3.2. Building Blocks
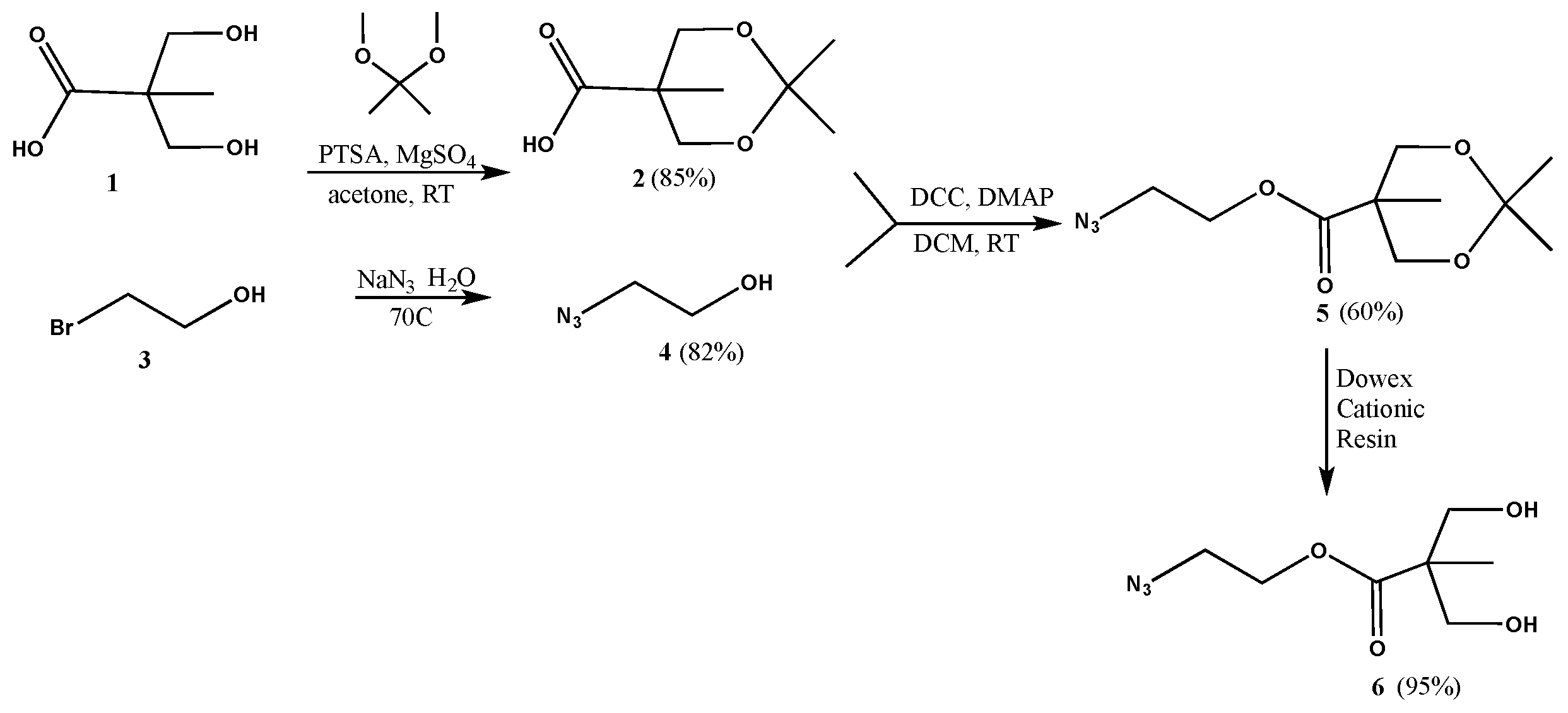
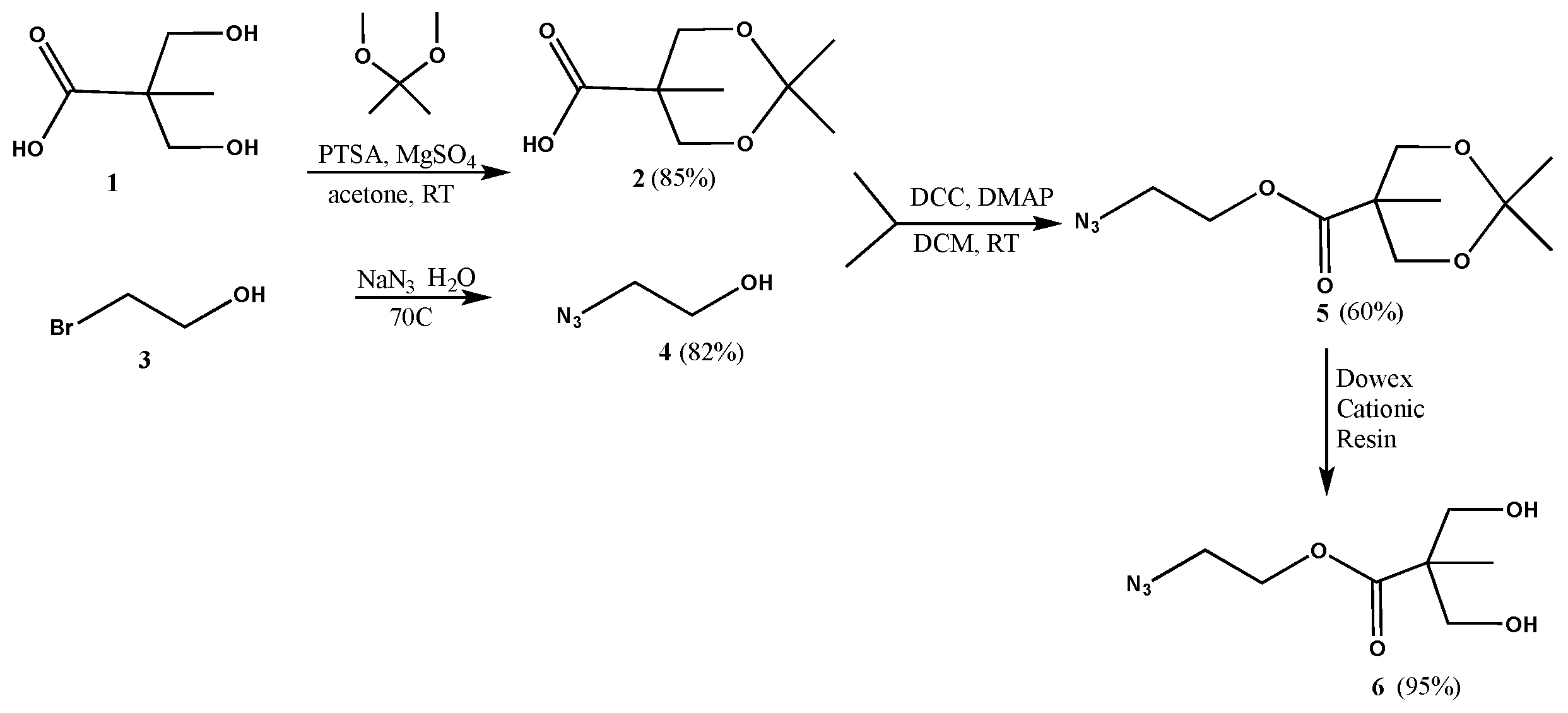
2,2,5-Trimethyl-1,3-dioxane-5-carboxylic acid (2): p-Toluenesulfonic acid (0.5975 g, 0.003141 mol) was added to a stirred solution of 2,2-bis(hydroxymethyl)propanoic acid (1, 8.4265 g, 0.06282 mol) in acetone (34 mL) in a 250 mL round bottom flask, under nitrogen. 2,2-Dimethoxypropane (9.8141 g, 0.09423 mol) and magnesium sulfate (0.7562 g, 0.006282 mol) were then added, and the reaction mixture was left to stir for 48 h. A solution of ammonia in dioxane (6.27 mL, 0.5 M) was added to neutralize the acid. The crude mixture was filtered, and the solvent evaporated to yield the product as a white powder (9.30 g, 0.0534 mol, 85% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.20 (s, 3H, -CO-C-CH3), 1.40 (s, 3H, -O-C-CH3), 1.43 (s, 3H, -O-C-CH3), 3.67 (t, 2H, -O-CH2-C-CO-), 4.18 (d, 2H, -O-CH2-C-CO-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 18.42 (-CO-C-CH3), 22.03 (-O-C-CH3), 25.07 (-O-C-CH3), 41.71 (-CO-C-), 65.81 (-O-CH2-C-), 98.28 (-O-C-(CH3)2), 180.15 (-CO-) ppm.
Azidoethanol (4): A mixture of bromoethanol (3, 2.3742 g, 0.01900 mol) and sodium azide (2.4700 g, 0.03800 mol) in water (4 mL) was left stirring overnight at 70 °C. The reaction mixture was then extracted with DCM, the organic layer was isolated and dried with magnesium sulfate. The solvent was then evaporated to yield the product as yellow oil (1.36 g, 0.01561 mol, 82% yield). 1H-NMR (400 MHz, CDCl3): δ = 3.46 (t, 2H, N3-CH2-CH2-), 3.79 (q, 2H, N3-CH2-CH2-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 53.6 (N3-CH2-CH2-), 61.6 (N3-CH2-CH2-) ppm.
