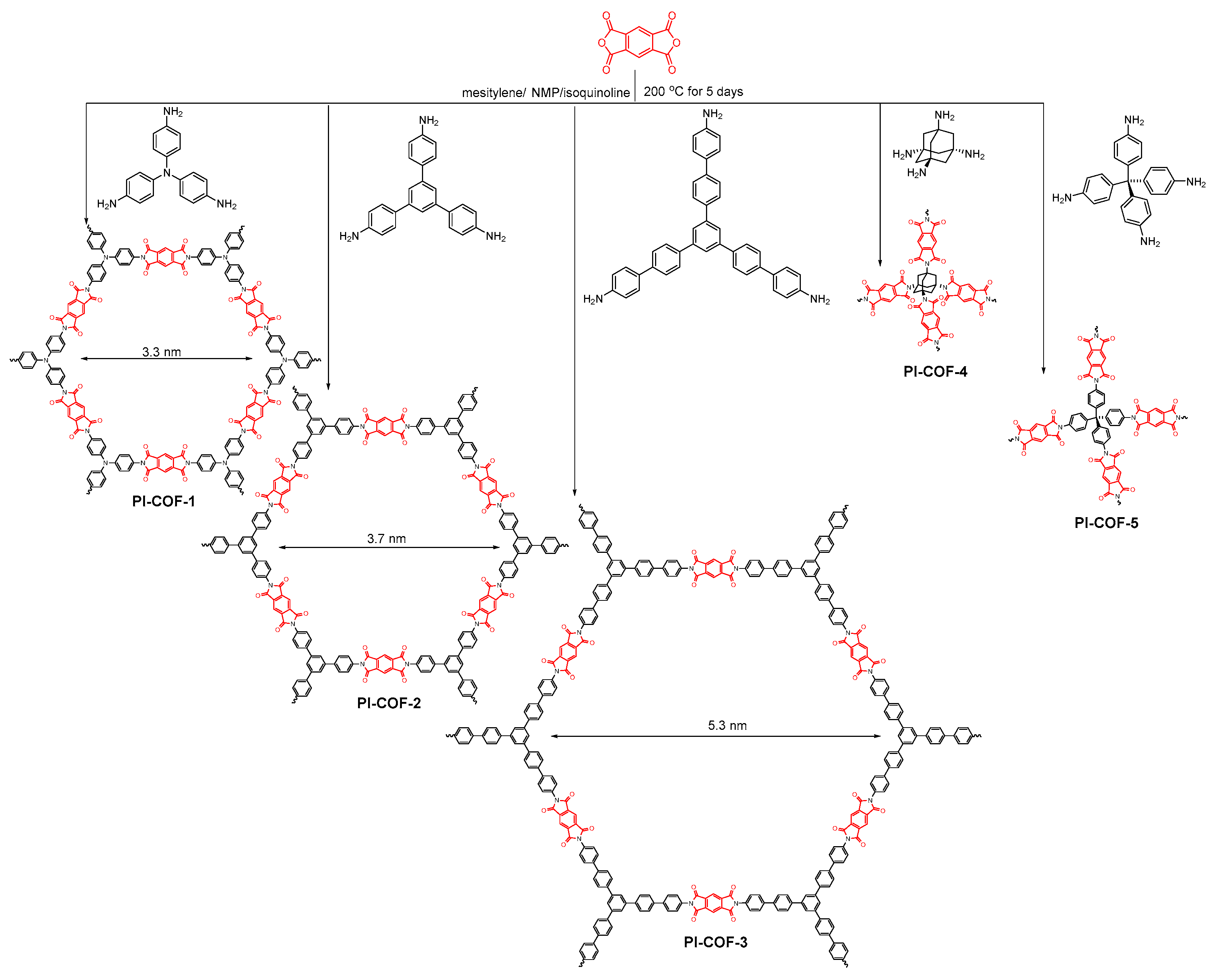
2.2.5. Imide-Linked COFs
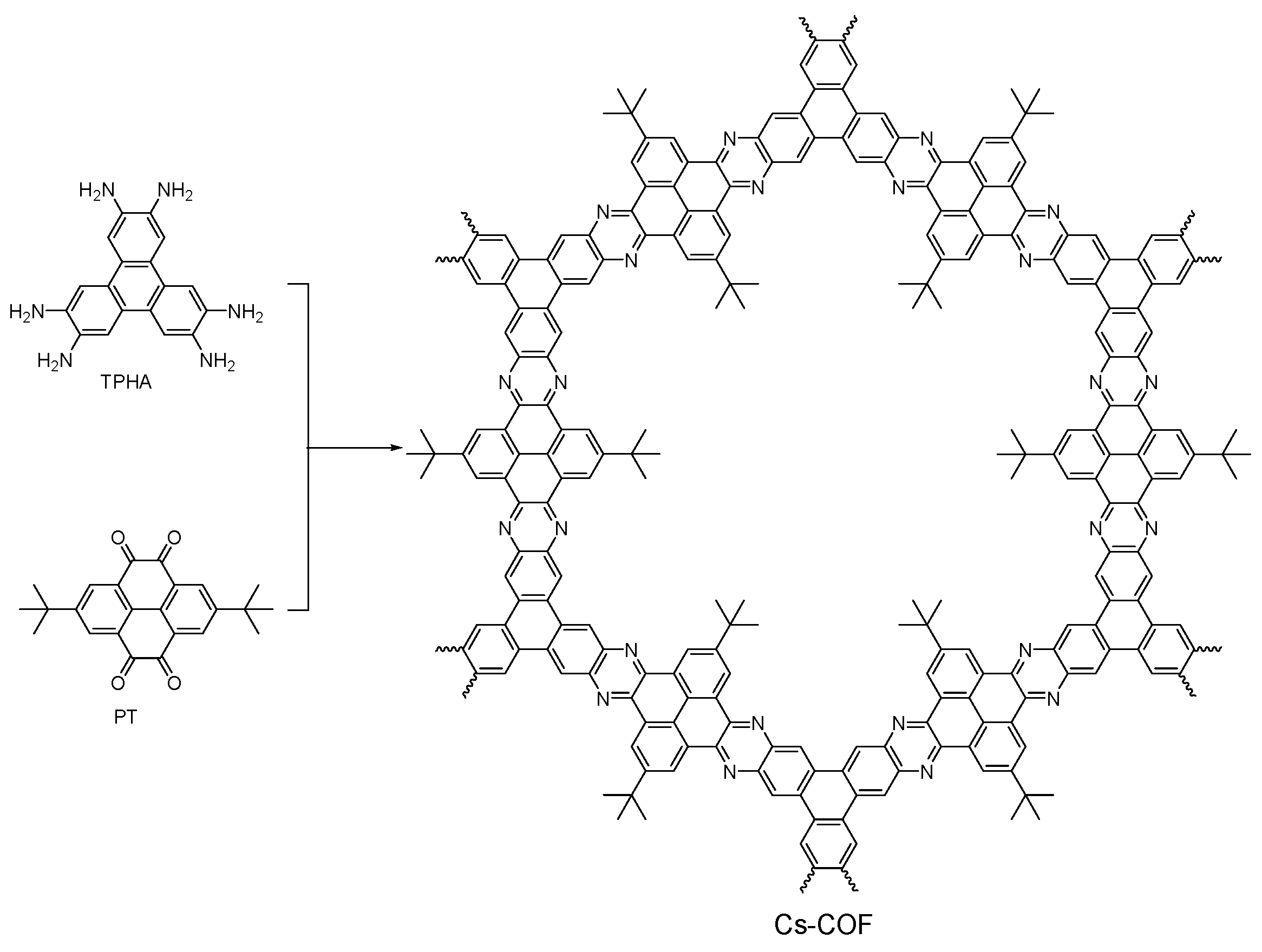
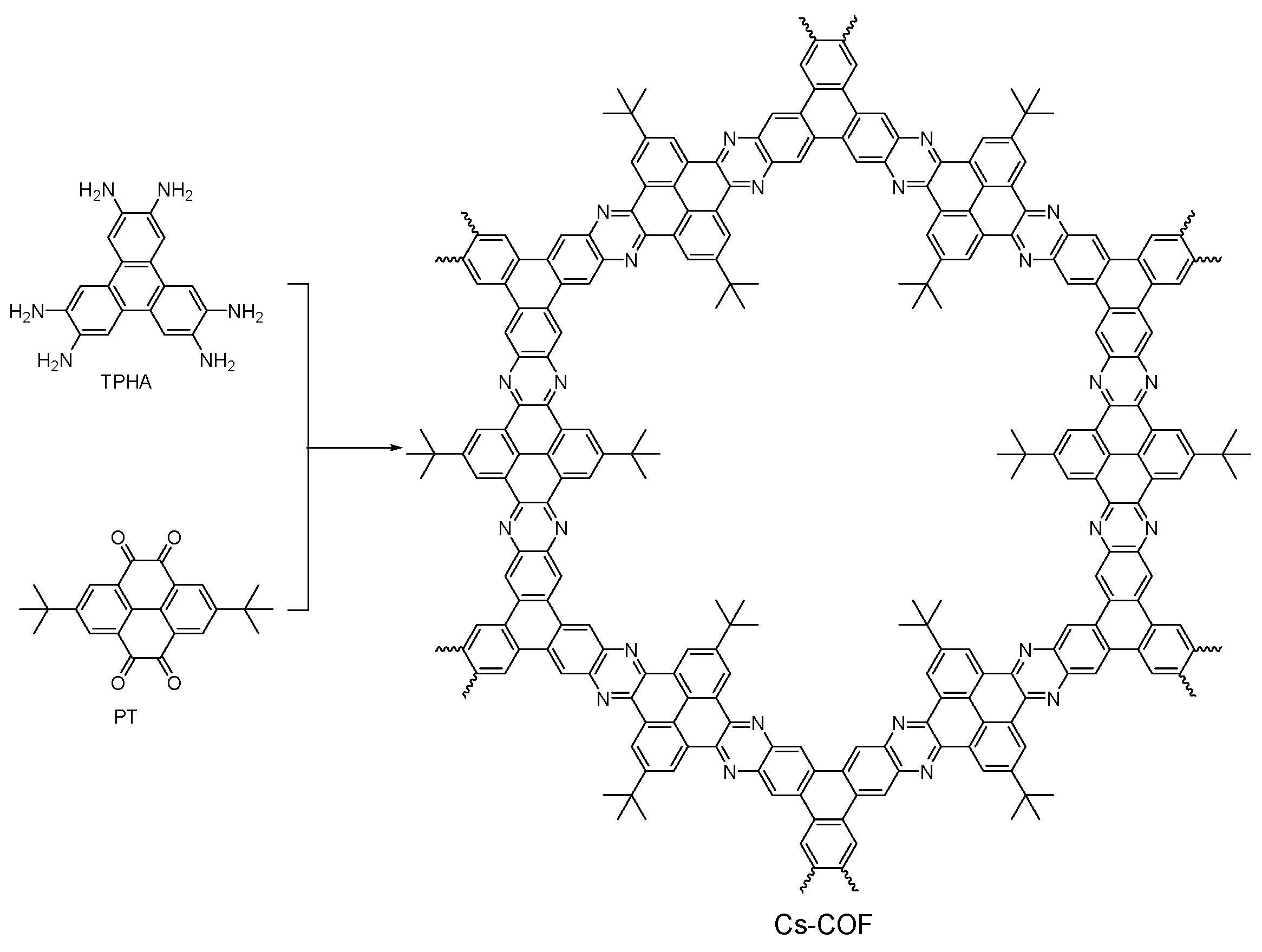
2.2.6. Phenazine-Linked COFs
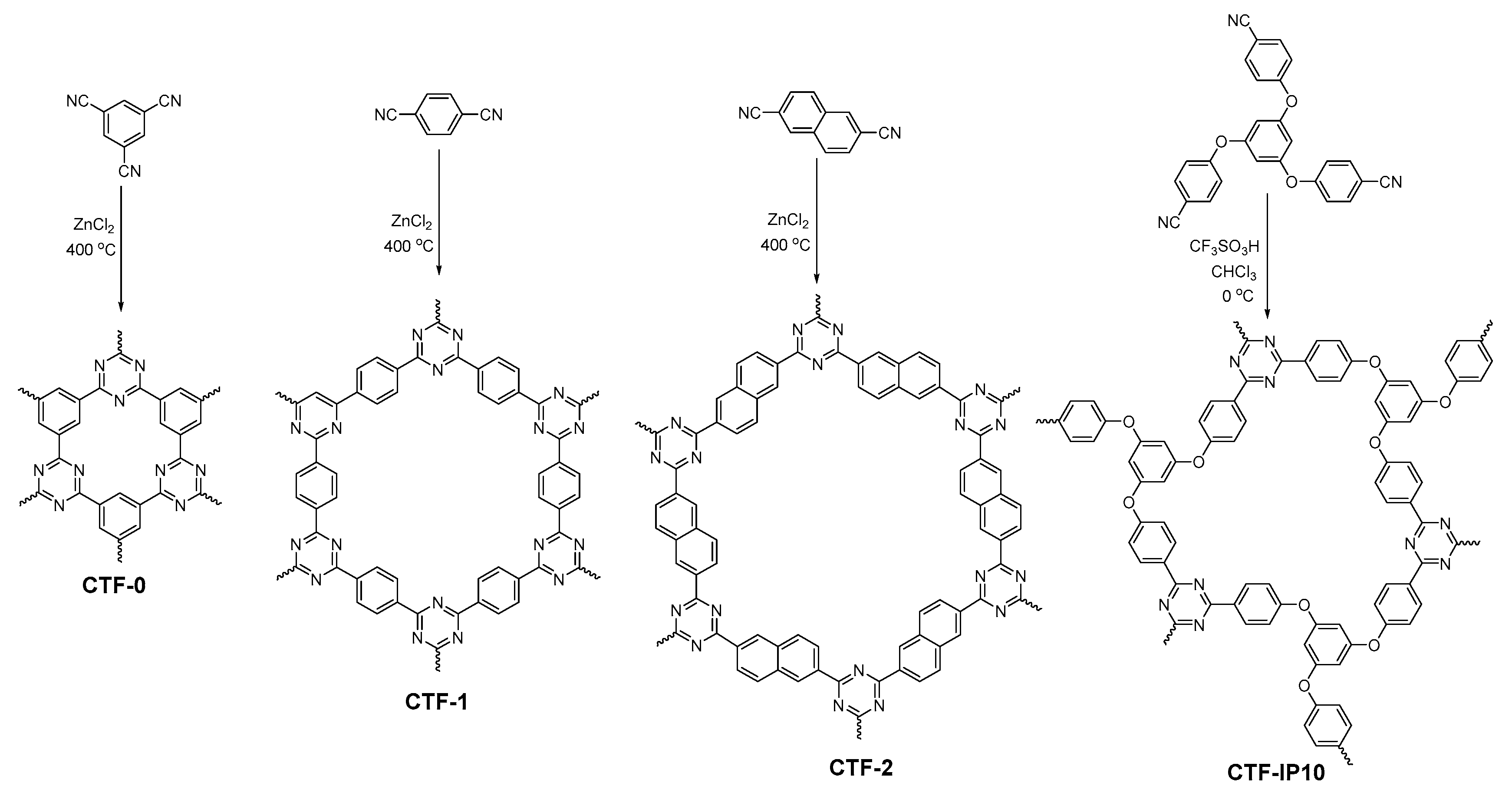
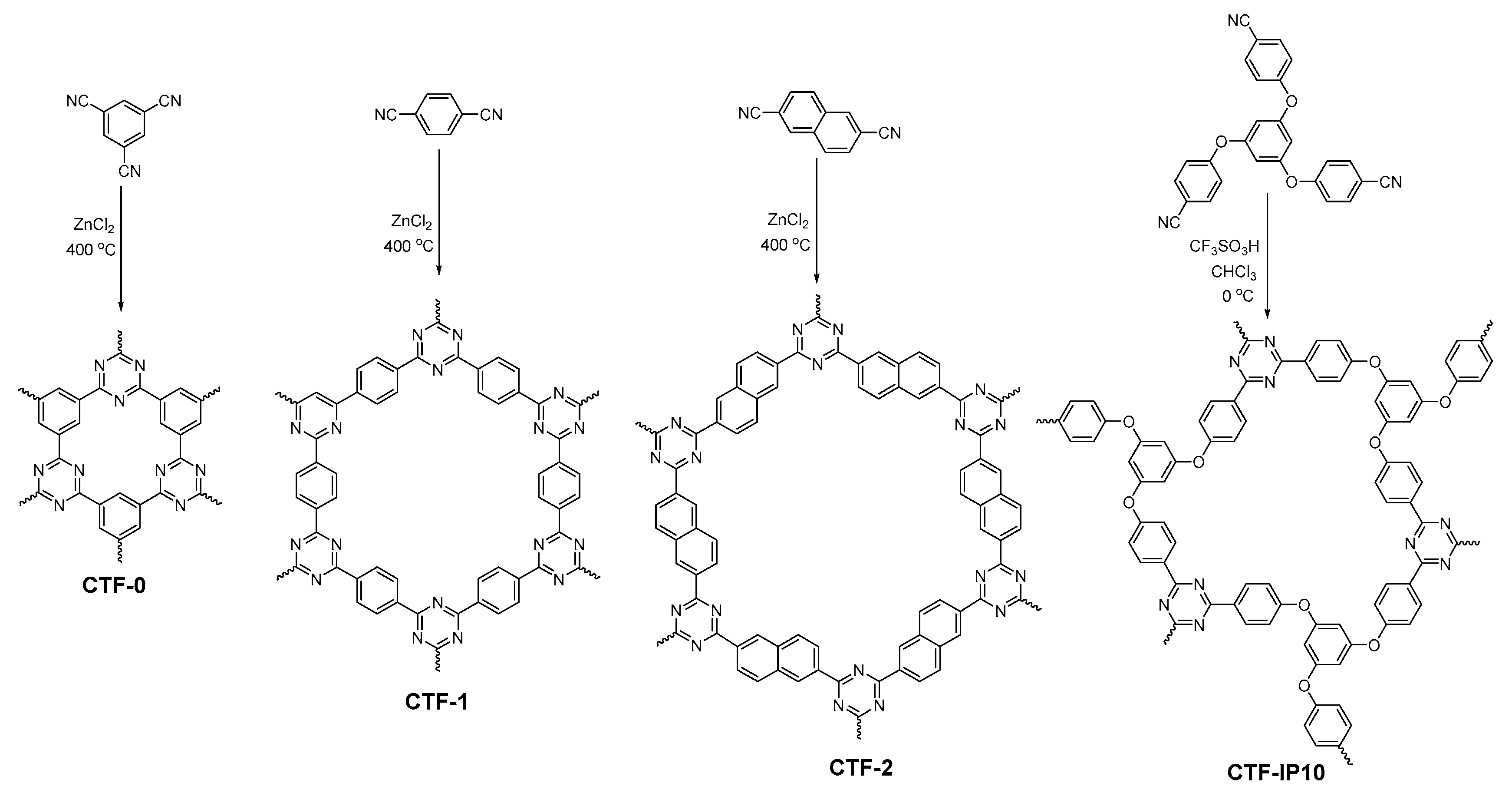
2.2.7. Triazine-Linked COFs
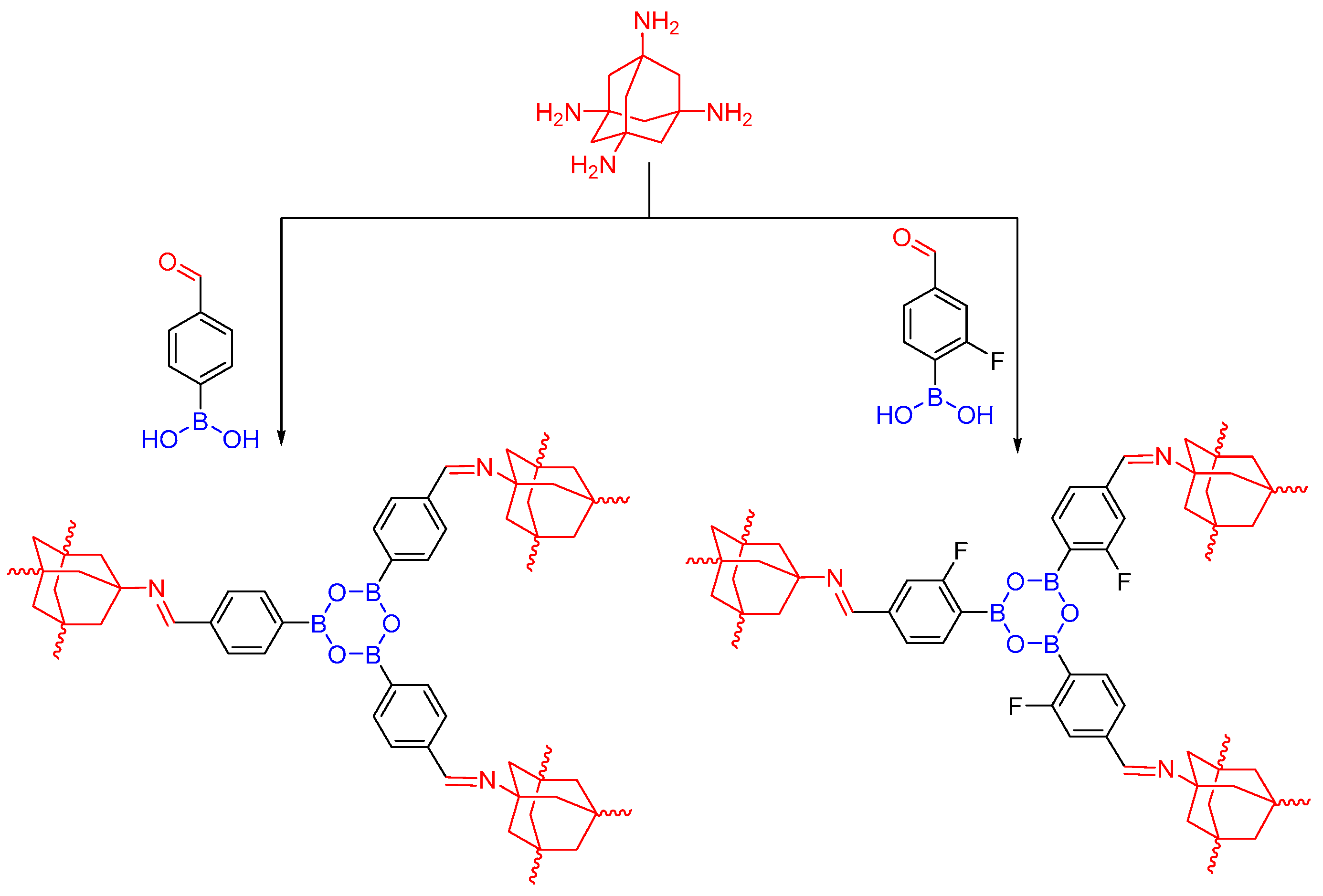
2.3. Hetero Linkages in One COF Structure
3. Gas Storage for Clean Energy Applications
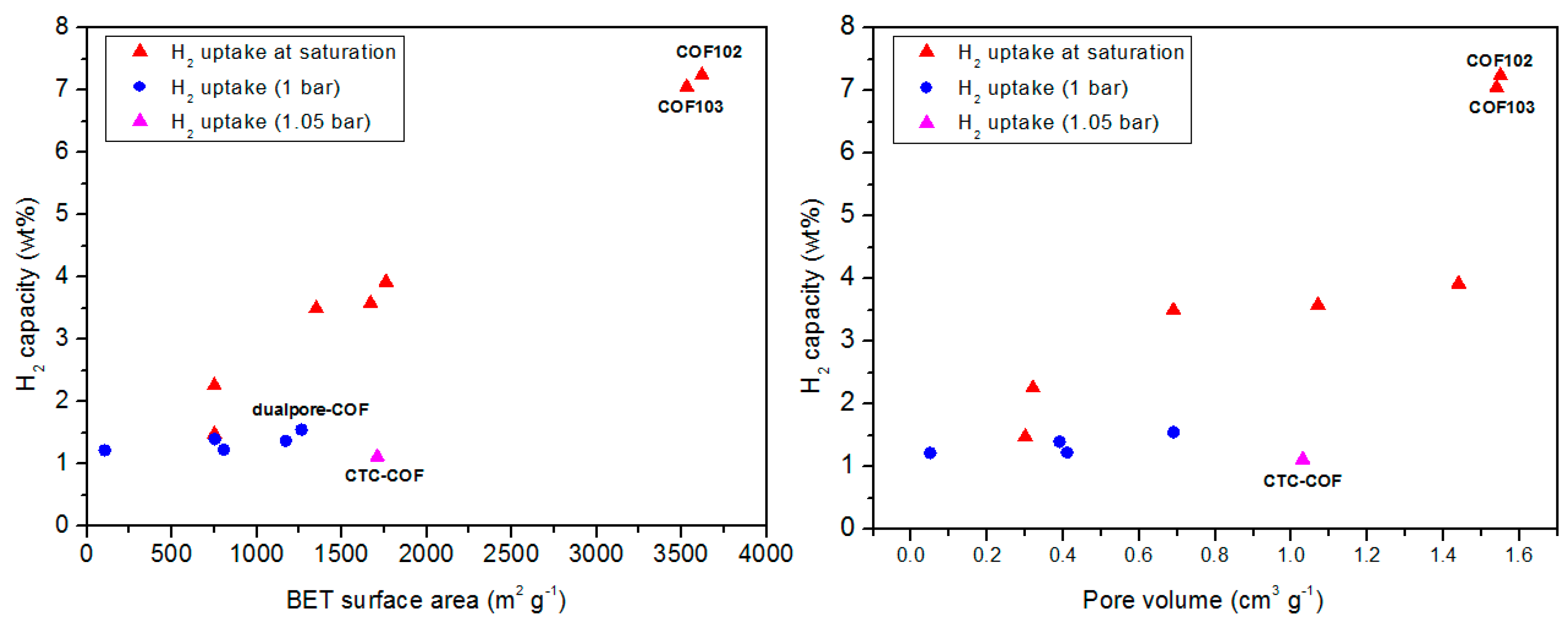
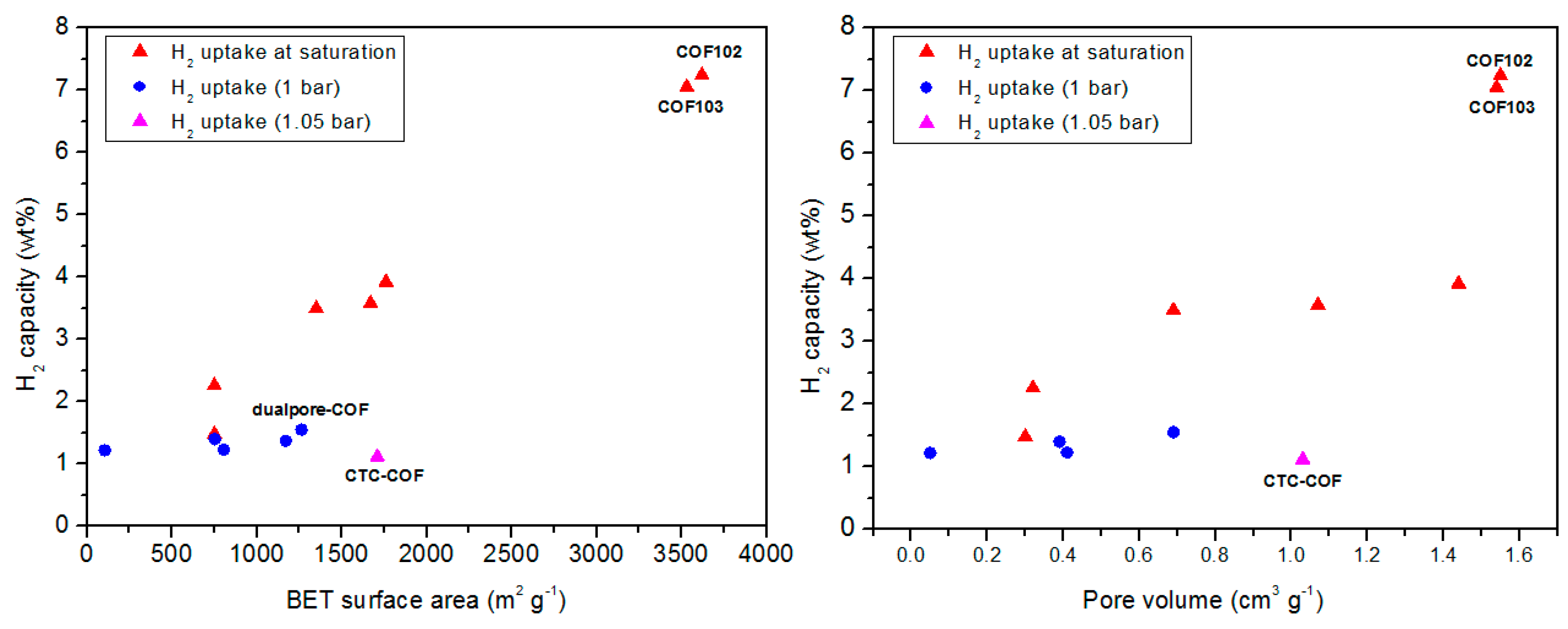
3.1. Hydrogen Storage
3.2. Methane Storage
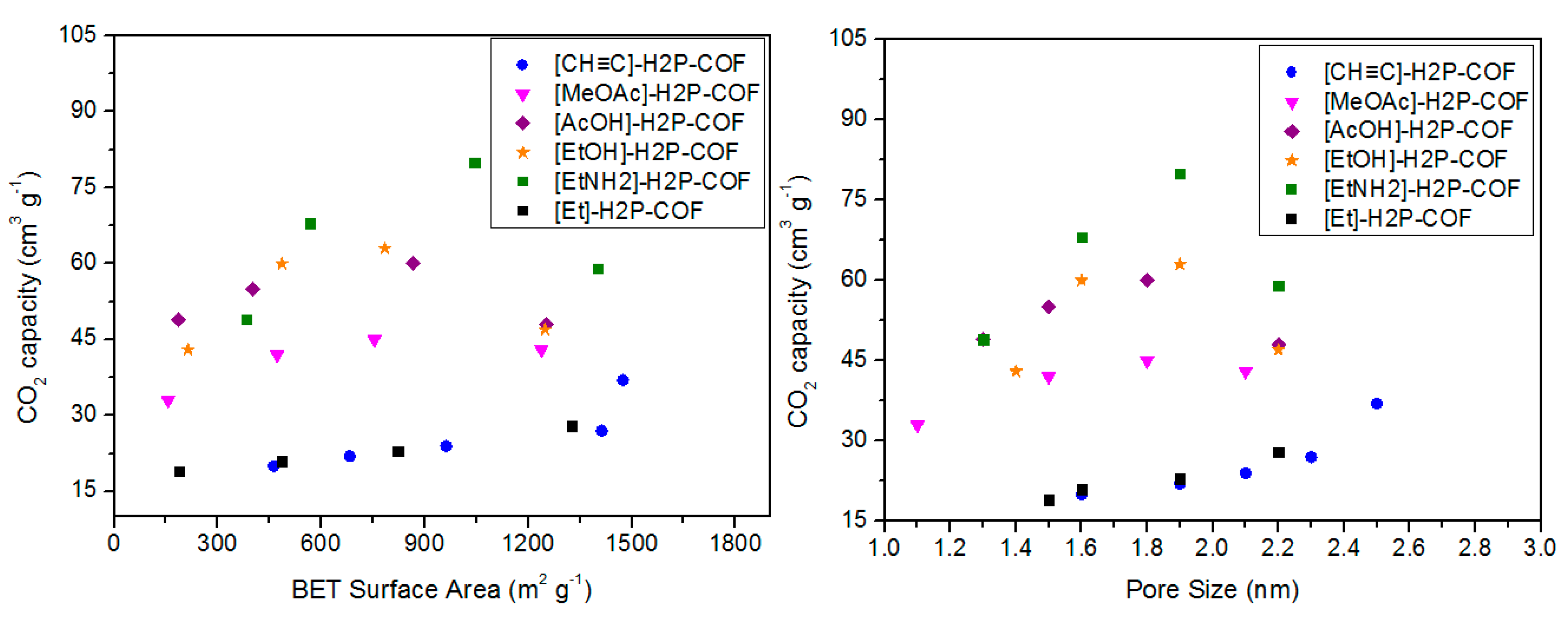
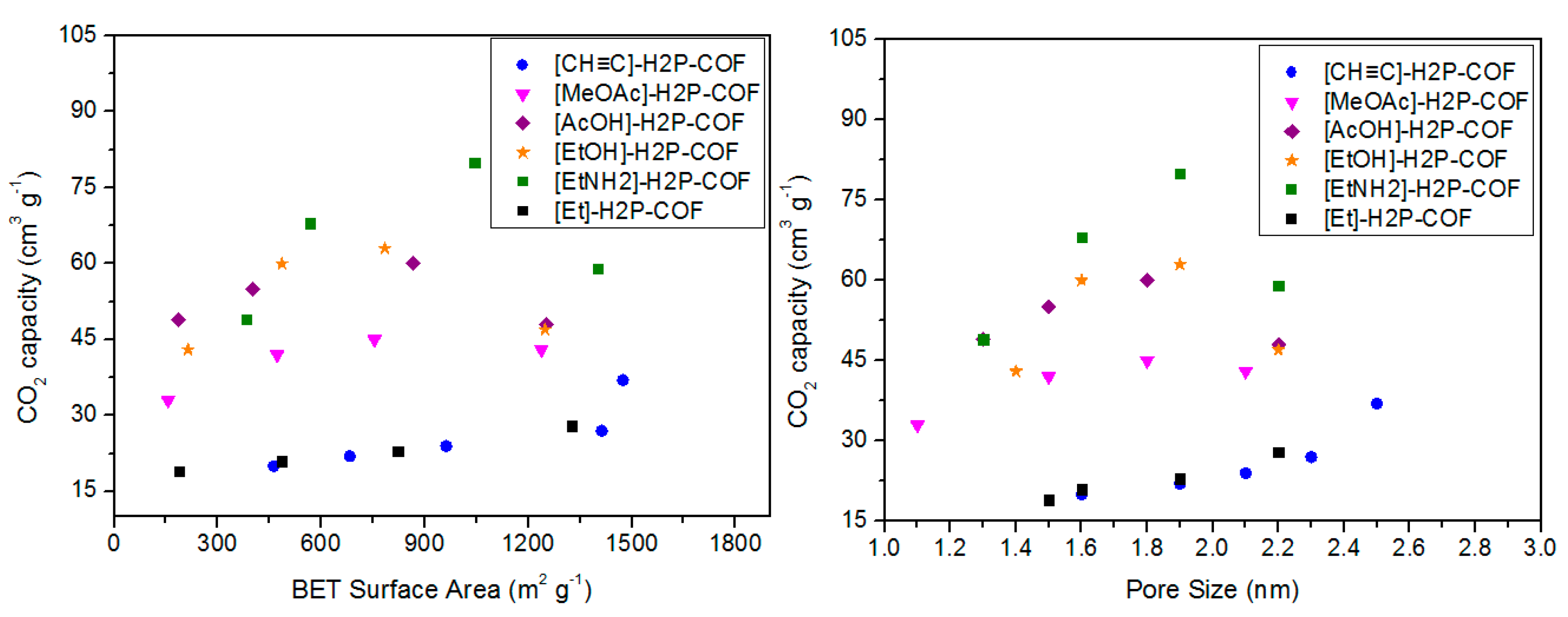
3.3. CO2 Capture
4. Perspectives and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Waller, P.J.; Gandara, F.; Yaghi, O.M. Chemistry of covalent organic frameworks. Acc. Chem. Res. 2015, 48, 3053–3063. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.Y.; Wang, W. Covalent organic frameworks (COFs): From design to applications. Chem. Soc. Rev. 2013, 42, 548–568. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Ding, X.S.; Jiang, D.L. Covalent organic frameworks. Chem. Soc. Rev. 2012, 41, 6010–6022. [Google Scholar] [CrossRef] [PubMed]
- Segura, J.L.; Mancheno, M.J.; Zamora, F. Covalent organic frameworks based on schiff-base chemistry: Synthesis, properties and potential applications. Chem. Soc. Rev. 2016, 45, 5635–5671. [Google Scholar] [CrossRef] [PubMed]
- Diercks, C.S.; Yaghi, O.M. The atom, the molecule, and the covalent organic framework. Science 2017, 355, 6328. [Google Scholar] [CrossRef] [PubMed]
- Rogge, S.M.J.; Bavykina, A.; Hajek, J.; Garcia, H.; Olivos-Suarez, A.I.; Sepulveda-Escribano, A.; Vimont, A.; Clet, G.; Bazin, P.; Kapteijn, F.; et al. Metal-organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 2017, 46, 3134–3184. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.J.; Cantrill, S.J.; Cousins, G.R.L.; Sanders, J.K.M.; Stoddart, J.F. Dynamic covalent chemistry. Angew. Chem. Int. Ed. 2002, 41, 898–952. [Google Scholar] [CrossRef]
- Roy, N.; Lehn, J.M. Dynamic covalent chemistry: A facile room-temperature, reversible, diels-alder reaction between anthracene derivatives and n-phenyltriazolinedione. Chem.-Asian J. 2011, 6, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, D.; Maris, T.; Wuest, J.D. Constructing monocrystalline covalent organic networks by polymerization. Nat. Chem. 2013, 5, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Cote, A.P.; Benin, A.I.; Ockwig, N.W.; O'Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, crystalline, covalent organic frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- El-Kaderi, H.M.; Hunt, J.R.; Mendoza-Cortes, J.L.; Cote, A.P.; Taylor, R.E.; O’Keeffe, M.; Yaghi, O.M. Designed synthesis of 3d covalent organic frameworks. Science 2007, 316, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.L.; Clowes, R.; Ritchie, L.K.; Cooper, A.I. Rapid microwave synthesis and purification of porous covalent organic frameworks. Chem. Mater. 