Recent Advances in Conotoxin Classification by Using Machine Learning Methods



Abstract
:
1. Introduction
2. Benchmark Datasets
2.1. Published Database Resources
2.2. Benchmark Dataset Construction
3. Conotoxin Sample Description Methods
3.1. Amino Acid Compositions and Dipeptide Compositions
3.2. Pseudo Amino Acid Composition
3.3. Hybrid Features
4. Feature Selection Techniques
4.1. Binomial Distribution
4.2. Relief Algorithm
4.3. F-Score Algorithm
4.4. Diffusion Map Reduction
4.5. Analysis of Variance
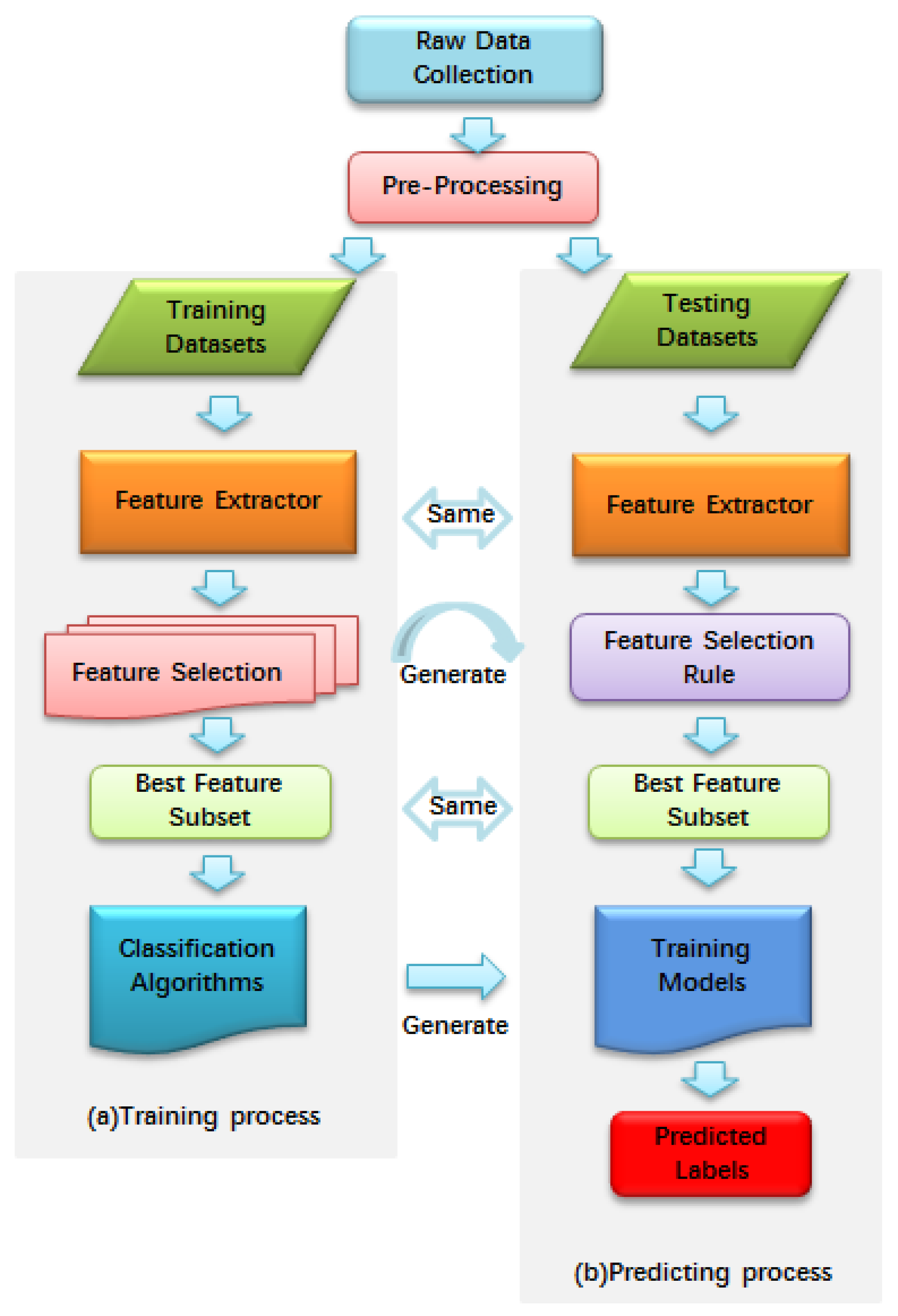
4.6. Feature Selection Process
5. Prediction Algorithms
5.1. Support Vector Machine
5.2. Profile Hidden Markov Models
5.3. K-Local Hyperplane Distance Nearest Neighbor Algorithm
5.4. Mahalanobis Discriminant
5.5. Radial Basis Function Network
5.6. Random Forest Algorithm
6. Prediction Accuracy
6.1. Commonly-Used Evaluation Metrics
6.2. Published Results
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kohn, A.J. The ecology of Conus in hawaii. Ecol. Monogr. 1959, 29, 47–90. [Google Scholar] [CrossRef]
- Daly, N.L.; Craik, D.J. Structural studies of conotoxins. IUBMB Life 2009, 61, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J. Conotoxins and their potential pharmaceutical applications. Drug Dev. 1999, 46, 219–234. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Phys. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Adams, D.J. Chemical modification of conotoxins to improve stability and activity. ACS Chem. Biol. 2007, 2, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Livett, B.G.; Gayler, K.R.; Khalil, Z. Drugs from the sea: Conopeptides as potential therapeutics. Curr. Med. Chem. 2004, 11, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.B.; Lopez-Vera, E.; Heimer de la Cotera, E.P.; Falcon, A.; Olivera, B.M.; Maillo, M. I-conotoxins in vermivorous species of the west atlantic: Peptide sr11a from Conus spurius. Peptides 2007, 28, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Vincler, M.; McIntosh, J.M. Targeting the alpha9alpha10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin. Ther. Targets 2007, 11, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Twede, V.D.; Miljanich, G.; Olivera, B.M.; Bulaj, G. Neuroprotective and cardioprotective conopeptides: An emerging class of drug leads. Curr. Opin. Drug Discov. Dev. 2009, 12, 231–239. [Google Scholar]
- Wang, Y.X.; Pettus, M.; Gao, D.; Phillips, C.; Scott Bowersox, S. Effects of intrathecal administration of ziconotide, a selective neuronal n-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain. Pain 2000, 84, 151–158. [Google Scholar] [CrossRef]
- Feng, W.H.; Zan, J.B.; Zhu, Y.P. Advances in study of structures and functions of conantokins. Zhejiang Da Xue Xue Bao Yi Xue Ban J. Zhejiang Univ. Med. Sci. 2007, 36, 204–208. [Google Scholar]
- Olivera, B.M.; Teichert, R.W. Diversity of the neurotoxic Conus peptides: A model for concerted pharmacological discovery. Mol. Interv. 2007, 7, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.E.; White, H.S.; Wilcox, K.S. The effect of cgx-1007 and ci-1041, novel nmda receptor antagonists, on nmda receptor-mediated epscs. Epilepsy Res. 2004, 59, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef] [PubMed]