2-Azidoethyl 2,2,5-trimethyl-1,3-dioxane-5-carboxylate (5): A solution of azidoethanol (4, 0.74 g, 0.008498 mol), 2,2,5-trimethyl-1,3-dioxane-5-carboxylic acid (2, 2.2204 g, 0.01275 mol) and DMAP (0.5191 g, 0.004249 mol) in anhydrous DCM (10 mL) was stirred for 5 min under nitrogen. DCC (2.1041 g, 0.01020 mol) was then added to the reaction mixture, and stirred under nitrogen, at room temperature, overnight. The precipitate was filtered off, and the solvent was evaporated to yield a residue that was purified by column chromatography (1:7 ethyl acetate–hexanes) to yield the product as a white solid (1.25 g, 0.005139 mol, 60% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.21 (s, 3H, -CO-C-CH3), 1.39 (s, 3H, -O-C-CH3), 1.44 (s, 3H, -O-C-CH3), 3.49 (t, 2H, N3-CH2-CH2-), 3.68 (d, 2H, -O-CH2-C-CO-), 4.21 (d, 2H, -O-CH2-C-CO-), 4.33 (t, 2H, N3-CH2-CH2-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 18.5 (-CO-C-CH3), 22.3 (-O-C-CH3), 24.9 (-O-C-CH3), 42.0 (-C-CH3), 49.8 (N3-CH2-CH2-), 63.6 (N3-CH2-CH2-), 65.9 (-O-CH2-C-), 98.1 (-C-CH3), 174.0 (-CO-) ppm.
2-Azidoethyl 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate (6): A spoonful of Dowex cationic resin was added to a solution of 5 (0.301 g, 0.001728 mol) in methanol (6 mL). The mixture was left stirring overnight at room temperature. The supernatant was decanted off and the resin was rinsed several times with methanol. The supernatant was then combined with rinsing and the solvent was evaporated to yield the product as colorless oil (0.238 g, 0.001171 mol, 95% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.09 (s, 3H, -CO-C-CH3), 2.47 (s, 2H, -OH), 3.41 (m, 2H, N3-CH2-CH2-), 3.50 (dd, 2H, -C-CH2-OH), 3.80 (dd, 2H, -C-CH2-OH), 4.28 (t, 2H, N3-CH2-CH2-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 17.0 (CH3-C-), 49.3 (CH3-C), 49.8 (N3-CH2-CH2-), 63.5 (N3-CH2-CH2-), 68.2 (CH2-OH), 175.5 (-CO-) ppm.
Generation 0 (7): Anhydrous DMF (25 mL) was added by syringe to a round bottom flask containing pentaerythritol (2 g, 0.01469 mol) and KOH (12.5 g, 0.2228 mol), with stirring, under nitrogen. The mixture was stirred for 5 min, under nitrogen, at 0 °C. A solution of propargyl bromide (20 g, 0.1681 mol) in toluene (80%) was then added dropwise to the reaction mixture over 30 min. The reaction was heated at 40 °C overnight, under nitrogen. Water (100 mL) was added to the mixture, which was then extracted with diethyl ether (3 × 50 mL). The organic layers were isolated, combined, and washed with water (3 × 50 mL) and brine (3 × 50 mL). The organic layer was isolated and dried with sodium sulfate. The solvent was removed to yield an orange oil which was purified by column chromatography (2:8 ethyl acetate–hexanes) to give the product as an orange-brown solid (1.7468 g, 0.006058 mol, 84% yield). 1H-NMR (400 MHz, CDCl3): δ = 2.40 (t, 4H, CH-C-CH2-), 3.53 (s, 8H, -C-CH2-O-), 4.12 (d, 8H, CH-C-CH2-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 44.7 (-C-CH2-O-), 58.7 (CH-C-CH2), 69.0 (-C-CH2-O-), 74.0 (CH-C-CH2), 80.0 (CH-C-CH2) ppm. ESI-MS: m/z = 311.1 [M + Na+].
Protected Generation 1 (pG1, 9): A solution of CuSO4·5H2O (0.0191 g, 0.00007669 mol) in water (0.5 mL) was added to a round bottom flask containing a stirred solution of 7 (0.0768 g, 0.0002663 mol) and (5) (0.2683 g, 0.001103 mol) in tetrahydrofuran (THF) (0.5 mL). Sodium ascorbate (0.0317 g, 0.0001598 mol) was added, and the mixture was allowed to react overnight. The product was purified by column chromatography (100% methanol) to yield a yellow oil (0.2778 g, 0.0002202 mol, 83% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.00 (s, 3H, -CO-C-CH3), 1.22 (s, 3H, -O-C-(CH3)2), 1.39 (s, 3H, -O-C-(CH3)2), 3.44 (s, 2H, -C-CH2-O-CH2), 3.63 (dd, 2H, -CO-C-CH2-O-), 4.07 (dd, 2H, -CO-C-CH2-O-), 4.51 (s, 2H, -O-CH2-C-N-), 4.54 (t, 2H, -N-CH2-CH2-O-), 4.72 (s, 2H, -N-CH2-CH2-O-), 8.04 (s, 1H, -C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 17.19 (-C-CH3), 20.52 (-O-C-(CH3)2), 24.93 (-O-C-(CH3)2), 41.78 (-C-C-CH3), 45.11 (-C-CH2-O-), 48.90 (-N-CH2-CH2-O-), 62.63 (-N-CH2-CH2-O-), 64.06 (-CH2-O-CH2-C-N-), 65.47 (-C-CH2-O-C-), 68.65 (-C-CH2-O-CH2-C-N-), 97.97 (-O-C-(CH3)2), 124.07 (-C-CH-N-), 145.00 (-C-N-), 173.80 (-C-O-CH2-) ppm. MALDI-MS: m/z = 1267.3 [M + Li+].
Generation 1 (G1, 8). A solution of CuSO4·5H2O (0.0191 g, 0.00007669 mol) in water (0.5 mL) was added to a round bottom flask containing a stirred solution of 7 (0.0768 g, 0.0002663 mol) and 6 (0.2597 g, 0.001278 mol) in THF (0.5 mL). Sodium ascorbate (0.0317 g, 0.0001598 mol) was then added and the mixture was allowed to react overnight. The product was purified by column chromatography (100% methanol) to yield an orange oil (0.1479 g, 0.0001343 mol, 50% yield).
G1 (8) from pG1 (9): Dowex Cationic Resin (6.72 g) was added to a solution of pG1 (9, 9.69 g, 0.0077 mol) in methanol (485 mL). The mixture was then left stirring overnight. The resin was then filtered off and the solvent evaporated to yield 8 as a viscous orange oil (6.68 g, 0.0061 mol, 79% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.08 (s, 3H, -CO-C-CH3), 3.45 (s, 2H, -C-CH2-O-), 3.61 (q, 4H, -C-CH2-OH), 4.51 (t, 4H, -O-CH2-C-N-), 4.52 (s, 2H, -N-CH2-CH2-O-), 4.70 (t, 2H, -N-CH2-CH2-O-), 8.04 (s, 1H, -C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 17.40 (-C-CH3), 46.53 (C-CH2-O-), 50.42 (-C-CH3), 51.78 (-N-CH2-CH2-O-), 63.86 (-N-CH2-CH2-O-), 65.40 (-O-CH2-C-N-), 65.85 (-C-CH2-OH-), 70.09 (C-CH2-O-), 125.77 (-C-CH-N-), 146.35 (-O-CH2-C-N-), 176.06 (-C-CO-O-) ppm. MALDI-MS: m/z = 1107.6 [M + Li+].
G1-ester-acetylne (10). DMAP (2.97 g, 0.0243 mol) was added to a stirred mixture of G1 (8, 6.68 g, 0.0061 mol) in anhydrous DCM (267 mL) in a 500 mL round bottom flask under nitrogen. DCC (15.02 g, 0.0728 mol), pyridine (133 mL) and 4-pentynoic acid (7.14 g, 0.0728 mol) were added, and the reaction mixture was then left stirring, under nitrogen, overnight. The crude mixture was filtered and the solvent evaporated. The product was purified by column chromatography (ethyl acetate) to yield the product as a yellow oil (7.39 g, 0.0042 mol, 70% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.19 (s, 3H, -C-CH3), 2.26 (t, 2H, -C-CH), 2.46–2.53 (m, 8H, -O-C-CH2-CH2-C-), 3.45 (s, 2H, -C-CH2-O-CH2-), 4.22 (q, 4H, -C-(CH2-O-C-)2), 4.54 (s, 2H, -C-CH2-O-CH2-C), 4.55 (t, 2H, -CH-N-CH2-CH2-), 4.73 (t, 2H, -C-CH-N-CH2-), 8.01 (s, 1H, -N-CH-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 15.10 (-CH2-C-CH), 18.20 (-C-CH3), 34.31 (-CH2-CH2-C-CH), 46.57 (-C-CH2-O-CH2-C-N-), 47.70 (-C-CH3), 50.22 (-N-CH2-CH2-O-), 64.51 (-N-CH2-CH2-O-), 65.59 (-C-CH2-O-CH2-C-N-), 66.61 (-C-CH2-O-C-), 70.19 (-C-CH2-O-CH2-C-N-), 70.55 (-C-CH), 83.54 (-C-CH), 125.49 (-C-CH-N-), 146.51 (-C-CH-N-), 172.84 (-O-C-CH2-), 173.72 (O-C-C-) ppm. MALDI-MS: m/z = 1747.5 [M + Li+].