2009, 21, 204–206. [Google Scholar] [CrossRef]
- Cote, A.P.; El-Kaderi, H.M.; Furukawa, H.; Hunt, J.R.; Yaghi, O.M. Reticular synthesis of microporous and mesoporous 2d covalent organic frameworks. J. Am. Chem. Soc. 2007, 129, 12914–12915. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Guo, J.; Kim, J.; Ihee, H.; Jiang, D.L. A photoconductive covalent organic framework: Self-condensed arene cubes composed of eclipsed 2d polypyrene sheets for photocurrent generation. Angew. Chem. Int. Ed. 2009, 48, 5439–5442. [Google Scholar] [CrossRef] [PubMed]
- Tilford, R.W.; Mugavero, S.J.; Pellechia, P.J.; Lavigne, J.J. Tailoring microporosity in covalent organic frameworks. Adv. Mater. 2008, 20, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Lanni, L.M.; Tilford, R.W.; Bharathy, M.; Lavigne, J.J. Enhanced hydrolytic stability of self-assembling alkylated two-dimensional covalent organic frameworks. J. Am. Chem. Soc. 2011, 133, 13975–13983. [Google Scholar] [CrossRef] [PubMed]
- Dogru, M.; Sonnauer, A.; Gavryushin, A.; Knochel, P.; Bein, T. A covalent organic framework with 4 nm open pores. Chem. Commun. 2011, 47, 1707–1709. [Google Scholar] [CrossRef] [PubMed]
- Spitler, E.L.; Colson, J.W.; Uribe-Romo, F.J.; Woll, A.R.; Giovino, M.R.; Saldivar, A.; Dichtel, W.R. Lattice expansion of highly oriented 2d phthalocyanine covalent organic framework films. Angew. Chem. Int. Ed. 2012, 51, 2623–2627. [Google Scholar] [CrossRef] [PubMed]
- Colson, J.W.; Mann, J.A.; DeBlase, C.R.; Dichtel, W.R. Patterned growth of oriented 2d covalent organic framework thin films on single-layer graphene. J. Polym. Sci. Polym. Chem. 2015, 53, 378–384. [Google Scholar] [CrossRef]
- Spitler, E.L.; Dichtel, W.R. Lewis acid-catalysed formation of two-dimensional phthalocyanine covalent organic frameworks. Nat. Chem. 2010, 2, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Spitler, E.L.; Giovino, M.R.; White, S.L.; Dichtel, W.R. A mechanistic study of lewis acid-catalyzed covalent organic framework formation. Chem. Sci. 2011, 2, 1588–1593. [Google Scholar] [CrossRef]
- Dogru, M.; Sonnauer, A.; Zimdars, S.; Doblinger, M.; Knochel, P.; Bein, T. Facile synthesis of a mesoporous benzothiadiazole-cof based on a transesterification process. Crystengcomm 2013, 15, 1500–1502. [Google Scholar] [CrossRef]