- Pallaghy, P.K.; Alewood, D.; Alewood, P.F.; Norton, R.S. Solution structure of robustoxin, the lethal neurotoxin from the funnel-web spider atrax robustus. FEBS Lett. 1997, 419, 191–196. [Google Scholar] [CrossRef]
- Savarin, P.; Guenneugues, M.; Gilquin, B.; Lamthanh, H.; Gasparini, S.; Zinn-Justin, S.; Menez, A. Three-dimensional structure of kappa-conotoxin pviia, a novel potassium channel-blocking toxin from cone snails. Biochemistry 1998, 37, 5407–5416. [Google Scholar] [CrossRef] [PubMed]
- Botana, L.M. Seafood and freshwater toxins. Phytochemistry 2000, 60, 549–550. [Google Scholar]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon Off. J. Int. Soc. Toxinol. 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Bulaj, G. Conotoxins—New vistas for peptide therapeutics. Curr. Pharm. Des. 2000, 6, 1249–1285. [Google Scholar] [CrossRef] [PubMed]
- Mouhat, S.; Jouirou, B.; Mosbah, A.; De Waard, M.; Sabatier, J.M. Diversity of folds in animal toxins acting on ion channels. Biochem. J. 2004, 378, 717–726. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Jones, R.M. Cone venom—From accidental stings to deliberate injection. Toxicon Off. J. Int. Soc. Toxinol. 2001, 39, 1447–1451. [Google Scholar] [CrossRef]
- Rajendra, W.; Armugam, A.; Jeyaseelan, K. Toxins in anti-nociception and anti-inflammation. Toxicon Off. J. Int. Soc. Toxinol. 2004, 44, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Bhavna, R.; Mohan Babu, R.; Ramakumar, S. Pseudo amino acid composition and multi-class support vector machines approach for conotoxin superfamily classification. J. Theor. Biol. 2006, 243, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.B.; McDougal, O.M. The m-superfamily of conotoxins: A review. Cell. Mol. Life Sci. CMLS 2010, 67, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Corpuz, G.P.; Jacobsen, R.B.; Jimenez, E.C.; Watkins, M.; Walker, C.; Colledge, C.; Garrett, J.E.; McDougal, O.; Li, W.; Gray, W.R.; et al. Definition of the m-conotoxin superfamily: Characterization of novel peptides from molluscivorous Conus venoms. Biochemistry 2005, 44, 8176–8186. [Google Scholar] [CrossRef] [PubMed]
- Baldomero, M.; Olivera, B.M. Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar]
- Lewis, R.J. Conotoxins as selective inhibitors of neuronal ion channels, receptors and transporters. IUBMB Life 2004, 56, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of neuronal alpha7-nachr by alpha-conotoxin imi explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Mahdavi, S.; Kuyucak, S. Computational study of binding of mu-conotoxin giiia to bacterial sodium channels navab and navrh. Biochemistry 2016, 55, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, Q.Z. Predicting conotoxin superfamily and family by using pseudo amino acid composition and modified mahalanobis discriminant. Biochem. Biophys. Res. Commun. 2007, 354, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.X.; Song, J.; Shen, H.B.; Kong, X. Predcsf: An integrated feature-based approach for predicting conotoxin superfamily. Protein Pept. Lett. 2011, 18, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.; Wolfsheimer, S.; Nuel, G.; Khuri, S. Conotoxin protein classification using free scores of words and support vector machines. BMC Bioinform. 