Protected Generation 2 (pG2, 11). A solution of CuSO4·5H2O (0.1463 g, 0.000586 mol) in water (10 mL) was added to a round bottom flask containing a stirred solution of 10 (1.70 g, 0.0009766 mol) and 5 (2.2807 g, 0.009375 mol) in THF (10 mL). Sodium ascorbate (0.2322 g, 0.001172 mol) was added and the mixture was allowed to react overnight. The product mixture was evaporated, dissolved in a minimum of methanol and run through a silica plug to remove the copper salts. The product was then purified by dialysis (MWCO = 1000 Da, methanol) to yield a brown solid (2.05 g, 0.000556 mol, 57% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.02 (s, 6H, CH3-C-O-CH2-C-CH3), 1.10 (s, 3H, C-C-O-CH2-C-CH3), 1.28 (s, 6H, CH3-C-O-CH2-C-CH3), 1.39 (s, 6H, CH3-C-O-CH2-C-CH3), 2.68 (t, 4H, -O-C-CH2-CH2-), 2.94 (t, 4H, t, 4H, -O-C-CH2-CH2-), 3.43 (s, 2H, -C-CH2-O-CH2-C-N-), 3.64 (dd, 4H, -C-CH2-O-C-CH3), 4.07 (dd, 4H, -C-CH2-O-C-CH3), 4.10 (s, 4H, -C-CH2-O-C-CH2), 4.50 (m, 8H, -N-CH2-CH2-O-C-C-CH2-O-C-CH3, -O-CH2-C-CH-N-CH2-CH2-), 4.68 (m, 6H, -N-CH2-CH2-O-C-C-CH2-O-C-CH3, -O-CH2-C-CH-N-CH2-CH2-), 7.85 (s, 1H, -O-CH2-C-CH-N-), 8.02 (s, 2H, -C-CH2-CH2-C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 18.10, 18.68, 21.85, 26.49, 34.24, 43.24, 46.57, 47.67, 50.03, 50.17, 50.27, 64.15, 64.48, 65.59, 66.51, 67.04, 70.22, 99.44, 124.20, 125.51, 146.45, 147.69, 173.37, 173.68, 175.31 ppm. MALDI-MS: m/z = 3710.1[M + Na+].
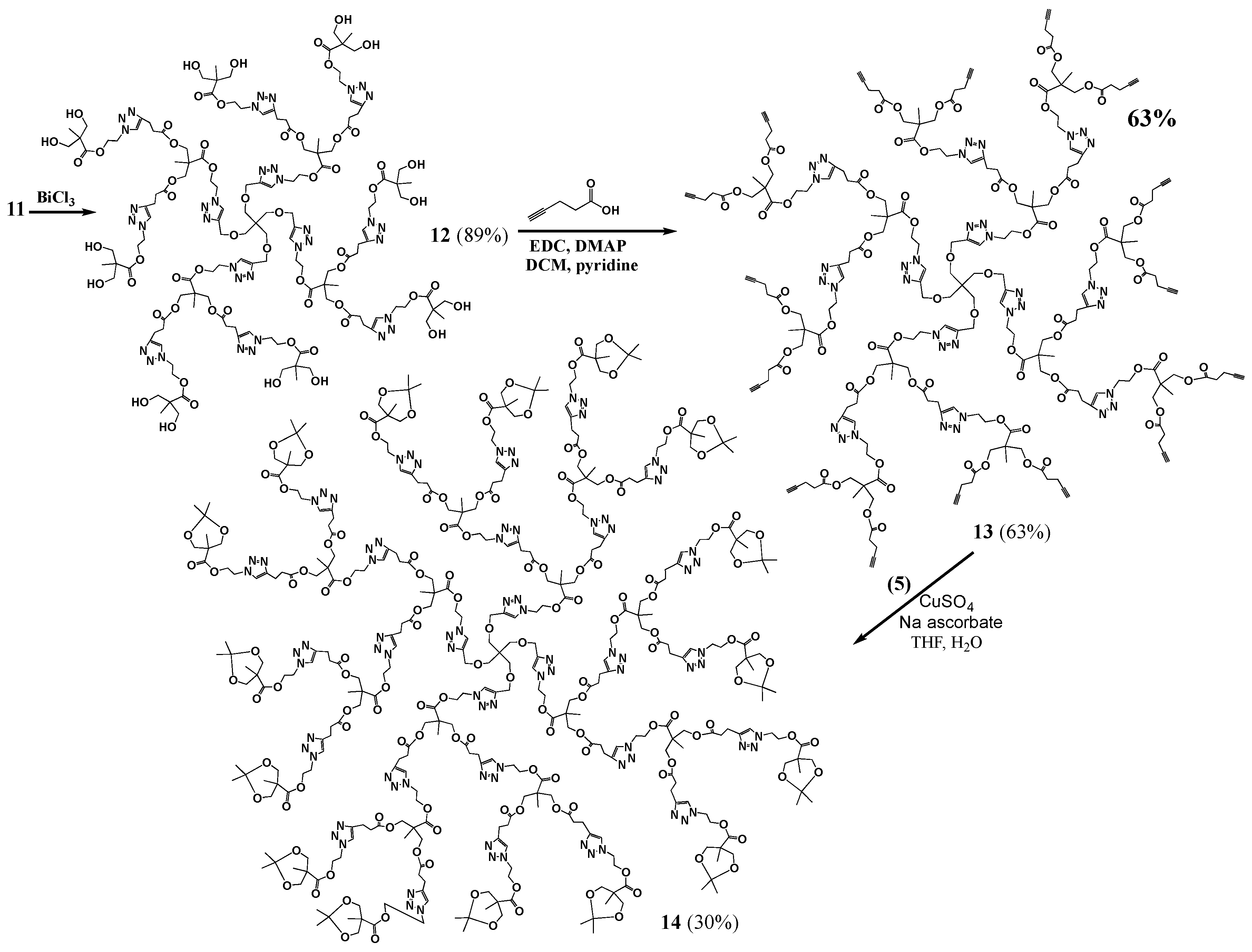
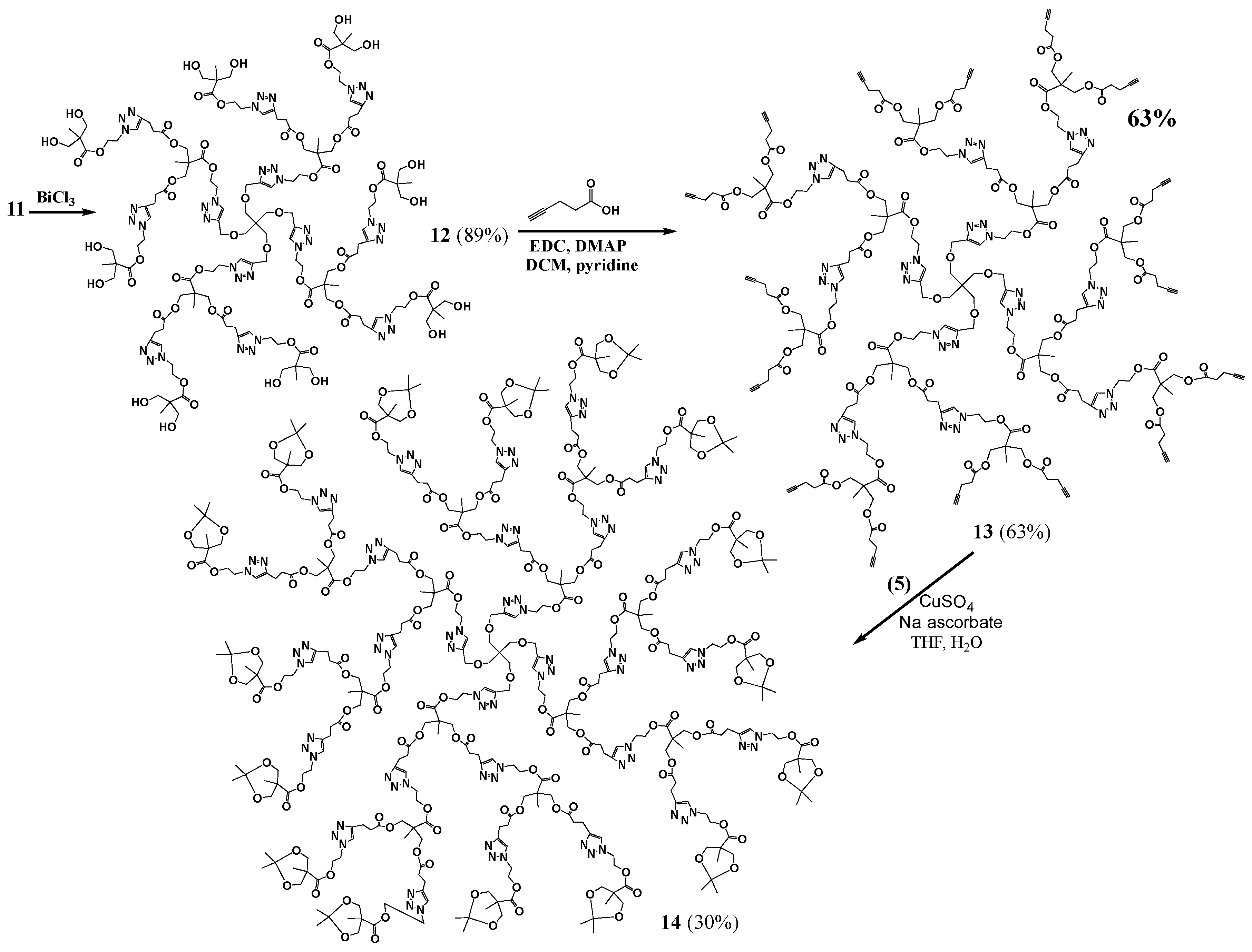
Generation 2 (G2, 12): BiCl3 (0.0009 g, 0.00000285 mol) and a drop of water were added to a stirred solution of pG2 (11, 0.025 g, 0.00000678 mol) in acetonitrile (1.1 mL). The reaction mixture was left stirring for 60 h at 45 °C. The product mixture was evaporated, dissolved in methanol, and filtered to remove the bismuth salts. The solvent was evaporated to yield a brown oil (0.020 g, 0.00000604 mol, 89% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.10 (s, 9H, -C-CH3), 2.69 (t, 4H, -C-CH2-CH2-C-N-), 2.95 (t, 4H, -C-CH2-CH2-C-N-), 3.43 (s, 2H, -C-CH2-O-CH2-C-N-), 3.60 (q, 8H, -C-CH2-OH), 4.10 (s, 4H, -C-CH2-O-C-), 4.48 (m, 8H, -N-CH2-CH2-O-C-C-CH2-OH, -O-CH2-C-CH-N-CH2-CH2-O-), 4.66 (m, 4H, -N-CH2-CH2-O-C-C-CH2-OH), 4.71 (s, 2H, -O-CH2-C-CH-N-CH2-CH2-O-, -O-CH2-C-CH-N-CH2-CH2-O-), 7.86 (s, 2H, -CH2-CH2-C-CH-N-), 8.03 (s, 1H, -O-CH2-C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = ppm 17.41, 18.08, 34.18, 46.53, 47.65, 49.99, 50.17, 50.34, 51.79, 63.91, 64.45, 65.51, 65.86, 66.51, 70.13, 124.35, 125.61, 146.37, 147.58, 173.47, 173.70, 176.08. MALDI-MS: m/z = 3388.5[M + K+].