- Calik, M.; Sick, T.; Dogru, M.; Doblinger, M.; Datz, S.; Budde, H.; Hartschuh, A.; Auras, F.; Bein, T. From highly crystalline to outer surface-functionalized covalent organic frameworks-a modulation approach. J. Am. Chem. Soc. 2016, 138, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yang, H.S.; Whiteley, J.M.; Wan, S.; Jin, Y.H.; Lee, S.H.; Zhang, W. Ionic covalent organic frameworks with spiroborate linkage. Angew. Chem. Int. Ed. 2016, 55, 1737–1741. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Romo, F.J.; Hunt, J.R.; Furukawa, H.; Klock, C.; O’Keeffe, M.; Yaghi, O.M. A crystalline imine-linked 3-d porous covalent organic framework. J. Am. Chem. Soc. 2009, 131, 4570–4571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Su, J.; Furukawa, H.; Yun, Y.F.; Gandara, F.; Duong, A.; Zou, X.D.; Yaghi, O.M. Single-crystal structure of a covalent organic framework. J. Am. Chem. Soc. 2013, 135, 16336–16339. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.X.; Li, Z.J.; Wei, L.; Ding, S.Y.; Zhang, Y.B.; Wang, W. A dynamic three-dimensional covalent organic framework. J. Am. Chem. Soc. 2017, 139, 4995–4998. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.Q.; Ding, H.M.; Yuan, D.Q.; Wang, B.S.; Wang, C. A pyrene-based, fluorescent three-dimensional covalent organic framework. J. Am. Chem. Soc. 2016, 138, 3302–3305. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.R.; Gu, S.; Zheng, J.; Zhuang, Z.B.; Qiu, S.L.; Yan, Y.S. 3D microporous base-functionalized covalent organic frameworks for size-selective catalysis. Angew. Chem. Int. Ed. 2014, 53, 2878–2882. [Google Scholar] [CrossRef] [PubMed]
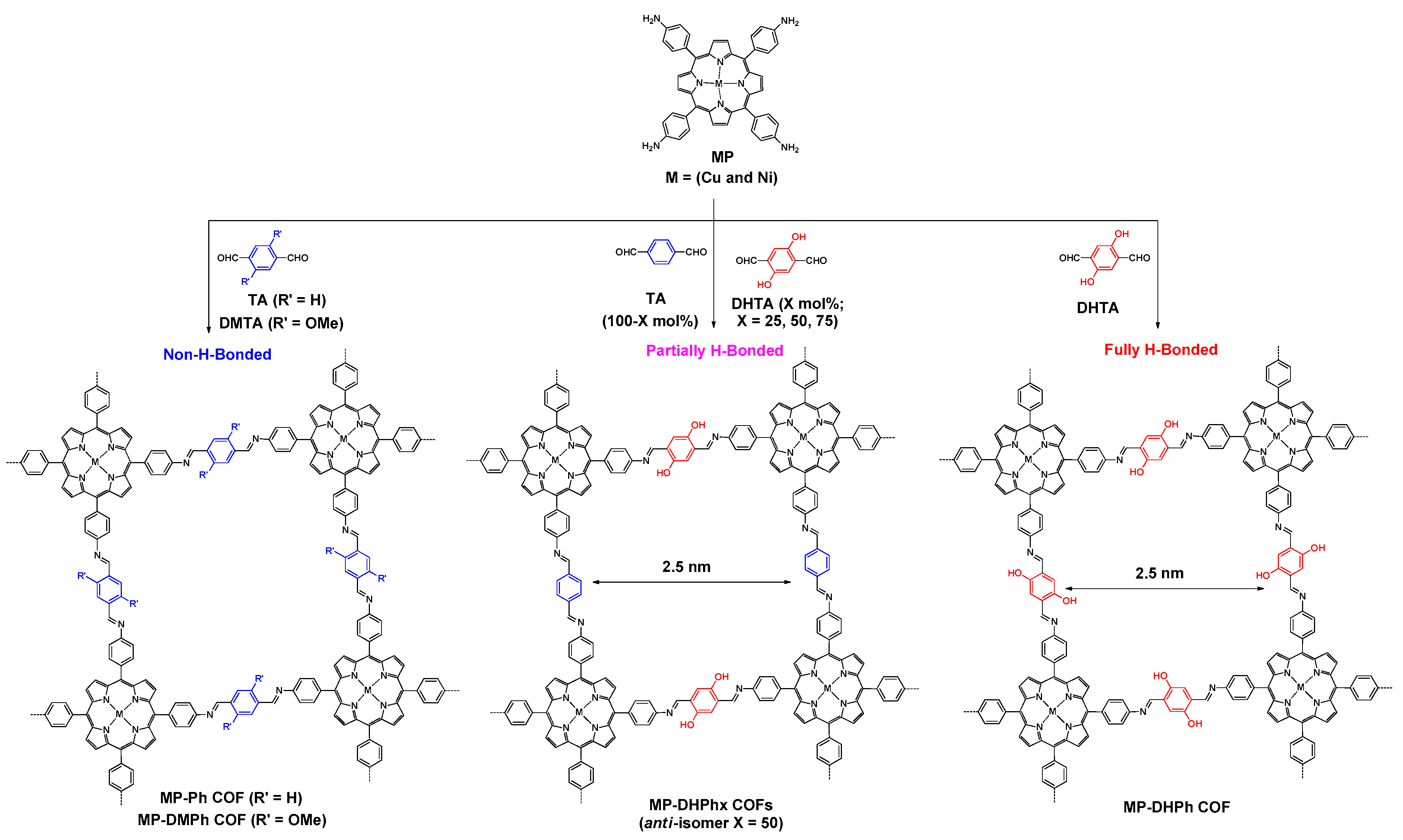
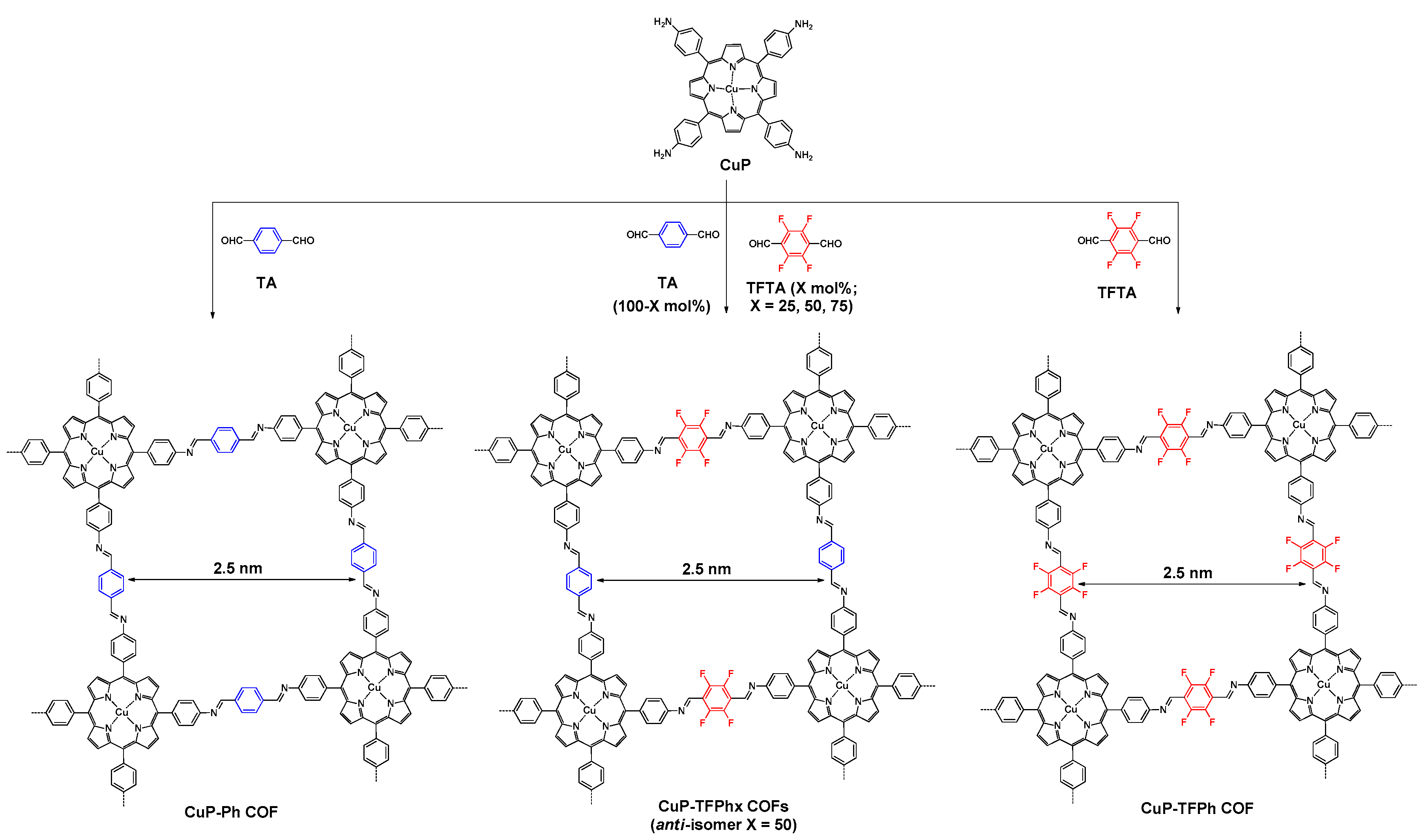
- Kandambeth, S.; Shinde, D.B.; Panda, M.K.; Lukose, B.; Heine, T.; Banerjee, R. Enhancement of chemical stability and crystallinity in porphyrin-containing covalent organic frameworks by intramolecular hydrogen bonds. Angew. Chem. Int. Ed. 2013, 52, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
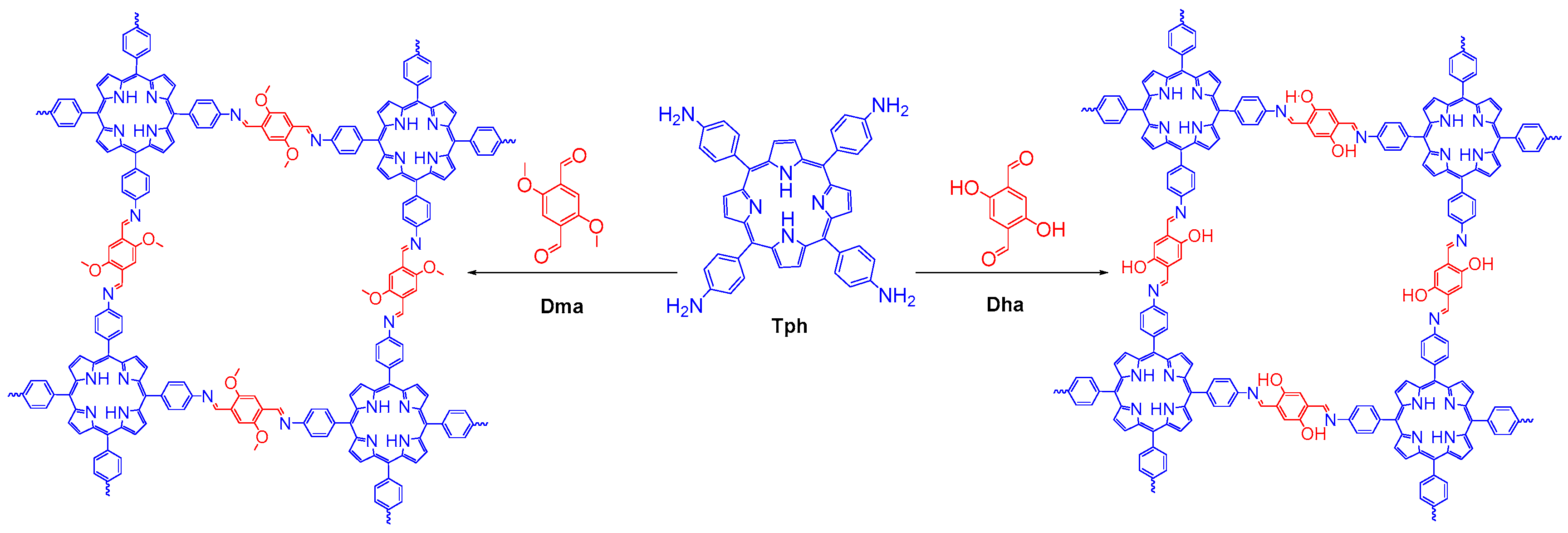
- Chen, X.; Addicoat, M.; Jin, E.; Zhai, L.; Xu, H.; Huang, N.; Guo, Z.; Liu, L.; Irle, S.; Jiang, D. Locking covalent organic frameworks with hydrogen bonds: General and remarkable effects on crystalline structure, physical properties, and photochemical activity. J. Am. Chem. Soc. 2015, 137, 3241–3247. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Addicoat, M.; Irle, S.; Nagai, A.; Jiang, D. Control of crystallinity and porosity of covalent organic frameworks by managing interlayer interactions based on self-complementary π-electronic force. J. Am. Chem. Soc. 2013, 135, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tao, S.S.; Jiang, D.L. Proton conduction in crystalline and porous covalent organic frameworks. Nat. Mater. 2016, 15, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Ascherl, L.; Sick, T.; Margraf, J.T.; Lapidus, S.H.; Calik, M.; Hettstedt, C.; Karaghiosoff, K.; Doblinger, M.; Clark, T.; Chapman, K.W.; et al. Molecular docking sites designed for the generation of highly crystalline covalent organic frameworks. Nat. Chem. 2016, 8, 310–316. [Google Scholar] [CrossRef]
- Auras, F.; Ascherl, L.; Haldmioun, A.H.; Margraf, J.T.; Hanusch, F.C.; Reuter, S.; Bessinger, D.; Doblinger, M.; Hettstedt, C.; Karaghiosoff, K.; et al. Synchronized offset stacking: A concept for growing large-domain and highly crystalline 2d covalent organic frameworks. J. Am. Chem. Soc. 2016, 138, 16703–16710. [Google Scholar] [CrossRef] [PubMed]
- Kandambeth, S.; Mallick, A.; Lukose, B.; Mane, M.V.; Heine, T.; Banerjee, R. Construction of crystalline 2d covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 2012, 134, 19524–19527. [Google Scholar] [CrossRef] [PubMed]
- Biswal, B.P.; Chandra, S.; Kandambeth, S.; Lukose, B.; Heine, T.; Banerjeet, R. Mechanochemical synthesis of chemically stable isoreticular covalent organic frameworks. J. Am. Chem. Soc. 2013, 135, 5328–5331. [Google Scholar] [CrossRef] [PubMed]
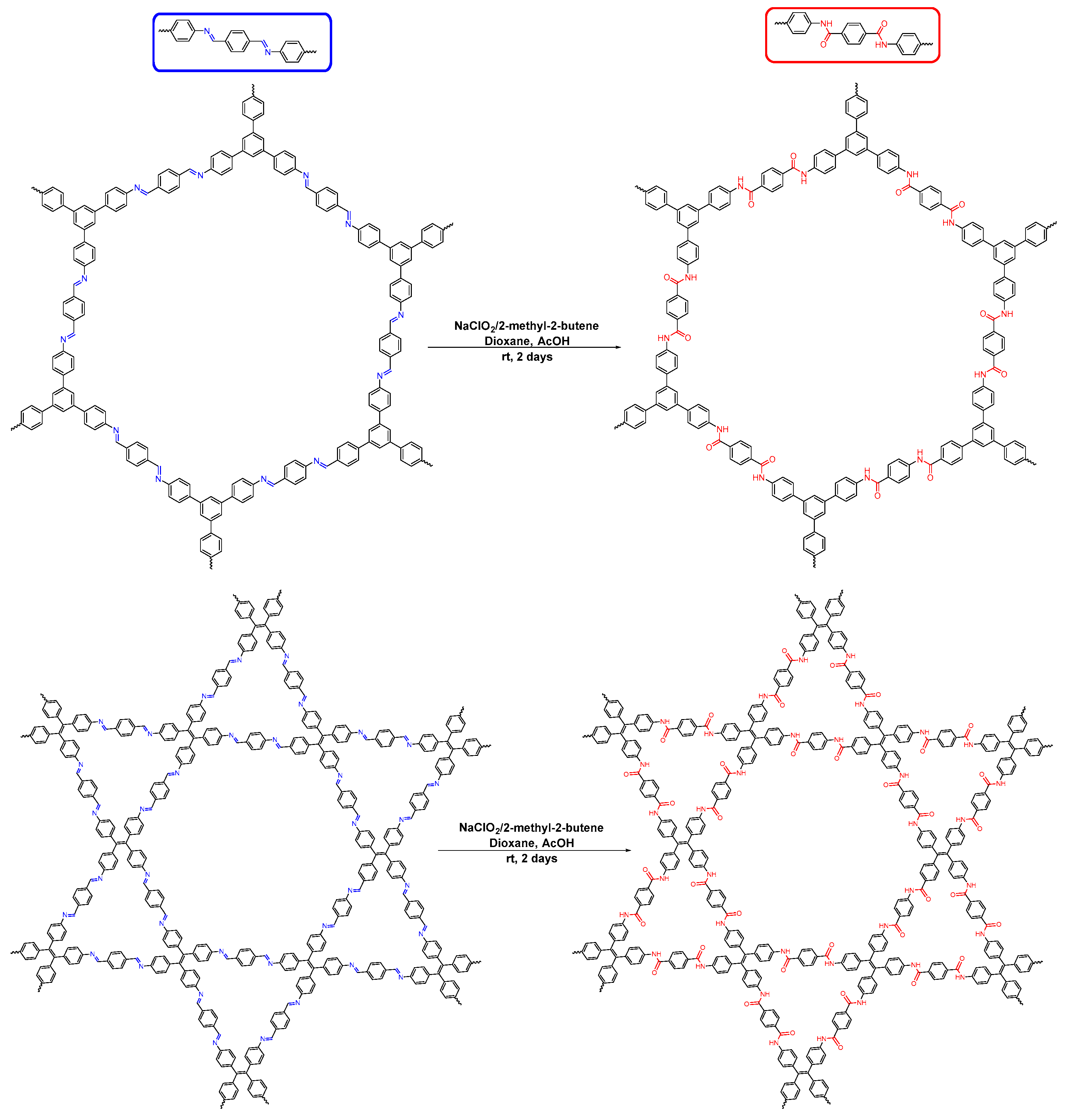
- Waller, P.J.; Lyle, S.J.; Popp, T.M.O.; Diercks, C.S.; Reimer, J.A.; Yaghi, O.M. Chemical conversion of linkages in covalent organic frameworks. J. Am. Chem. Soc. 2016, 138, 15519–15522. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Romo, F.J.; Doonan, C.J.; Furukawa, H.; Oisaki, K.; Yaghi, O.M. Crystalline covalent organic frameworks with hydrazone linkages. J. Am. Chem. Soc. 2011, 133, 11478–11481. [Google Scholar] [CrossRef] [PubMed]
- Bunck, D.N.; Dichtel, W.R. Bulk synthesis of exfoliated two-dimensional polymers using hydrazone-linked covalent organic frameworks. J. Am. Chem. Soc. 2013, 135, 14952–14955. [Google Scholar] [CrossRef] [PubMed]
- Stegbauer, L.; Schwinghammer, K.; Lotsch, B.V. A hydrazone-based covalent organic framework for photocatalytic hydrogen production. Chem. Sci. 2014, 5, 2789–2793. [Google Scholar] [CrossRef]
- Ding, S.Y.; Dong, M.; Wang, Y.W.; Chen, Y.T.; Wang, H.Z.; Su, C.Y.; Wang, W. Thioether-based fluorescent covalent organic framework for selective detection and facile removal of mercury(ii). J. Am. Chem. Soc. 2016, 138, 3031–3037. [Google Scholar] [CrossRef] [PubMed]