2011, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Nazar Zaki, F.S. Conotoxin protein classification using pairwise comparison and amino acid composition. In Proceedings of the Genetic & Evolutionary Computation Conference, Dublin, Ireand, 12–16 July 2011; Volume 5540, pp. 323–330. [Google Scholar]
- Yin, J.B.; Fan, Y.X.; Shen, H.B. Conotoxin superfamily prediction using diffusion maps dimensionality reduction and subspace classifier. Curr. Protein Pept. Sci. 2011, 12, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Laht, S.; Koua, D.; Kaplinski, L.; Lisacek, F.; Stocklin, R.; Remm, M. Identification and classification of conopeptides using profile hidden markov models. Biochim. Biophys. Acta 2012, 1824, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Brauer, A.; Laht, S.; Kaplinski, L.; Favreau, P.; Remm, M.; Lisacek, F.; Stocklin, R. Conodictor: A tool for prediction of conopeptide superfamilies. Nucleic Acids Res. 2012, 40, W238–W241. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Laht, S.; Kaplinski, L.; Stocklin, R.; Remm, M.; Favreau, P.; Lisacek, F. Position-specific scoring matrix and hidden markov model complement each other for the prediction of conopeptide superfamilies. Biochim. Biophys. Acta 2013, 1834, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Gowd, K.H.; Dewan, K.K.; Iengar, P.; Krishnan, K.S.; Balaram, P. Probing peptide libraries from Conus achatinus using mass spectrometry and cdna sequencing: Identification of delta and omega-conotoxins. J. Mass Spectrom. JMS 2008, 43, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.F.; Ding, C.; Guo, S.H.; Ding, H.; Chen, W.; Lin, H. Prediction of the types of ion channel-targeted conotoxins based on radial basis function network. Toxicol. Int. J. Publ. Assoc. BIBRA. 2013, 27, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Deng, E.Z.; Yuan, L.F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.C. Ictx-type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. BioMed Res. Int. 2014, 2014, 286419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Gao, R.; Yang, R.; Song, Q. Using the smote technique and hybrid features to predict the types of ion channel-targeted conotoxins. J. Theor. Biol. 2016, 403, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zheng, Y.; Tang, H. Identifying the types of ion channel-targeted conotoxins by incorporating new properties of residues into pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 3981478. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Wang, X.; Zhang, Y. Predicting the types of ion channel-targeted conotoxins based on avc-svm model. BioMed Res. Int. 2017, 2017, 2929807. [Google Scholar]
- He, B.; Chai, G.; Duan, Y.; Yan, Z.; Qiu, L.; Zhang, H.; Liu, Z.; He, Q.; Han, K.; Ru, B.; et al. Biopanning data bank. Nucleic Acids Res. 2016, 44, D1127–D1132. [Google Scholar] [CrossRef] [PubMed]