G2-ester-acetylene (13). DMAP (0.1586 g, 0.001298 mol) was added to a stirred mixture of 12 (0.2732 g, 0.00008113 mol) in anhydrous DCM (6.6 mL), under nitrogen. 1-ethyl-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) (0.3732 g, 0.001947 mol), pyridine (3.3 mL) and 4-pentynoic acid (0.1911 g, 0.001947 mol) were added to the flask. The reaction mixture was left stirring, under nitrogen, overnight. The crude product mixture was then washed with methanol, dissolved in chloroform and filtered to yield the product as brown oil (0.27 g, 0.0000581 mol, 63% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.12 (s, 3H, -N-C-CH2-CH2-C-O-CH2-C-CH3), 1.20 (s, 6H, -CH-C-CH2-CH2-C-O-CH2-C-CH3), 1.98 (s, 4H, -C-CH), 2.50 (m, 16H, -C-CH2-CH2-C-CH), 2.70 (t, 4H, -O-C-CH2-CH2-C-N-), 2.97 (t, 4H, -O-C-CH2-CH2-C-N-), 3.42 (s, 2H, -C-CH2-O-CH2-), 4.10 (m, 4H, -C-CH2-O-C-CH2-CH2-C-N-), 4.21 (m, 8H, -C-CH2-O-C-CH2-CH2-C-CH), 4.51 (m, 8H, -CH2-CH2-C-N-N-N-CH2-CH2-O-, -C-CH2-O-CH2-C-CH-N-CH2-CH2-), 4.59 (m, 4H, -C-CH2-O-CH2-C-CH-N-CH2-CH2-), 4.65 (m, 2H, -C-CH2-O-CH2-C-CH-N-CH2-CH2-), 7.48 (s, 2H, -O-C-CH2-CH2-C-CH-N-), 7.78 (s, 1H, -CH2-O-CH2-C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = ppm 14.26, 17.58, 17.68, 20.76, 33.11, 33.15, 43.19, 45.17, 46.27, 46.29, 48.67, 63.10, 64.75, 65.10, 65.25, 69.02, 69.34, 77.20, 82.26, 121.87, 123.31, 145.29, 146.22, 171.09, 171.98, 172.09, 172.14. MALDI-MS: m/z = 4669.1 [M + Na+].
pG3 (14). A solution of CuSO4·5H2O (0.0081 g, 0.000586 mol) in water (2 mL) was added to a round bottom flask containing a stirred solution of 13 (0.1262 g, 0.00002715 mol) and 5 (0.1268 g, 0.0005212 mol) in THF (2 mL). Sodium ascorbate (0.0129 g, 0.00006515 mol) was added and the mixture was allowed to react overnight. The product mixture was evaporated, dissolved in a minimum of methanol and run through a silica plug to remove the copper salts. The product was precipitated out of chloroform with hexanes, and dried to yield a yellow oil (0.067 g, 0.00000785 mol, 30% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.04 (s, 12H, CH3-C-O-CH2-C-CH3), 1.11 (s, 3H, C-C-O-CH2-C-CH3), 1.12 (s, 6H, C-C-O-CH2-C-CH3), 1.33 (s, 12H, CH3-C-O-CH2-C-CH3), 1.41 (s, 12H, CH3-C-O-CH2-C-CH3), 2.69 (t, 12H, -O-C-CH2-CH2-), 2.97 (t, 12H, -O-C-CH2-CH2-), 3.40 (s, 2H, C-CH2-O-CH2-), 3.62 (dd, 8H, -C-CH2-O-C-CH3), 4.10 (s, 12H, -C-CH2-O-C-CH2), 4.12 (dd, 8H, -C-CH2-O-C-CH3), 4.40–4.75 (m, 30H, -CH2-CH2-C-N-N-N-CH2-CH2-O-, -C-CH2-O-CH2-C-CH-N-CH2-CH2-, -N-CH2-CH2-O-C-C-CH2-O-C-CH3), 7.56 (s, 2H, -CH-N-CH2-CH2-O-C-C-CH2-O-C-CH2-), 7.61 (s, 4H, -CH-N-CH2-CH2-O-C-C-CH2-O-C-CH3), 7.88 (s, 1H, -CH2-O-CH2-C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = ppm 17.48, 18.12, 20.71, 20.86, 26.22, 33.05, 41.98, 46.19, 48.68, 48.99, 62.82, 62.94, 65.04, 65.85, 68.41, 77.20, 98.05, 122.22, 146.71, 171.86, 171.98, 172.05, 173.70. MALDI-MS: m/z = 8580.6 [M + K+].