- Dalapati, S.; Jin, S.B.; Gao, J.; Xu, Y.H.; Nagai, A.; Jiang, D.L. An azine-linked covalent organic framework. J. Am. Chem. Soc. 2013, 135, 17310–17313. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.P.; Feng, X.; Zou, Y.C.; Zhang, Y.W.; Xia, H.; Liu, X.M.; Mu, Y. A 2d azine-linked covalent organic framework for gas storage applications. Chem. Commun. 2014, 50, 13825–13828. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.P.; Zhi, Y.F.; Feng, X.; Ding, X.S.; Zou, Y.C.; Liu, X.M.; Mu, Y. An azine-linked covalent organic framework: Synthesis, characterization and efficient gas storage. Chem.-Eur. J. 2015, 21, 12079–12084. [Google Scholar] [CrossRef] [PubMed]
- Alahakoon, S.B.; Thompson, C.M.; Nguyen, A.X.; Occhialini, G.; McCandless, G.T.; Smaldone, R.A. An azine-linked hexaphenylbenzene based covalent organic framework. Chem. Commun. 2016, 52, 2843–2845. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.S.; Haase, F.; Stegbauer, L.; Savasci, G.; Podjaski, F.; Ochsenfeld, C.; Lotsch, B.V. A tunable azine covalent organic framework platform for visible light-induced hydrogen generation. Nat. Commun. 2015, 6, 8508–8516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.B.; Li, N.; Liu, J.F.; Zhang, W.; Zhang, Z.B.; Zhou, N.C.; Zhu, X.L. A novel azobenzene covalent organic framework. Crystengcomm 2014, 16, 6547–6551. [Google Scholar] [CrossRef]
- Nagai, A.; Chen, X.; Feng, X.; Ding, X.S.; Guo, Z.Q.; Jiang, D.L. A squaraine-linked mesoporous covalent organic framework. Angew. Chem. Int. Ed. 2013, 52, 3770–3774. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.R.; Wang, J.H.; Gu, S.; Kaspar, R.B.; Zhuang, Z.B.; Zheng, J.; Guo, H.X.; Qiu, S.L.; Yan, Y.S. 3d porous crystalline polyimide covalent organic frameworks for drug delivery. J. Am. Chem. Soc. 2015, 137, 8352–8355. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.R.; Zhuang, Z.B.; Gu, S.; Kaspar, R.B.; Zheng, J.; Wang, J.H.; Qiu, S.L.; Yan, Y.S. Designed synthesis of large-pore crystalline polyimide covalent organic frameworks. Nat. Commun. 2014, 5, 4503–4510. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Xu, Y.H.; Guo, Z.Q.; Jiang, D.L. Supercapacitive energy storage and electric power supply using an aza-fused pi-conjugated microporous framework. Angew. Chem. Int. Edit. 2011, 50, 8753–8757. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, Y.H.; Jin, S.B.; Chen, L.; Kaji, T.; Honsho, Y.; Addicoat, M.A.; Kim, J.; Saeki, A.; Ihee, H.; et al. Conjugated organic framework with three-dimensionally ordered stable structure and delocalized pi clouds. Nat. Commun. 2013, 4, 2736–2743. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Antonietti, M.; Thomas, A. Porous, covalent triazine-based frameworks prepared by ionothermal synthesis. Angew. Chem. Int. Ed. 2008, 47, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Forget, A.; Su, D.; Thomas, A.; Antonietti, M. From microporous regular frameworks to mesoporous materials with ultrahigh surface area: Dynamic reorganization of porous polymer networks. J. Am. Chem. Soc. 2008, 130, 13333–13337. [Google Scholar] [CrossRef] [PubMed]
- Bojdys, M.J.; Jeromenok, J.; Thomas, A.; Antonietti, M. Rational extension of the family of layered, covalent, triazine-based frameworks with regular porosity. Adv. Mater. 2010, 22, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.J.; Bojdys, M.J.; Dawson, R.; Laybourn, A.; Khimyak, Y.Z.; Adams, D.J.; Cooper, A.I. Porous, fluorescent, covalent triazine-based frameworks via room-temperature and microwave-assisted synthesis. Adv. Mater. 2012, 24, 2357–2361. [Google Scholar] [CrossRef] [PubMed]
- Katekomol, P.; Roeser, J.; Bojdys, M.; Weber, J.; Thomas, A. Covalent triazine frameworks prepared from 1,3,5-tricyanobenzene. Chem. Mater. 2013, 25, 1542–1548. [Google Scholar] [CrossRef]
- Karmakar, A.; Kumar, A.; Chaudhari, A.K.; Samanta, P.; Desai, A.V.; Krishna, R.; Ghosh, S.K. Bimodal functionality in a porous covalent triazine framework by rational integration of an electron-rich and -deficient pore surface. Chem.-Eur. J. 2016, 22, 4931–4937. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.F.; Zou, R.Y.; Luo, Z.; Zhang, H.C.; Yao, X.; Ma, X.; Zou, R.Q.; Zhao, Y.L. Covalent organic frameworks formed with two types of covalent bonds based on orthogonal reactions. J. Am. Chem. Soc. 2015, 137, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Pan, Q.Y.; Ma, Y.C.; Guan, X.Y.; Xue, M.; Fang, Q.R.; Yan, Y.S.; Valtchev, V.; Qiu, S.L. Three-dimensional covalent organic frameworks with dual linkages for bifunctional cascade catalysis. J. Am. Chem. Soc. 2016, 138, 14783–14788. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Yaghi, O.M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 2009, 131, 8875–8883. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Lan, J.; Wang, W.; Smit, B. Lithium-doped 3D covalent organic frameworks: High-capacity hydrogen storage materials. Angew. Chem. Int. Ed. 2009, 48, 4730–4733. [Google Scholar] [CrossRef] [PubMed]
- Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Hydrogen storage in lithium-functionalized 3-D covalent-organic framework materials. J. Phys. Chem. C 2009, 113, 21253–21257. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, K.X.; Teng, B.; Zhang, T.; Han, Y. A perfluorinated covalent triazine-based framework for highly selective and water-tolerant CO2 capture. Energy Environ. Sci. 2013, 6, 3684–3692. [Google Scholar] [CrossRef]