- Ru, B.; Huang, J.; Dai, P.; Li, S.; Xia, Z.; Ding, H.; Lin, H.; Guo, F.; Wang, X. Mimodb: A new repository for mimotope data derived from phage display technology. Molecules 2010, 15, 8279–8288. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ru, B.; Zhu, P.; Nie, F.; Yang, J.; Wang, X.; Dai, P.; Lin, H.; Guo, F.B.; Rao, N. Mimodb 2.0: A mimotope database and beyond. Nucleic Acids Res. 2012, 40, D271–D277. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.Y.; Lai, H.Y.; Yang, H.; Zhang, C.J.; Yang, H.; Wei, H.H.; Chen, X.X.; Zhao, Y.W.; Su, Z.D.; Li, W.C.; et al. Pro54db: A database for experimentally verified sigma-54 promoters. Bioinformatics 2017, 33, 467–469. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The rcsb protein data bank: Integrative view of protein, gene and 3d structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [PubMed]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. Conoserver: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. Conoserver, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Xu, Y.; Fang, X.; Zheng, C.; Liu, B. Protein fold recognition based on sparse representation based classification. Artif. Intell. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using chou’s pseudo amino acid composition with feature selection technique. Mol. Biosyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-one: A web server for generating various modes of pseudo components of DNA, rna, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Mathura, V.S.; Kolippakkam, D. Apdbase: Amino acid physico-chemical properties database. Bioinformation 2005, 1, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Leise, T.L. Wavelet-based analysis of circadian behavioral rhythms. Methods Enzymol. 2015, 551, 95–119. [Google Scholar] [PubMed]
- Ding, C.; Yuan, L.F.; Guo, S.H.; Lin, H.; Chen, W. Identification of mycobacterial membrane proteins and their types using over-represented tripeptide compositions. J. Proteom. 2012, 77, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.K.; Street, W.N.; Menczer, F. Feature selection in data mining. Data Min. Oppor. Chall. 2003, 9, 80–105. [Google Scholar]
- Rocchi, L.; Chiari, L.; Cappello, A. Feature selection of stabilometric parameters based on principal component analysis. Med. Biol. Eng. Comput. 2004, 42, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Lin, H.; Ding, H. Predicting ion channels and their types by the dipeptide mode of pseudo amino acid composition. J. Theor. Biol. 2011, 269, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kira, K.; Rendell, L.A. He feature selection problem: Traditional methods and a new algorithm. In Proceedings of the Tenth National Conference on Artificial Intelligence, San Jose, CA, USA, 12–16 July 1992; pp. 129–134. [Google Scholar]
- Sun, Y. Iterative relief for feature weighting: Algorithms, theories, and applications. IEEE Trans. Pattern Anal. Mach. Intell. 2007, 29, 1035–1051. [Google Scholar] [CrossRef] [PubMed]
- Lafon, S.; Lee, A.B. Diffusion maps and coarse-graining: A unified framework for dimensionality reduction, graph partitioning, and data set parameterization. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Xie, S.; Lin, Z.; Wu, M.; Ju, Y. Finding the best classification threshold in imbalanced classification. Big Data Res. 2016, 5, 2–8. [Google Scholar] [CrossRef]