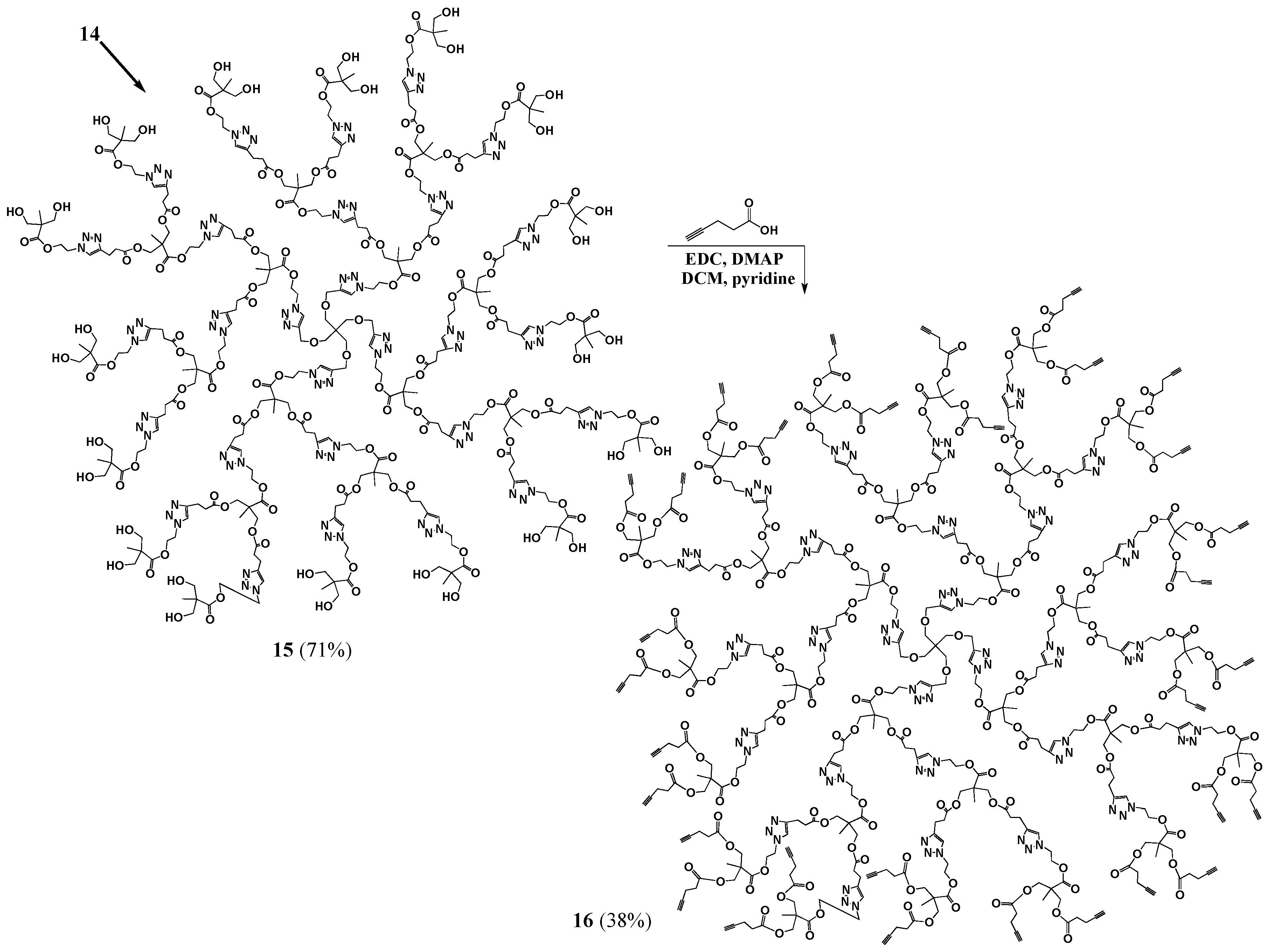
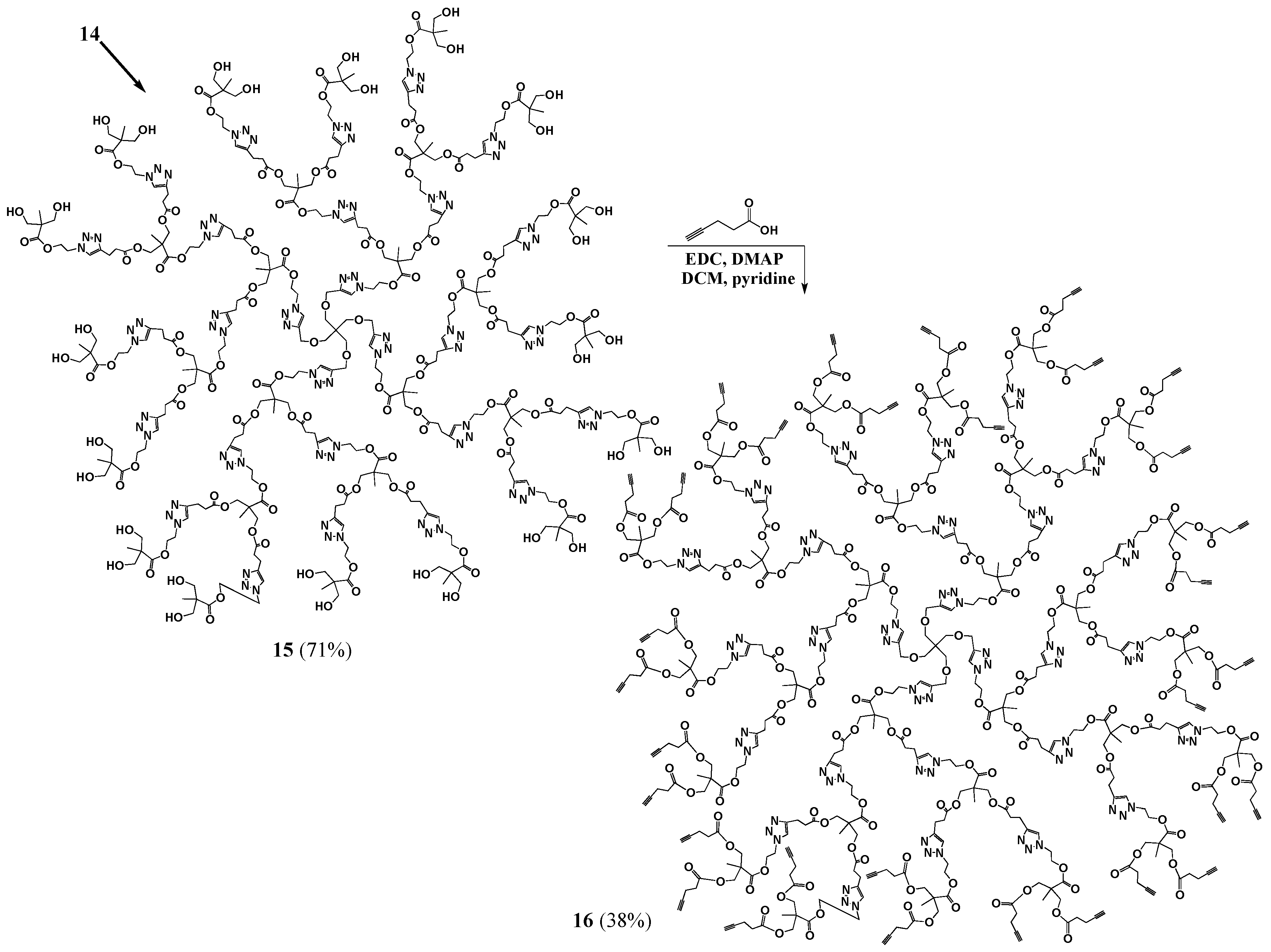
G3 (15). Dowex Cationic Resin (0.0629 g) was added to a solution of pG3 (14, 0.1531 g, 0.0000179 mol) in methanol (8.5 mL). The mixture was left stirring for three days at 45 °C. The resin was then filtered off and the solvent evaporated to yield the product as a yellow oil (0.0983 g, 0.0000124 mol, 71% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.09 (s, 21H, -C-CH3), 2.69 (m, 12H, -O-C-CH2-CH2-C-N-), 2.95 (m, 12H, -O-C-CH2-CH2-C-N-), 3.43 (s, 2H, -C-CH2-O-CH2-C-), 3.60 (q, 16H, -C-CH2-OH), 4.10 (m, 12H, -C-CH2-O-C-), 4.48 (m, 16H, -C-CH2-O- CH2-C-N-, -N-CH2-CH2-O-), 4.66 (m, 14H, -N-CH2-CH2-O-), 7.82–8.07 (m, 7H, -C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, DMSO-d6): δ = ppm 16.65, 16.85, 16.89, 20.38, 32.69, 45.77, 45.80, 48.05, 48.34, 48.53, 50.24, 51.25, 62.24, 62.94, 63.06, 63.68, 63.84, 64.82, 122.31, 122.44, 123.98, 144.08, 145.22, 145.26, 171.50, 171.83, 174.20. MALDI-MS: m/z = 7894.2 [M + H+].
G3-ester-acetylene (16). DMAP (0.0266 g, 0.0002181 mol) was added to a stirred mixture of G3 (0.0713 g, 0.000006814 mol) in anhydrous DCM (3.3 mL), under nitrogen. EDC (0.0627 g, 0.0003271 mol), pyridine (1.7 mL) and 4-pentynoic acid (0.0796 g, 0.0003271 mol) were added and the reaction mixture was left stirring, under nitrogen, overnight. The crude product mixture was dissolved in DCM and filtered to remove the salts. The product was then precipitated out of DCM with hexanes. Ether and hexanes washes were then performed to obtain a light brown oil (38% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.07–1.24 (m, 32H, -CH3), 1.99 (s, 8H, -CH), 2.43–2.55 (m, 32H, -CO-CH2-CH2-C-CH), 2.71 (m, 12H, -CO-CH2-CH2-C-N-), 2.96 (m, 12H, -CO-CH2-CH2-C-N-), 3.42 (s, 2H, -C-CH2-O-CH2-C-N-), 4.11 (s, 12H, CH3-C-CH2-O-CO-CH2-CH2-C-N-), 4.17–4.26 (m, 16H, CH3-C-CH2-O-CO-CH2-CH2-C-CH), 4.49–4.66 (m, 30H, -N-CH2-CH2-O-, -C-CH2-O-CH2-C-N-), 7.48–7.80 (m, 7H, -C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = ppm 14.29, 17.29, 17.62, 17.72, 20.9, 29.69, 33.14, 43.60, 45.22, 46.33, 48.68, 48.98, 62.95, 63.14, 64.82 (-C-CH2-O-CH2-C-N-N-N-CH2-CH2-), 64.97, 65.14, 65.28, 65.80, 69.39, 77.21, 82.30, 121.91, 121.98, 122.38, 146.18, 146.25, 146.29, 171.12, 172.01, 172.05 172.13, 172.15, 172.18. MALDI-MS: m/z = 10,000–11,000 (theoretical mass = 10,456).