- Huang, N.; Chen, X.; Krishna, R.; Jiang, D. Two-dimensional covalent organic frameworks for carbon dioxide capture through channel-wall functionalization. Angew. Chem. Int. Ed. 2015, 54, 2986–2990. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Krishna, R.; Jiang, D. Tailor-made pore surface engineering in covalent organic frameworks: Systematic functionalization for performance screening. J. Am. Chem. Soc. 2015, 137, 7079–7082. [Google Scholar] [CrossRef] [PubMed]



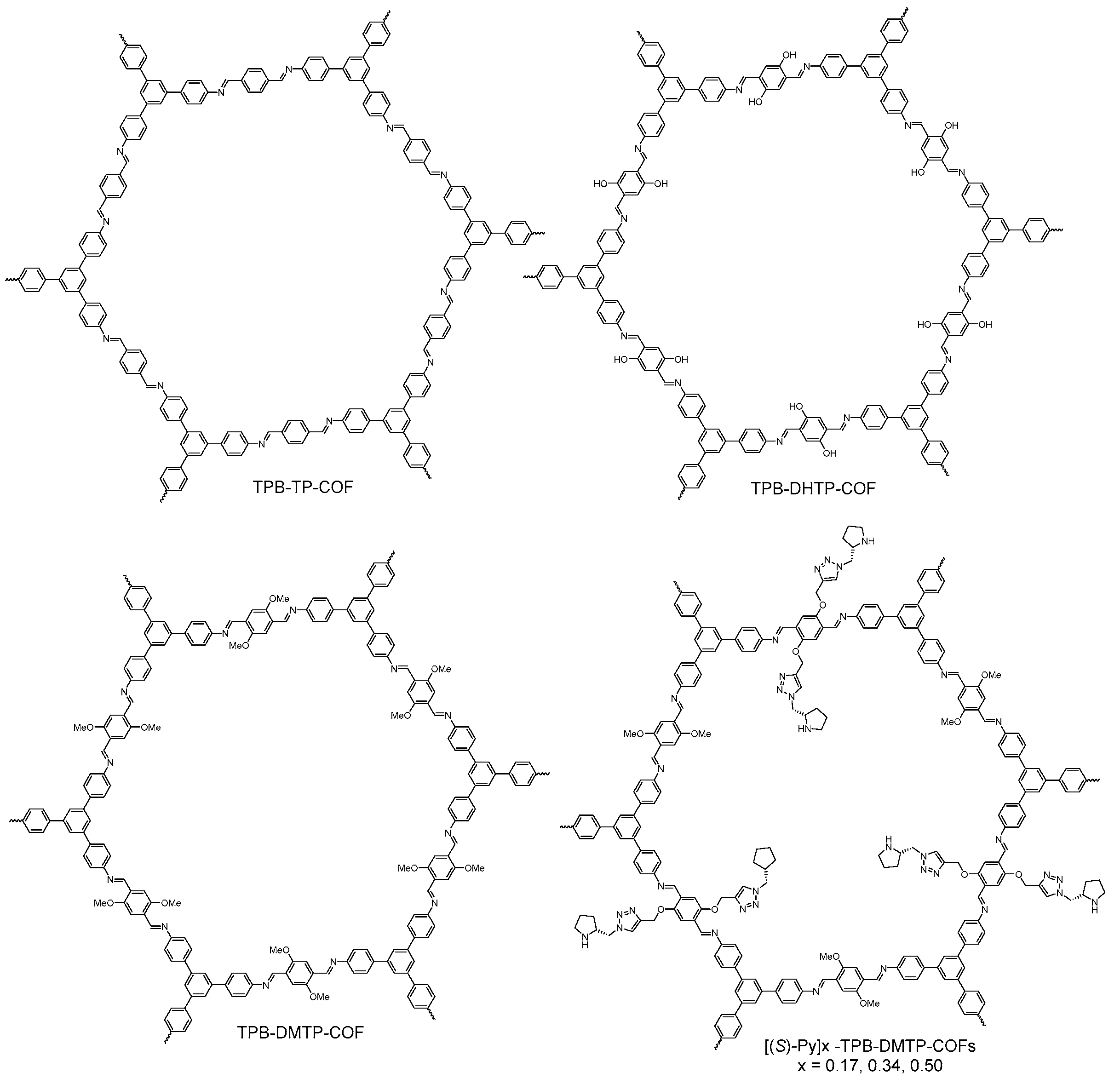

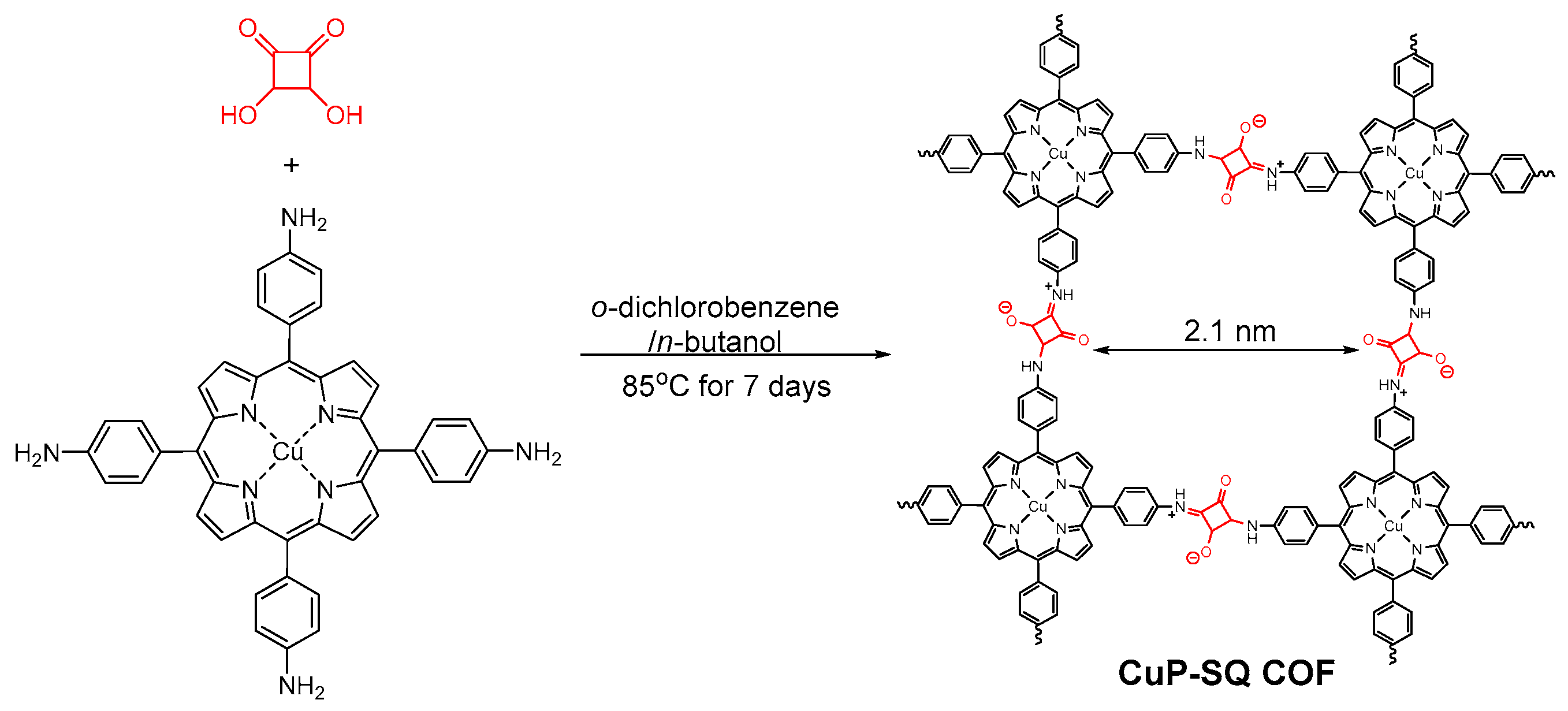


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
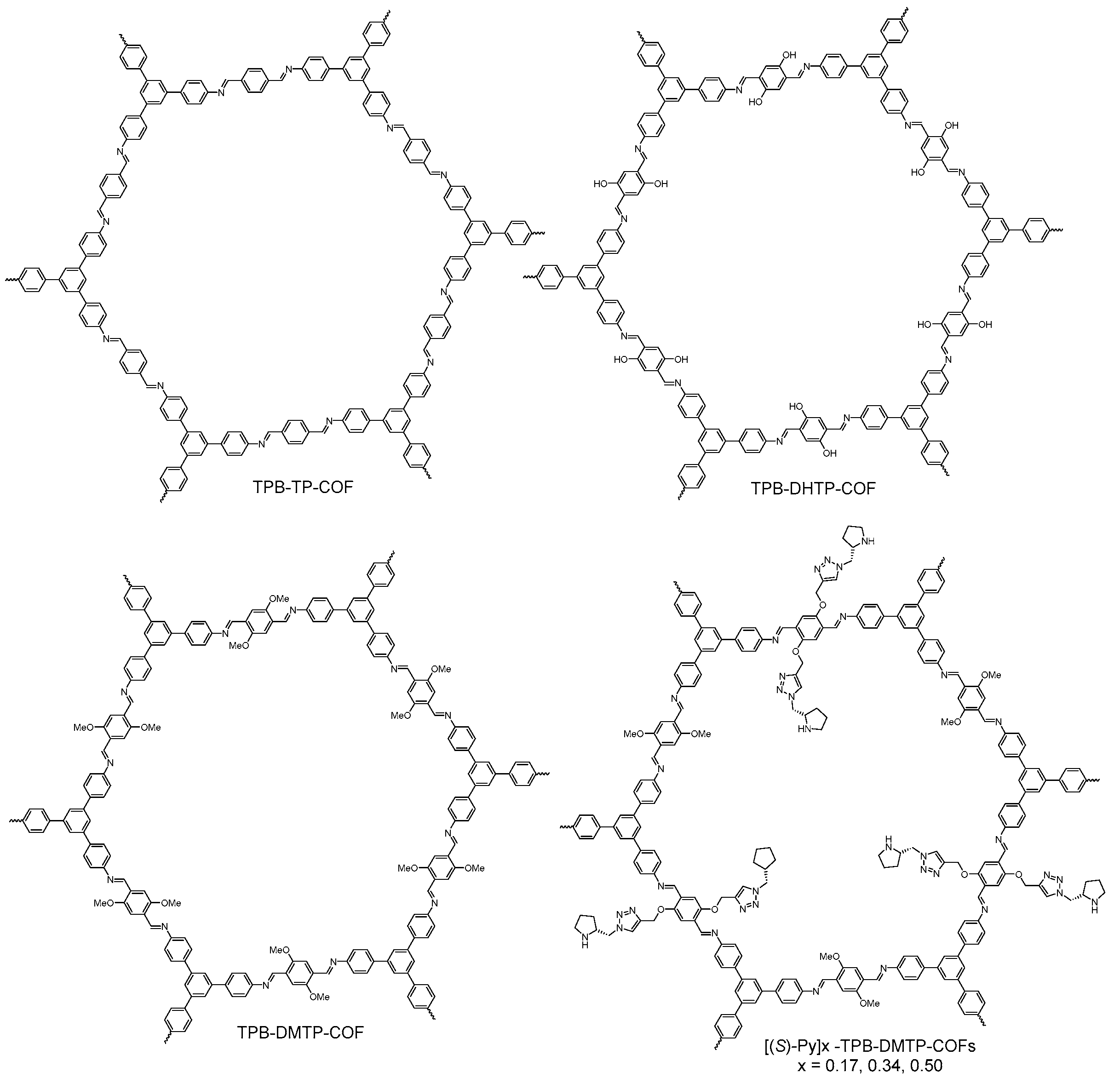{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
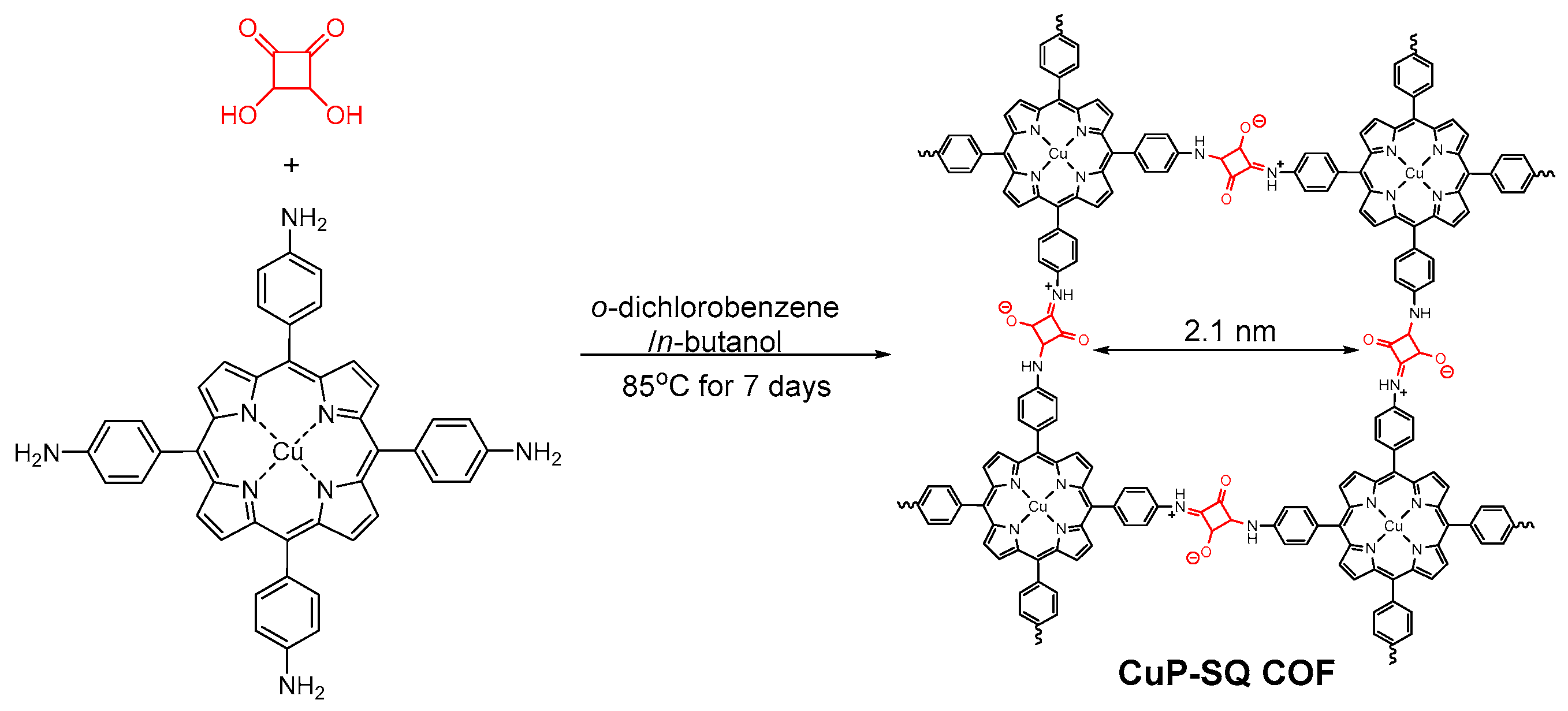{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COF Materials | BET Surface Area (m2·g−1) | Pore Size (Å) | Pore Volume (Vp, cm3·g−1) | H2 Uptake (wt %) | Ref. |
|---|---|---|---|---|---|
| COF-1 | 750 | 9 | 0.3 | 1.48 | [62] |
| COF-5 | 1670 | 27 | 1.07 | 3.58 | |
| COF-6 | 750 | 6.4 | 0.32 | 2.26 | |
| COF-8 | 1350 | 18.7 | 0.69 | 3.5 | |
| COF-10 | 1760 | 34.1 | 1.44 | 3.92 | |
| COF-102 | 3620 | 11.5 | 1.55 | 7.24 | |
| COF-103 | 3530 | 12.5 | 1.54 | 7.05 | |
| COF-1 | 628 | 9 | 0.36 | 1.28 (1 bar) | |
| COF-11Å | 105 | 11 | 0.05 | 1.22 (1 bar) | |
| COF-14Å | 805 | 14 | 0.41 | 1.23(1 bar) | |
| COF-16Å | 753 | 16 | 0.39 | 1.4 (1 bar) | |
| COF-18Å | 1263 | 18 | 0.69 | 1.55 (1 bar) | |
| CTC-COF | 1710 | 22.6 | 1.03 | 1.12 (1.05 bar) | |
| dualpore-COF | 1771 | 7.3, 25.2 | 3.189 (P/P° 0.99) | 1.37 (1 bar) |
| COF Materials | BET Surface Area (m2·g−1) | Pore Size (Å) | Pore Volume (Vp, cm3·g−1) | CH4 Uptake (mg·g−1) a | Ref. |
|---|---|---|---|---|---|
| COF-1 | 750 | 9 | 0.3 | 40 | [62] |
| COF-5 | 1670 | 27 | 1.07 | 89 | |
| COF-6 | 750 | 6.4 | 0.32 | 65 | |
| COF-8 | 1350 | 18.7 | 0.69 | 87 | |
| COF-10 | 1760 | 34.1 | 1.44 | 80 | |
| COF-102 | 3620 | 11.5 | 1.55 | 187 | |
| COF-103 | 3530 | 12.5 | 1.54 | 175 | |
| ILCOF-1 | 2723 | 23 | 1.21 | 129 (L·L−1) |
| COF Materials | BET Surface Area (m2·g−1) | Pore Size (nm) | Pore Volume (Vp, cm3·g−1) | CO2 Uptake (cm3·g−1) | Ref. | |
|---|---|---|---|---|---|---|
| Low a | High b | |||||
| COF-1 | 750 | 0.9 | 0.3 | 51 | 117 | [39] |
| COF-5 | 1670 | 2.7 | 1.07 | 31 | 443 | |
| COF-6 | 750 | 0.64 | 0.32 | 85 | 158 | |
| COF-8 | 1350 | 1.87 | 0.69 | 33 | 321 | |