- Ding, H.; Li, D. Identification of mitochondrial proteins of malaria parasite using analysis of variance. Amino Acids 2015, 47, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zou, P.; Zhang, C.; Chen, R.; Chen, W.; Lin, H. Identification of apolipoprotein using feature selection technique. Sci. Rep. 2016, 6, 30441. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Tang, H.; Li, W.C.; Wu, H.; Chen, W.; Ding, H.; Lin, H. Identification of bacterial cell wall lyases via pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 1654623. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, H.; Chen, X.X.; Zhang, C.J.; Zhu, P.P.; Ding, H.; Chen, W.; Lin, H. Identification of secretory proteins in mycobacterium tuberculosis using pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 5413903. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tang, H.; Chen, W.; Lin, H. Predicting human enzyme family classes by using pseudo amino acid composition. Curr. Proteom. 2016, 13, 99–104. [Google Scholar] [CrossRef]
- Zhao, Y.W.; Lai, H.Y.; Tang, H.; Chen, W.; Lin, H. Prediction of phosphothreonine sites in human proteins by fusing different features. Sci. Rep. 2016, 6, 34817. [Google Scholar] [CrossRef] [PubMed]
- Vapnik, V.N.; Vapnik, V. Statistical Learning Theory; John Wiley and Sons Inc.: New York, NY, USA, 1998. [Google Scholar]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liang, Z.Y.; Tang, H.; Chen, W. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.C. IRNA-pseu: Identifying rna pseudouridine sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar] [PubMed]
- Lai, H.Y.; Chen, X.X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169–28175. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Lin, H. Methyrna: A web server for identification of n6-methyladenosine sites. J. Biomol. Struct. Dyn. 2017, 35, 683–687. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Kang, J.; Ru, B.; Ding, H.; Zhou, P.; Huang, J. Sabinder: A web service for predicting streptavidin-binding peptides. BioMed Res. Int. 2016, 2016, 9175143. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Nie, F.; Kang, J.; Ding, H.; Zhou, P.; Huang, J. Nieluter: Predicting peptides eluted from hla class i molecules. J. Immunol. Methods 2015, 422, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Ru, B.; Pa, T.H.; Nie, F.; Lin, H.; Guo, F.B.; Huang, J. Phd7faster: Predicting clones propagating faster from the ph.D.-7 phage display peptide library. J. Bioinform. Comput. Biol. 2014, 12, 1450005. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.; Liu, F.; Wang, X.; Chen, J.; Chou, K.C. Identification of real microrna precursors with a pseudo structure status composition approach. PLoS ONE 2015, 10, e0121501. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ju, Y.; Zou, Q. Protein folds prediction with hierarchical structured svm. Curr. Proteom. 2016, 13, 79–85. [Google Scholar] [CrossRef]