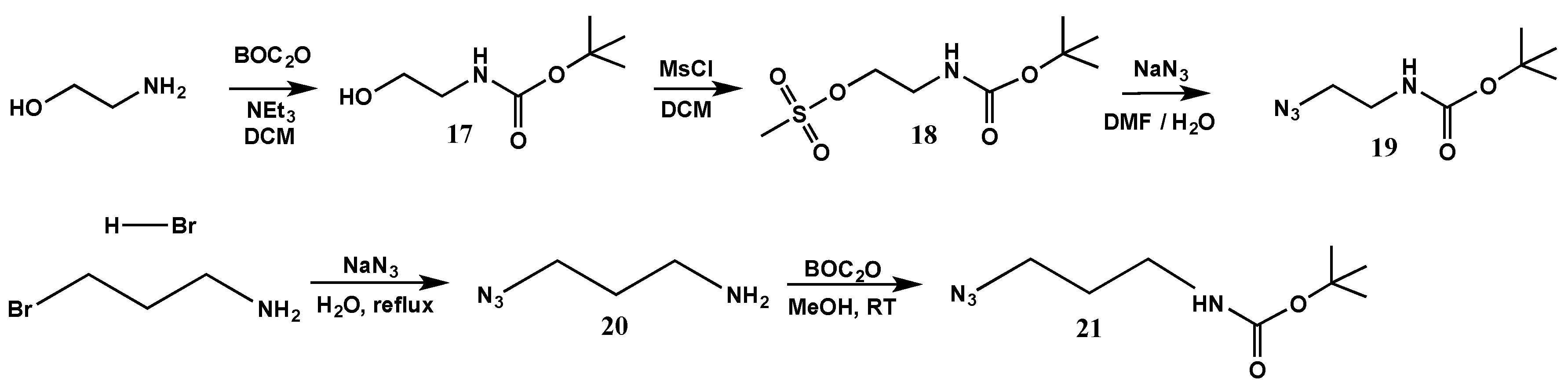
Amine functionalization: tert-butyl (2-hydroxyethyl)carbamate (17). Di-tert-butyl dicarbonate (BOC2O) (7.86 g, 0.036 mol) was added slowly in a portionwise fashion to a stirred solution of ethanolamine (2.0 g, 0.033 mol) in DCM and triethylamine (5 mL), at 0 °C. The mixture was allowed to react overnight, at room temperature. The crude mixture was washed with an aqueous potassium carbonate solution, followed by an aqueous hydrochloric acid solution, brine, and finally water. The organic phase was isolated, dried with magnesium sulfate, and the solvent evaporated to yield the product as a colourless oil (3.70 g, 0.023 mol, 70% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.47 (s, 9H, -C-(CH3)3), 3.26 (s, 2H, -CH2-NH-), 3.67 (s, 2H, -CH2-OH), 5.04 (s, 1H, -NH-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 28.6 (s, -C-(CH3)3), 43.4 (s, -CH2-NH-), 62.8 (s, -CH2-OH), 79.9 (C-(CH3)3), 157.1 (-NH-CO-O-) ppm.
2-((tert-Butoxycarbonyl)amino)ethyl methanesulfonate (18). Methanesulfonyl chloride (9.42 g, 0.0822 mol) was added very slowly in a dropwise fashion to a stirred solution of (17) (12.8713 g, 0.0798 mol) in anhydrous DCM (500 mL) and triethylamine (20 mL) at 0 °C, under nitrogen. The reaction was left stirring, under nitrogen at room temperature overnight. The crude mixture was then washed with water (2 × 250 mL) and brine (2 × 250 mL). The organic layer was isolated, dried with magnesium sulfate and the solvent evaporated to yield the product as a yellow oil (17.95 g, 0.075 mol, 94% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.44 (s, 9H, -C-(CH3)3), 3.03 (s, 3H, -S-CH3), 3.46 (m, 2H, -CH2-NH-), 4.28 (m, 2H, -CH2-O-), 4.92 (s, 1H, -NH-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 28.6 ( -C-(CH3)3), 37.6 (-CH2-NH-), 40.2 (-S-CH3), 69.1 (-CH2-O-), 80.2 (-C-(CH3)3) 158 (-NH-CO-O-) ppm.
tert-Butyl (2-azidoethyl)carbamate (19): Sodium azide (27.30 g, 0.4199 mol) was added to a stirred solution of (18) (19.98 g, 0.0835 mol) in DMF (100 mL) and water (100 mL), under nitrogen. The reaction was left stirring, at 80 °C overnight, under nitrogen. The crude product mixture was extracted with ethyl acetate (3 × 100 mL), the organic layers were isolated and combined and washed with water (3 × 100 mL) and brine (2 × 100 mL). The organic layer was isolated, dried with magnesium sulfate and the solvent evaporated to yield the product as a yellow oil (9.93 g, 0.0533 mol, 64% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.44 (s, 9H, -C-(CH3)3), 3.28 (m, 2H, -CH2-NH-), 3.40 (m, 2H, N3-CH2-), 4.82 (s, 1H, -NH-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 28.3 (-C-(CH3)3), 40.0 (-CH2-NH-), 51.2 (-CH2-N3), 79.8 (-C-(CH3)3), 155.70 (-NH-CO-O-) ppm.
3-Azidopropylamine (20). A solution of sodium azide (3.39 g, 0.05215 mol) in water (11 mL) was added to a stirred solution of 3-bromopropylamine hydrobromide (3.42 g, 0.01566 mol) in water (8 mL). The reaction was refluxed overnight, and 2/3 of the solvent was then evaporated and the crude mixture was placed in an ice bath. Potassium hydroxide (4.2 g, 0.0749 mol) and ether (50 mL) were added, the aqueous phase was isolated and washed with ether (3 × 35 mL). The organic phases were isolated, combined and dried with magnesium sulfate to yield the product as a yellow oil (1.38 g, 0.0138 mol, 88% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.42 (s, 2H, -NH2), 1.72 (q, 2H, -CH2-CH2-CH2-), 2.79 (t, 2H, NH2-CH2-CH2-CH2-N3), 3.36 (t, 2H, NH2-CH2-CH2-CH2-N3) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 30.54 (-CH2-CH2-CH2-), 36.13 (N3-CH2-CH2-CH2-NH2), 51.12 (N3-CH2-CH2-CH2-NH2) ppm.
tert-Butyl (3-azidopropyl)carbamate (21). A solution of BOC2O (1.0899 g, 0.004994 mol) in methanol (6 mL) was added dropwise to a solution of (20) (0.500 g, 0.004994 mol) in triethylamine (3.48 mL) and methanol (20 mL). The reaction was left stirring, overnight at room temperature. The product mixture was filtered, and the solvent evaporated. The mixture was purified by column chromatography (chloroform) to yield the product as a yellow oil (0.5457 g, 0.00273 mol, 55% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.41(s, 9H, -C-(CH3)3), 1.74 (q, 2H, -CH2-CH2-CH2-), 3.17 (t, 2H, -CH2-CH2-CH2-N3), 3.33 (t, 2H, -CH2-CH2-CH2-N3), 4.75 (s, 1H, -NH-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 28.39 (C-(CH3)3), 29.87 (-CH2-CH2-CH2-), 48.13 (N3-CH2-CH2-CH2-NH-), 53.93 (N3-CH2-CH2-CH2-NH-), 79.74 (-C-(CH3)3), 155.47 (-NH-COO-) ppm.
G0-NHBOC. A solution of CuSO4·5H2O (0.0754 g, 0.000302 mol) in water (2.5 mL) was added to a round bottom flask containing a stirred solution of 7 (0.300 g, 0.00104 mol) and 19 (0.8528 g, 0.00458 mol) in THF (2.5 mL). Sodium ascorbate (0.1236 g, 0.000624 mol) was added and the mixture was allowed to react overnight. The product mixture was evaporated and purified by column chromatography (acetone) to yield an orange solid (0.9041 g, 0.000875 mol, 97% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.36 (s, 9H, -C-(CH3)3), 3.41 (s, 2H, -C-CH2-O-), 3.55 (q, 2H, -N-CH2-CH2-NH-), 4.43 (t, 2H, -N-CH2-CH2-NH-), 4.47 (s, 2H, -C-CH2-O-CH2-), 5.62 (t, 1H, -NH-), 7.56 (s, 1H,-C-CH-N- ) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 28.66 (-C-(CH3)3), 40.29 (-CH-N-CH2-CH2-), 46.47 (-C-CH2-O-CH2-C-N-), 48.52 (-CH-N-CH2-CH2-), 65.44 (-C-CH2-O-CH2-C-N-), 70.25 (-C-CH2-O-CH2-C-N-), 80.21 (-C-(CH3)3), 126.40 (-CH-), 147.20 (-C-CH-), 154.93 (-CO-) ppm. ESI-MS: m/z = 1056.5 [M + Na+].
