4.1. General Remarks
MeCN, pyridine and dichloromethane were dried over 3Å molecular sieves and triethylamine over CaH2. NMR spectra were recorded using a 500 MHz instrument. The chemical shifts for 1H and 13C NMR resonances are given in parts of million from the residual signal of the deuterated solvents (CD3OD and CD3CN). 31P NMR resonance shifts are compared to external H3PO4. Mass spectra were recorded using electrospray ionization (ESI-TOF).
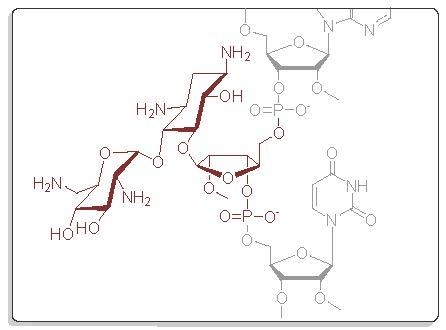
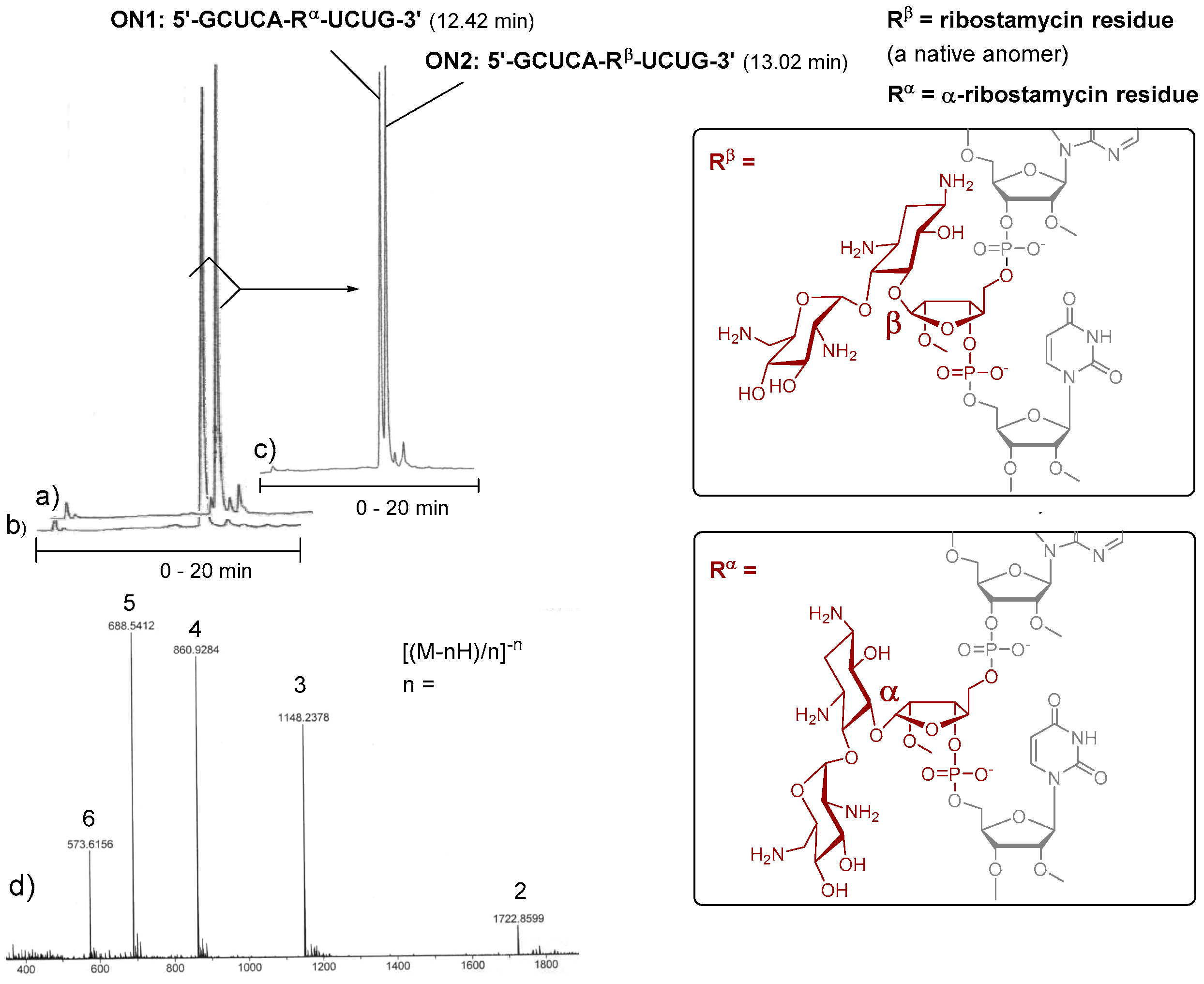
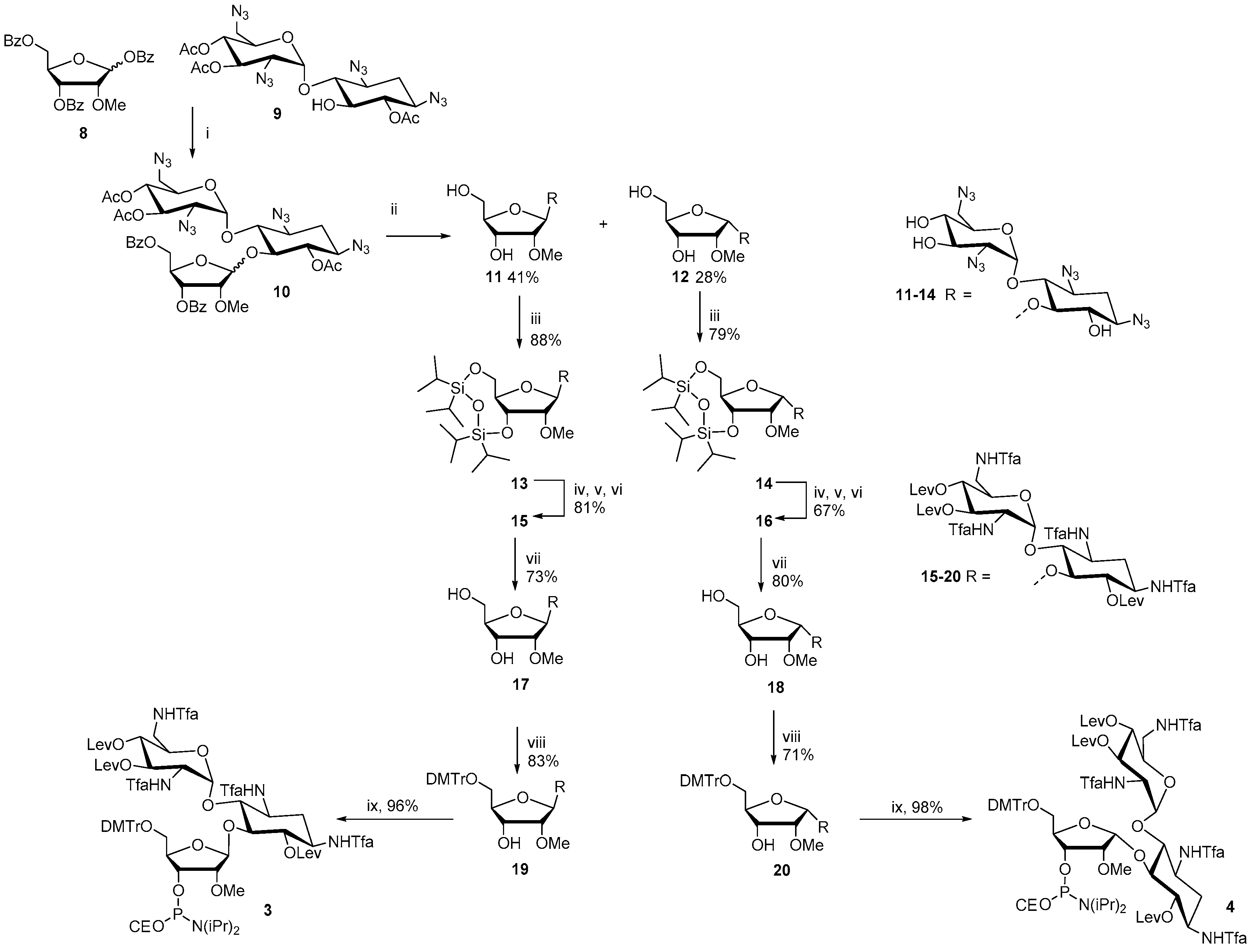
1,3,2′,6′-Tetraazido-2′′-O-methyl ribostamycin (
11 and 12). 6,3′,4′-Tri-
O-acetyl-1,3,2′,6′-tetraazido neamine (
9, 1.6 g, 2.9 mmol) [
25] and 1,3,5-di-
O-benzoyl-2-
O-methyl-α/β-d-ribofuranose (
8, 3.4 g, 7.1 mmol) were dissolved in dichloromethane (20 mL) and the mixture was cooled down to 0 °C. Trimethylsilyl trifluoromethanesulfonate (0.58 mL, 0.32 mmol) was slowly added to the mixture