| COF-10 | 1760 | 3.41 | 1.44 | 27 | 514 | |
| COF-102 | 3620 | 1.15 | 1.55 | 34 | 611 | |
| COF-103 | 3530 | 1.25 | 1.54 | 38 | 606 | |
| FCTF-1 | 662 | 0.46,0.54 | - | 105 | [65] | |
| FCTF-1-600 | 1535 | 0.46,0.59 | - | 124 | ||
| CTF-1 | 746 | 0.54 | 55 | |||
| CTF-1-600 | 1553 | - | 80 | |||
| [HO]25%-H2P-COF | 1054 | 2.5 | 27 | [66] | ||
| [HO]50%-H2P-COF | 1089 | 2.5 | 23 | |||
| [HO]75%-H2P-COF | 1153 | 2.5 | 26 | |||
| [HO]100%-H2P-COF | 1284 | 2.5 | 32 | |||
| [HO2C]25%-H2P-COF | 786 | 2.2 | 49 | |||
| [HO2C]50%-H2P-COF | 673 | 1.9 | 68 | |||
| [HO2C]75%-H2P-COF | 482 | 1.7 | 80 | |||
| [HO2C]100%-H2P-COF | 364 | 1.4 | 89 | |||
| [CH≡C]0-H2P-COF | 1474 | 2.5 | 0.75 | 37 | [67] | |
| [CH≡C]25-H2P-COF | 1413 | 2.3 | 0.71 | 27 | ||
| [CH≡C]50-H2P-COF | 962 | 2.1 | 0.57 | 24 | ||
| [CH≡C]75-H2P-COF | 683 | 1.9 | 0.42 | 22 | ||
| [CH≡C]100-H2P-COF | 462 | 1.6 | 0.28 | 20 | ||
| [Et]25-H2P-COF | 1326 | 2.2 | 0.55 | 28 | ||
| [Et]50-H2P-COF | 821 | 1.9 | 0.48 | 23 | ||
| [Et]75-H2P-COF | 485 | 1.6 | 0.34 | 21 | ||
| [Et]100-H2P-COF | 187 | 1.5 | 0.18 | 19 | ||
| [MeOAc]25-H2P-COF | 1238 | 2.1 | 0.51 | 43 | ||
| [MeOAc]50-H2P-COF | 754 | 1.8 | 0.42 | 45 | ||
| [MeOAc]75-H2P-COF | 472 | 1.5 | 0.31 | 42 | ||
| [MeOAc]100-H2P-COF | 156 | 1.1 | 0.14 | 33 | ||
| [AcOH]25-H2P-COF | 1252 | 2.2 | 0.52 | 48 | ||
| [AcOH]50-H2P-COF | 866 | 1.8 | 0.45 | 60 | ||
| [AcOH]75-H2P-COF | 402 | 1.5 | 0.32 | 55 | ||
| [AcOH]100-H2P-COF | 186 | 1.3 | 0.18 | 49 | ||
| [EtOH]25-H2P-COF | 1248 | 2.2 | 0.56 | 47 | ||
| [EtOH]50-H2P-COF | 784 | 1.9 | 0.43 | 63 | ||
| [EtOH]75-H2P-COF | 486 | 1.6 | 0.36 | 60 | ||
| [EtOH]100-H2P-COF | 214 | 1.4 | 0.19 | 43 | ||
| [EtNH2]25-H2P-COF | 1402 | 2.2 | 0.58 | 59 | ||
| [EtNH2]50-H2P-COF | 1044 | 1.9 | 0.5 | 80 | ||
| [EtNH2]75-H2P-COF | 568 | 1.6 | 0.36 | 68 | ||
| [EtNH2]100-H2P-COF | 382 | 1.3 | 0.21 | 49 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Zhang, Y.-B. Crystallization of Covalent Organic Frameworks for Gas Storage Applications. Molecules 2017, 22, 1149. https://doi.org/10.3390/molecules22071149
Zhu L, Zhang Y-B. Crystallization of Covalent Organic Frameworks for Gas Storage Applications. Molecules. 2017; 22(7):1149. https://doi.org/10.3390/molecules22071149
Chicago/Turabian StyleZhu, Lijuan, and Yue-Biao Zhang. 2017. "Crystallization of Covalent Organic Frameworks for Gas Storage Applications" Molecules 22, no. 7: 1149. https://doi.org/10.3390/molecules22071149
APA StyleZhu, L., & Zhang, Y.-B. (2017). Crystallization of Covalent Organic Frameworks for Gas Storage Applications. Molecules, 22(7), 1149. https://doi.org/10.3390/molecules22071149