- Chang, C.C.; Hsu, C.W.; Lin, C.J. The analysis of decomposition methods for support vector machines. IEEE Trans. Neural Netw. 2000, 11, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Pedrycz, W. Advances in kernel methods: Support vector learning. Neurocomputing 2002, 47, 303–304. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput. Biol. 2008, 4, e1000069. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Eddy, S.R. Nhmmer: DNA homology search with profile hmms. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Yu, M.; Jiang, L.; Duan, Y.; Huang, J. Hmmcas: A web tool for the identification and domain annotations of cas proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Boudaren, E.M.Y.; Pieczynski, W. Dempster-shafer fusion of multisensor signals in nonstationary markovian context. EURASIP J. Adv. Signal Process. 2012, 2012, 134. [Google Scholar] [CrossRef]
- Boudaren, M.E.; Monfrini, E.; Pieczynski, W. Unsupervised segmentation of random discrete data hidden with switching noise distributions. IEEE Signal Process. Lett. 2012, 19, 619–622. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Vincent, P.; Bengio, Y. K-local hyperplane and convex distance nearest neighbor algorithms. Adv. Neural Inf. Process. Syst. 2002, 14, 985–992. [Google Scholar]
- Mahalanobis, P.C. On the generalised distance in statistics. Proc. Natl. Inst. Sci. India 1936, 2, 49–55. [Google Scholar]
- Lin, H. The modified mahalanobis discriminant for predicting outer membrane proteins by using chou’s pseudo amino acid composition. J. Theor. Biol. 2008, 252, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Luo, L. Use of tetrapeptide signals for protein secondary-structure prediction. Amino Acids 2008, 35, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Ou, Y.Y.; Lee, T.Y.; Gromiha, M.M. Prediction of transporter targets using efficient rbf networks with pssm profiles and biochemical properties. Bioinformatics 2011, 27, 2062–2067. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, J.; Xuan, P.; Zou, Q. Bp neural network could help improve pre-mirna identification in various species. BioMed Res. Int. 2016, 2016, 9565689. [Google Scholar] [CrossRef] [PubMed]
- Witten, I.H.; Frank, E. Data Mining: Practical Machine Learning Tools and Techniques; MorganKaufmann: SanFrancisco, CA, USA, 2005. [Google Scholar]
- Zhang, C.J.; Tang, H.; Li, W.C.; Lin, H.; Chen, W.; Chou, K.C. Iori-human: Identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 2016, 7, 69783–69793. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Ju, Y.; Zou, Q. Prediction of g protein-coupled receptors with svm-prot features and random forest. Scientifica 2016, 2016, 8309253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zou, Q.; Liu, B.; Liu, X. Exploratory predicting protein folding model with random forest and hybrid features. Curr. Proteom. 2014, 11, 289–299. [Google Scholar] [CrossRef]