and the reaction was stirred at 0 °C for 2 h and then at room temperature for 2 h under a nitrogen atmosphere. Dichloromethane (50 mL) and saturated NaHCO
3 (20 mL) were added to the mixture. The organic layer was separated, washed with saturated NaCl, dried over Na
2SO
4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (30% EtOAc in petroleum ether) to give 2.13 g (81%) of
10 as colourless oil (anomeric mixture, β:α = 1:2). The peracylated ribostamycine (
10) was dissolved in 0.1 mol L
−1 methanolic sodium methoxide (5.0 mL). The mixture was stirred at ambient temperature for 1 h, neutralized by addition of strongly acidic cation-exchange resin, and filtered. The filtrate was evaporated to dryness and purified by silica gel chromatography (1. EtOAc, 2. 10% MeOH in CH
2Cl
2) to give 0.55 g (41%) of
11 (β anomer) and 0.37 g (28%) of
12 (α anomer).
11:
1H NMR (500 MHz, CD
3OH): δ 5.89 (d, 1H,
J = 3.8 Hz), 5.45 (b, 1H), 4.21–4.17 (m, 2H), 3.92–3.89 (m, 2H), 3.82–3.79 (m, 2H), 3.70–3.63 (m, 3H), 3.58–3.52/m, 2H), 3.54 (s, 3H), 3.48–3.42 (m, 3H), 3.39 (m, 1H), 3.13 (dd, 1H,
J = 10.6 Hz & 3.8 Hz), 2.24 (m, 1H), 1.40 (m, 1H);
13C (125 MHz, CD
3OH): δ 105.5, 96.7, 84.6, 84.0, 83.1, 76.0, 75.6, 71.8, 71.2, 70.7, 69.8, 63.1, 62.2, 60.6, 59.9, 57.3, 51.2, 31.6; HRMS (ESI-TOF)
m/
z: [M + Na]
+ calcd. for C
18H
28N
12NaO
10 595.1949, found 595.1930.
12:
1H NMR (500 MHz, CD
3OH): δ 5.84 (d, 1H,
J = 3.9 Hz), 5.54 (d, 1H,
J = 4.6 Hz), 4.27–4.24 (m, 3H), 3.87 (dd, 1H,
J = 5.0 Hz, both), 3.81 (dd, 1H,
J = 10.0 Hz & 9.1 Hz), 3.70–3.61 (m, 4H), 3.55 (s, 3H), 3.55–3.43 (m, 5H), 3.38 (m, 1H), 3.24 (dd, 1H,
J = 10.5 Hz & 4.0 Hz), 2.25 (ddd, 1H,
J = 12.9 Hz, 4.4 Hz & 4.4 Hz), 1.44 (ddd, 1H,
J = 12.3 Hz, each);
13C (125 MHz, CD
3OH): δ 103.0, 97.4, 86.6, 84.6, 81.1, 77.8, 74.9, 71.8, 71.5, 71.1, 68.3, 64.0, 61.8, 60.1, 59.5, 57.6, 51.1, 31.6; HRMS (ESI-TOF)
m/
z: [M + Na]
+ calcd. for C
18H
28N
12NaO
10 595.1949, found 595.1929.