G1-NHBOC. A solution of CuSO4·5H2O (0.0081 g, 0.00003232 mol) in water (2 mL) was added to a round bottom flask containing a stirred solution of 10 (0.0987 g, 0.0000567 mol) and 20 (0.1655 g, 0.0006804 mol) in THF (2 mL). Sodium ascorbate (0.0135 g, 0.00006804 mol) was added and the mixture was allowed to react overnight. The product mixture was evaporated and purified by column chromatography (acetone) to yield a sticky yellow solid (0.095 g, 0.0000284 mol, 50% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.09 (s, 3H, -C-C-CH3), 1.42 (s, 18H, -C-(CH3)3), 2.03 (m, 4H, -N-CH2-CH2-CH2-NH-), 2.68 (t, 4H, -O-C-CH2-CH2-C-N-), 2.94 (t, 4H, -O-C-CH2-CH2-C-N-), 3.05 (t, 4H, -N-CH2-CH2-CH2-NH-), 3.42 (s, 2H, -C-CH2-O-), 4.09 (q, 4H, CH3-C-CH2-O-), 4.38 (t, 4H, -N-CH2-CH2-CH2-NH-), 4.51 (m, 4H, -CH2-O-CH2-C-CH-N-CH2-), 4.71 (t, 2H, -N-CH2-CH2-O-), 6.69 (t, 2H, -NH-), 7.77 (s, 2H, -CH-N-CH2-CH2-CH2-), 8.03 (s, 1H, -CH2-O-CH2-C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CDCl3): δ = 18.08 (CH3-C-CH2-), 21.84 (-CH2-CH2-C-N-), 28.87 (-C-(CH3)3), 31.67 (-CH2-CH2-NH-), 34.22 (-CH2-CH2-C-N-), 38.52 (-CH2-NH-), 46.55 (-C-CH2-O-CH2-C-N), 47.67 (-CH2-C-CH3), 49.90 (-N-CH2-CH2-O-), 50.16 (-N-CH2-CH2-CH2-NH-), 64.45 (-N-CH2-CH2-O-), 65.58 (-C-CH2-O-CH2-C-N), 66.48 (-CH2-C-CH3), 70.18 (-C-CH2-O-CH2-C-N), 80.12 (-C-(CH3)3), 123.73 (-C-CH-N-CH2-CH2-CH2-), 125.52 (-C-CH2-O-CH2-C-CH-N-), 146.45 (-C-CH2-O-CH2-C-CH-N-), 147.43 (-C-CH-N-CH2-CH2-CH2-), 158.45 (-CO-O-C-(CH3)3), 173.43 (-O-CO-CH2-), 173.67 (-O-CO-C-) ppm. MALDI-MS: m/z = 3365.1 [M + Na+].
G2-NHBOC. A solution of CuSO4·5H2O (0.0059 g, 0.00002349 mol) in water (1 mL) was added to a round bottom flask containing a stirred solution of 13 (0.091 g, 0.00001958 mol) and 20 (0.0941 g, 0.0004698 mol) in THF (1 mL). Sodium ascorbate (0.0093 g, 0.00004698 mol) was added and the mixture was allowed to react overnight. The product mixture was evaporated and filtered in DCM to remove the salts. The product was precipitated out of DCM using ether, isolated and dried to yield a viscous yellow oil (0.0429 g, 0.000005463 mol, 28% yield). 1H-NMR (400 MHz, CDCl3): δ = 1.11 (s, 9H, -O-C-C-CH3), 1.41 (s, 36H, -C-(CH3)3), 2.04 (q, 8H, -N-CH2-CH2-CH2-NH-), 2.69 (t, 12H, -O-C-CH2-CH2-C-N-), 2.97 (t, 12H, -O-C-CH2-CH2-C-N-), 3.10 (m, 8H, -N-CH2-CH2-CH2-NH-), 3.46 (s, 2H, -C-CH2-O-CH2-), 4.10 (m, 12H, CH3-C-CH2-O-C-), 4.36 (t, 8H, -N-CH2-CH2-CH2-NH-), 4.48 (m, 8H, -CH2-CH2-C-N-N-N-CH2-CH2-O-), 4.58 (t, 4H, -CH2-O-CH2-C-N-N-N-CH2-CH2-O-), 4.65 (s, 2H, -C-CH2-O-CH2-), 5.14 (s, 4H, -NH-), 7.47 (s, 4H, -CH-N-CH2-CH2-CH2-NH-), 7.54 (s, 2H, -C-CH2-CH2-C-CH-N-CH2-CH2-O-), 7.83 (s, 1H, -O-CH2-C-CH-N-) ppm. 13C{1H} NMR (300 MHz, CDCl3): δ = 17.58 (-CH2-C-CH3), 20.76 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 20.81 (-CH2-C-CH-N-CH2-CH2-CH2-), 28.36 (-C-(CH3)3), 30.62 (-N-CH2-CH2-CH2-), 33.17 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 33.28 (-CH2-CH2-C-CH-N-CH2-CH2-CH2-), 37.36 (-N-CH2-CH2-CH2-), 45.18 (-C-CH2-O-CH2-C-N-), 46.26 (-C-CH2-O-CH2-C-CH-N-CH2-CH2-), 46.30 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 47.47 (-N-CH2-CH2-CH2-), 48.68 (-C-CH2-O-CO-CH2-CH2-C-CH-N-CH2-CH2-CH2-), 48.75 (-C-CH2-O-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 63.05 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 63.11 (-C-CH2-O-CH2-C-CH-N-CH2-CH2-), 64.75 (-C-CH2-O-CH2-C-N-), 65.14 (-C-CH2-O-CO-CH2-CH2-C-CH-N-CH2-CH2-CH2-), 69.05 (-C-CH2-O-CH2-C-N-), 70.50 (-C-CH2-O-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 79.32 (-C-(CH3)3), 121.67 (-C-CH-N-CH2-CH2-CH2-), 122.10 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 123.50 (-C-CH2-O-CH2-C-CH-N-), 146.01 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 146.15 (-C-CH-N-CH2-CH2-CH2-), 149.93 (-C-CH2-O-CH2-C-CH-N-), 156.12 (-CO-O-C-(CH3)3), 171.96 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 172.01 (-CO-CH2-CH2-C-CH-N-CH2-CH2-CH2-), 172.12 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-CO-), 172.15 (-C-CH2-O-CH2-C-CH-N-CH2-CH2-O-CO-) ppm. MALDI-MS: m/z = 7881.9 [M + Na+].