- Liu, B.; Long, R.; Chou, K.C. Idhs-el: Identifying dnase i hypersensitive-sites by fusing three different modes of pseu-do nucleotide composition into an ensemble learning framework. Bioinformatics 2016, 32, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Zhang, C.T. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.; Liu, F.; Wang, X.; Chou, K.C. Imirna-psedpc: Microrna precursor identification with a pseudo distance-pair composition approach. J. Biomol. Struct. Dyn. 2016, 34, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Metz, C.E. Some practical issues of experimental design and data analysis in radiological roc studies. Investig. Radiol. 1989, 24, 234–245. [Google Scholar] [CrossRef]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden markov model speed heuristic and iterative hmm search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
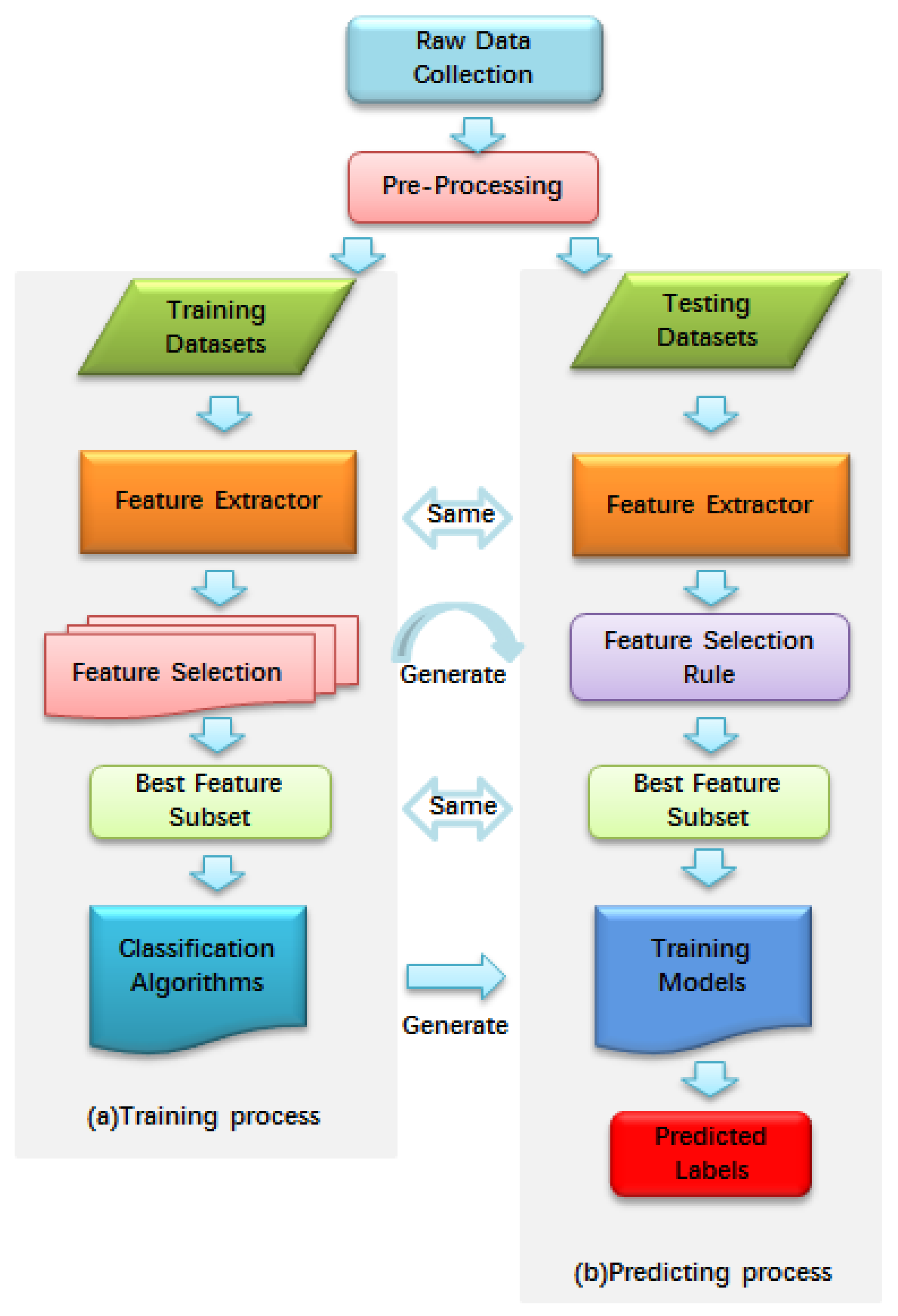{kind=link}
| Superfamily | Total Number | Reference | ||||
| A | M | O | T | |||
| S1 | 25 | 13 | 16 | 17 | 116 | [24,32,34,35] |
| S2 | 63 | 48 | 95 | 55 | 216 | [33,36] |
| Type of Ion Channel | Total Number | Reference | ||||
| K-Conotoxin | Na-Conotoxin | Ca-Conotoxin | ||||
| I1 | 24 | 43 | 45 | 112 | [41,42,44,45] | |
| I2 | 26 | 49 | 70 | 145 | [43] | |
| Superfamily Prediction | Reference | |||||||
| Dataset | Methods | A | M | O | T | AA | OA | |
| S1 | Multi-class SVMs | 0.840 | 0.923 | 0.869 | 0.941 | 0.893 | 0.881 | [24] |
| IDQD | 0.960 | 0.923 | 0.820 | 0.940 | 0.911 | 0.883 | [32] | |
| SVM-Freescore | 0.960 | 0.984 | 0.984 | 1 | 0.982 | 0.974 | [34] | |
| Toxin-AAM | 0.957 | 0.966 | 0.891 | 0.966 | 0.945 | 0.966 | [35] | |
| S2 | PredCFS | 0.960 | 0.984 | 0.984 | 1 | 0.982 | 0.903 | [33] |
| dHKNN | 0.957 | 0.966 | 0.891 | 0.966 | 0.945 | 0.919 | [36] | |
| Type of Ion Channel-Targeted Prediction | Reference | |||||||
| Dataset | Methods | K-Conotoxin | Na-Conotoxin | Ca-Conotoxin | AA | OA | ||
| I1 | RBF network | 0.917 | 0.884 | 0.889 | 0.897 | 0.893 | [41] | |
| iCTX-Type | 0.833 | 0.978 | 0.898 | 0.903 | 0.911 | [42] | ||
| Fscore-SVM | 0.917 | 0.953 | 0.953 | 0.942 | 0.946 | [44] | ||
| AVC-SVM | 0.931 | 0.942 | 0.892 | 0.922 | 0.920 | [45] | ||
| I2 | ICTCPred | 1 | 0.919 | 1 | 0.973 | 0.957 | [43] | |
| Name | Prediction Type | URL | Reference |
|---|---|---|---|
| PredCSF | Superfamily | http://www.csbio.sjtu.edu.cn/bioinf/PredCSF/ | [33] |
| ConoDictor | Superfamily | http://conco.ebc.ee | [38] |
| iCTX-Type | ion channel-target | http://lin.uestc.edu.cn/server/iCTX-Type | [42] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dao, F.-Y.; Yang, H.; Su, Z.-D.; Yang, W.; Wu, Y.; Hui, D.; Chen, W.; Tang, H.; Lin, H. Recent Advances in Conotoxin Classification by Using Machine Learning Methods. Molecules 2017, 22, 1057. https://doi.org/10.3390/molecules22071057
Dao F-Y, Yang H, Su Z-D, Yang W, Wu Y, Hui D, Chen W, Tang H, Lin H. Recent Advances in Conotoxin Classification by Using Machine Learning Methods. Molecules. 2017; 22(7):1057. https://doi.org/10.3390/molecules22071057
Chicago/Turabian StyleDao, Fu-Ying, Hui Yang, Zhen-Dong Su, Wuritu Yang, Yun Wu, Ding Hui, Wei Chen, Hua Tang, and Hao Lin. 2017. "Recent Advances in Conotoxin Classification by Using Machine Learning Methods" Molecules 22, no. 7: 1057. https://doi.org/10.3390/molecules22071057
APA StyleDao, F.-Y., Yang, H., Su, Z.-D., Yang, W., Wu, Y., Hui, D., Chen, W., Tang, H., & Lin, H. (2017). Recent Advances in Conotoxin Classification by Using Machine Learning Methods. Molecules, 22(7), 1057. https://doi.org/10.3390/molecules22071057