1,3,2′,6′-Tetraazido-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-2′′-O-methyl ribostamycin (13). 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (350 μL, 1.1 mmol) was added to a mixture of 11 (0.55 g, 0.96 mmol) in pyridine (3.0 mL). The mixture was stirred overnight at ambient temperature and then dichloromethane and saturated NaHCO3 were added. The organic layer was separated, washed with saturated NaCl, dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (5% MeOH in CH2Cl2) to yield 0.69g (88%) of the product (13) as white foam. 1H NMR (500 MHz, CD3OH): δ 6.10 (d, 1H, J = 3.8 Hz), 5.27 (s, 1H), 4.53 (dd, 1H, J = 9.0 Hz & 4.2 Hz), 4.20 (m, 1H), 4.12 (dd, 1H, J = 12.9 Hz & 1.9 Hz), 3.96 (dd, 1H, J = 12.8 Hz & 2.5 Hz), 3.92–3.86 (m, 3H), 3.67–3.62 (m, 2H), 3.59 (s, 3H), 3.57–3.41 (m, 4H), 3.31–3.24 (m, 2H), 3.07 (dd, 1H, J = 10.5 Hz & 3.8 Hz), 2.24 (m, 1H), 1.36 (ddd, J = 12.5 Hz, each), 1.16–1.06 (m, 28H); 13C (125 MHz, CD3OH): δ 107.8, 96.3, 85.9, 84.8, 80.4, 76.7, 75.2, 71.52, 71.50, 71.1, 70.3, 63.5, 60.9, 60.6, 60.1, 51.5, 31.8, 16.8, 16.62, 16.59, 16.5, 16.40, 16.37, 16.31, 16.29, 16.24, 16.16, 16.1, 13.5, 13.4, 13.1, 12.9, 12.5, 12.4; HRMS (ESI-TOF) m/z: [M + K]+ calcd. for C30H54KN12O11Si2 853.3211, found 853.3148.
1,3,2′,6′-Tetraazido-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-2′′-O-methyl-α1′′−5-ribostamycin (14). The α-anomer (14) was synthesized as described for 13 above. 0.37 g (0.64 mmol) of 12 gave 0.42 g (79%) of 14 as white foam. 1H NMR (500 MHz, CD3OH): δ 5.67 (d, 1H, J = 3.8 Hz), 5.43 (d, 1H, J = 4.1 Hz), 4.40 (dd, 1H, J = 7.7 Hz & 5.1 Hz), 4.28 (m, 1H), 4.20 (m, 1H), 4.04–4.00 (m, 2H), 3.96 (dd, 1H, J = 12.8 Hz & 4.4 Hz), 3.87 (dd, 1H, J = 10.3 Hz & 9.1 Hz), 3.66 (s, 3H), 3.64–3.61 (m, 2H), 3.56–3.43 (m, 5H), 3.40 (dd, 1H, J = 9.6 Hz & 9.2 Hz), 3.24 (dd, 1H, J = 10.5 Hz & 3.9 Hz), 2.24 (m, 1H), 1.46 (ddd, 1H, J = 12.3 Hz, each), 1.16–1.06 (m, 28H); 13C (125 MHz, CD3OH): δ 103.4, 97.9, 87.3, 81.4, 80.0, 77.7, 74.9, 72.0, 71.3, 71.14, 71.08, 63.4, 61.0, 59.9, 59.22, 59.18, 51.1, 31.5, 16.5, 16.42, 16.38, 16.30, 16.29, 16.2, 16.1; HRMS (ESI-TOF) m/z: [M + K]+ calcd. for C30H54KN12O11Si2 853.3211, found 853.3174.