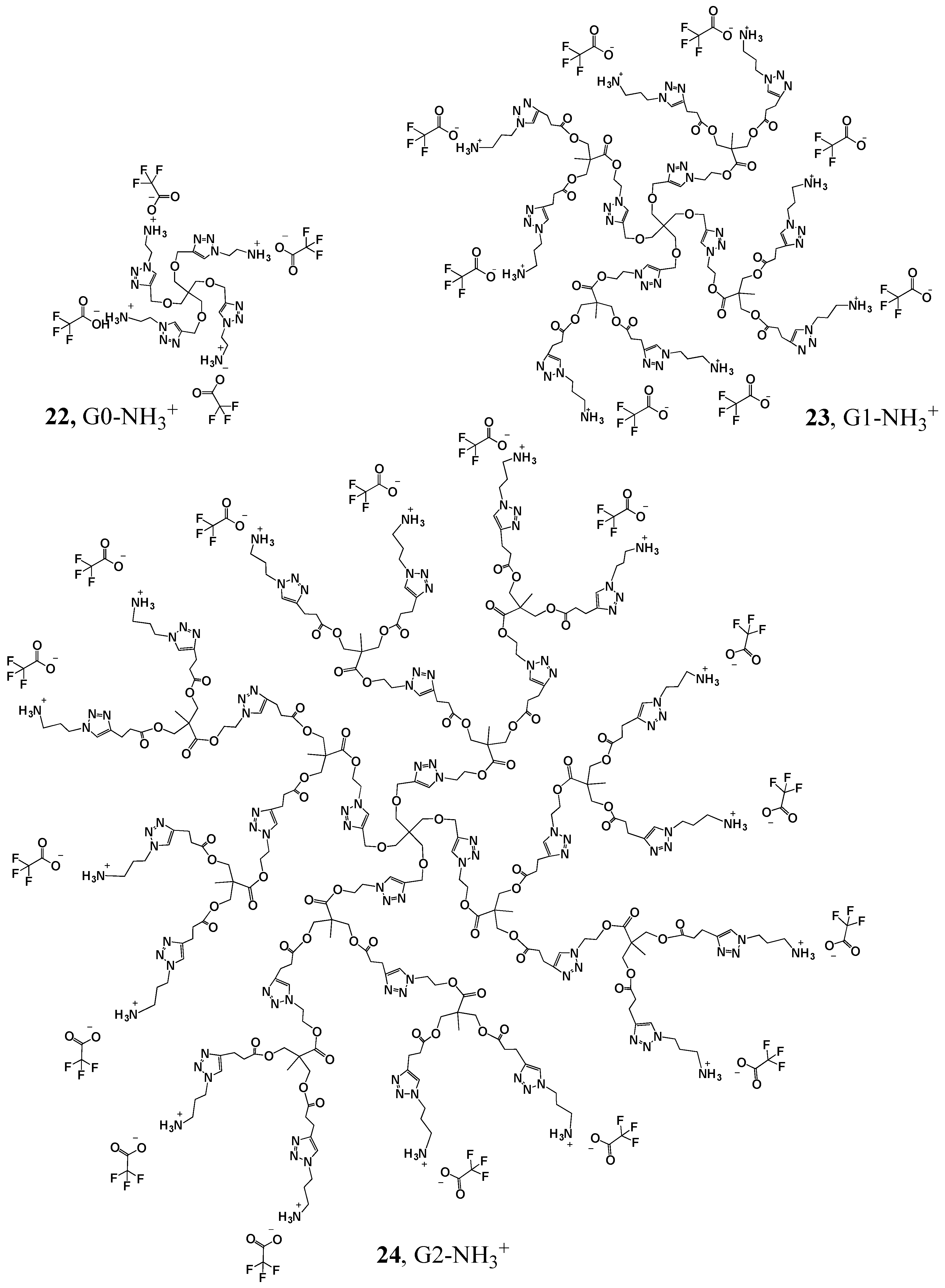
G0-NH3+ (22). TFA (5 mL) was added dropwise to a solution of G0-NHBOC (0.9041 g, 0.000875 mol) in DCM (5 mL). The reaction was left stirring for 5 min, the solvent was evaporated and the product was dissolved in methanol. The methanol was evaporated, bringing the remaining TFA with it. This methanol addition and evaporation cycle was then repeated several times to ensure that all remaining TFA had been evaporated. The product was obtained as a sticky orange solid (0.800 g, 0.000735 mol, 84% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.80 (s, 3H, -NH3), 3.47 (s, 2H, -C-CH2-O-), 3.59 (t, 2H, -N-CH2-CH2-NH3), 4.53 (s, 2H, -C-CH2-O-CH2-), 4.76 (t, 2H, -N-CH2-CH2-NH3), 8.06 (s, 1H, -C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 40.30 (-CH-N-CH2-CH2-), 46.48 (-C-CH2-O-CH2-C-N-), 48.54 (-CH-N-CH2-CH2-), 65.46 (-C-CH2-O-CH2-C-N-), 70.27 (-C-CH2-O-CH2-C-N-), 119.20 (-CF3), 126.40 (-CH-), 147.22 (-C-CH-), 162.95 (-CO-) ppm. ESI-MS: m/z = 633.3 [M − 4TFA + H+].
G1-NH3+ (23). TFA (0.5 mL) was added dropwise to a solution of G1-NHBOC (0.05 g, 0.000015 mol) in DCM (0.5 mL). The reaction was left stirring for 5 min, the solvent was evaporated and the product was dissolved in methanol. The methanol was then evaporated, bringing the remaining TFA with it. This methanol addition and evaporation cycle was then repeated several times to ensure that all remaining TFA had been evaporated. The product was obtained as a sticky yellow solid (0.041 g, 0.000012 mol, 80% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.10 (s, 3H, -C-CH3), 2.27 (q, 4H, -N-CH2-CH2-CH2-NH3), 2.69 (t, 4H, -O-C-CH2-CH2-C-N-), 2.95 (t, 4H, -O-C-CH2-CH2-C-N-), 3.00 (t, 4H, -N-CH2-CH2-CH2-NH3), 3.42 (s, 2H, -C-CH2-O-CH2-), 4.08 (s, 4H, CH3-C-CH2-O-), 4.51 (t, 8H, -CH2-O-CH2-C-), 4.71 (t, 2H,-N-CH2-CH2-O-C-, -N-CH2-CH2-CH2-NH3), 7.80 (s, 2H, -CH-N-CH2-CH2-CH2-), 8.04 (s, 1H,-CH2-O-CH2-C-CH-N-) ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 18.03 (CH3-C-CH2-), 21.74 (-CH2-CH2-C-N-), 29.18 (-CH2-CH2-NH3), 34.14 (-CH2-CH2-C-N-), 38.17 (-CH2-NH3), 46.55 (-C-CH2-O-CH2-C-N-), 47.63 (-CH2-C-CH3), 49.90 (-N-CH2-CH2-O-), 50.20 (-N-CH2-CH2-CH2-NH3-), 64.44 (-N-CH2-CH2-O-), 65.48 (-C-CH2-O-CH2-C-N-), 66.41 (-CH2-C-CH3), 70.15 (-C-CH2-O-CH2-C-N), 119.08 (-C-F3), 123.91 (-C-CH-N-CH2-CH2-CH2-), 125.64 (-C-CH2-O-CH2-C-CH-N-), 146.39 (-C-CH2-O-CH2-C-CH-N-), 147.69 (-C-CH-N-CH2-CH2-CH2-), 162.57 (-CO-O-C-F3), 173.46 (-O-CO-CH2-), 173.69 (-O-CO-C-) ppm. MALDI-MS: m/z = 2563.6 [M − 8TFA + Na+].
G2-NH3+ (24). TFA (1 mL) was added dropwise to a solution of G2-NHBOC (0.0429 g, 0.000005463 mol) in DCM (1 mL). The reaction was left stirring for 5 min, the solvent was evaporated and the product was dissolved in methanol. The methanol was then evaporated, bringing the remaining TFA with it. This methanol addition and evaporation cycle was repeated several times to ensure that all remaining TFA had been evaporated. The product was obtained as a sticky yellow solid (0.034 g, 0.00000421 mol, 77% yield). 1H-NMR (400 MHz, CD3OD): δ = 1.10 (s, 9H, -O-C-C-CH3), 2.27 (q, 8H, -N-CH2-CH2-CH2-NH3), 2.68 (t, 12H, -O-C-CH2-CH2-C-N-), 2.97 (t, 12H, -O-C-CH2-CH2-C-N-), 3.01 (m, 8H, -N-CH2-CH2-CH2-NH3), 3.41 (s, 2H, -C-CH2-O-CH2-), 4.08 (m, 12H, CH3-C-CH2-O-C-), 4.50 (m, 8H, -CH2-CH2-C-N-N-N-CH2-CH2-O-, -N-CH2-CH2-CH2-NH3), 4.66 (t, 4H, -CH2-O-CH2-C-N-N-N-CH2-CH2-O-), 4.70 (s, 2H, -C-CH2-O-CH2-), 4.88 (s, 12H, -NH3), 7.82 (s, 4H, -CH-N-CH2-CH2-CH2-NH-), 7.86 (s, 2H, -C-CH2-CH2-C-CH-N-CH2-CH2-O-), 8.07 (s, 1H, -O-CH2-C-CH-N-)ppm. 13C{1H}-NMR (300 MHz, CD3OD): δ = 16.55 (-CH2-CH2-CH2-N-N-N-C-CH2-CH2-CO-O-CH2-C-CH3), 16.63 (O-CH2-CH2-N-N-N-C-CH2-CH2-CO-O-CH2-C-CH3), 20.30 (-CH2-C-CH-N-CH2-CH2-CH2-), 20.35 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 27.71 (-N-CH2-CH2-CH2-), 32.70 (-CO-CH2-CH2-C-N-), 36.72 (-N-CH2-CH2-CH2-), 45.05 (-C-CH2-O-CH2-C-N-), 46.17 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 46.19 (-C-CH2-O-CH2-C-CH-N-CH2-CH2-), 46.85 (-N-CH2-CH2-CH2-), 48.73 (CH3-C-CH2-O-CO-), 62.97 (-N-CH2-CH2-O-), 64.05 (-C-CH2-O-CH2-C-N-), 64.97 (-C-CH2-O-CO-CH2-CH2-C-CH-N-CH2-CH2-CH2-), 65.05 (-C-CH2-O-CO-CH2-CH2-C-CH-N-CH2-CH2-O-), 68.75 (-C-CH2-O-CH2-C-N-), 118.50 (-CF3), 122.48 (-C-CH-N-), 146.08 (-C-CH-N-), 161.39 (-CO-O-CF3), 171.98 (-CO-O-CH2-C-CH3), 172.24 (-CO-CH2-CH2-C-CH-N-CH2-CH2-O-CO-), 172.29 (-C-CH2-O-CH2-C-CH-N-CH2-CH2-O-CO-) ppm.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}