6,3′,4′-Tri-O-levulinoyl-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin (15). Trimethylphosphine (1 mol L−1 Me3P in toluene, 5.1 mL, 5.1 mmol) was added to a mixture of 13 (0.42 g, 0.51 mmol) in water–dioxane (1:4, v/v, 5.0 mL). The mixture was stirred at ambient temperature for 4 h under nitrogen and then concentrated ammonia (1.0 mL) was added. After overnight reaction, the mixture was evaporated to dryness and the residue was coevaporated with pyridine. The residue was dissolved in methanol (4.0 mL) and triethylamine (2.0 mL), and methyl trifluoroacetate (0.41 mL, 4.1 mmol) was added to the mixture. The reaction was stirred at ambient temperature for 5 h. Saturated NaHCO3 was added and the mixture was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and evaporated to dryness. The residue was dissolved in pyridine (5.0 mL) and then freshly prepared levulinic anhydride (0.66 g, 3.1 mmol) and a catalytic amount of DMAP were added to the mixture. After overnight stirring, methanol and saturated NaHCO3-were added to the mixture and the crude product was extracted with ethyl acetate. The combined organic layers were dried over NaSO4, filtered and evaporated to dryness. The residue was subjected to a silica gel chromatography (30% petroleum ether in ethyl acetate) to yield 0.58 g (81 %) of 15 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.05 (d, 1H, J = 3.7 Hz), 5.51 (dd, 1H, J = 10.1 Hz, both), 4.97–4.90 (m, 3H), 4.37 (d, 1H, J = 3.9 Hz), 4.34 (m, 1H), 4.26–4.11 (m, 2H), 4.08–4.03 (m, 2H), 3.95 (dd, 1H, J = 13.2 Hz & 1.5 Hz), 3.84–3.80 (m, 2H), 3.73 (m, 1H), 3.64 (dd, 1H, J = 14.6 Hz & 3.0 Hz), 3.67 (s, 3H), 3.59–3.43 (m, 2H), 2.83–2.41 (m, 12H), 2.17, 2.16 and 2.15 (3 × s, 9H), 2.04–1.95 (m, 2H), 1.14–1.06 (m, 28H); 13C (125 MHz, CD3OH): δ 206.4, 205, 9, 205.0, 170.9, 170, 8, 170.4, 156.4–155.6 (m), 118.0–111.0 (m), 107.6, 93.8, 82.9, 82.4, 78.7, 73.9, 73.8, 69.0, 68.7, 67.8, 65.4, 58.3, 56.6, 49.9, 49.2, 47.2, 46.7, 37.9, 35.7, 35.6, 35.5, 29.5, 26.7, 26.65, 26.62, 26.57, 26.1, 25.9, 25.8, 25.7, 15.02, 14.93, 14.92, 14.89, 14.88, 14.78, 14.76, 14.7, 14.6, 14.5, 11.9, 11.6, 11.5, 11.0, 10.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C53H76F12N4NaO21Si2 1411,4247, found 1411.4283.
6,3′,4′-Tri-O-levulinoyl-3′′,5′′-O-(tetraisopropyldisiloxane-1,3-diyl)-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α1′′−5-ribostamycin (16). The α-anomer (16) was synthesized as described for 15 above. 0.41 g (0.50 mmol) of 14 gave 0.42 g (59%) of 16 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.02 (d, 1H, J = 3.9 Hz), 5.24 (dd, 1H, J = 10.7 Hz & 9.6 Hz), 5.06 (dd, 1H, J = 10.1 Hz & 9.1 Hz), 5.02 (d, 1H, J = 3.9 Hz), 4.96 (dd, 1H, J = 9.9 Hz, both), 4.39 (dd, 1H, J = 10.9 Hz & 3.9 Hz), 4.25–4.19 (m, 2H), 4.13 (m, 1H), 4.04–4.00 (m, 2H), 3.97–3.91 (m, 2H), 3.88 (dd, 1H, J = 12.7 Hz & 2.8 Hz), 3.80 (dd, 1H, J = 12.7 Hz & 4.3 Hz), 3.66–3.58 (m, 2H), 3.54 (s, 3H), 3.55–3.50 (m, 1H), 2.85–2.75 (m, 6H), 2.71–2.40 (m, 6H), 2.17 (s, 3H), 2.15 (s, 2 × 3H), 2.04–1.95 (m, 2H), 1.14–1.05 (m, 28H); 13C (125 MHz, CD3OH): δ 207.8, 207.4, 207.2, 172.6, 172.02, 171.98, 158.4–156.8 (m), 119.5–111.5 (m), 102.9, 95.7, 85.3, 81.5, 79.7, 76.1, 74.4, 70.8, 70.2, 68.5, 67.5, 60.5, 58.5, 51.8, 48.8, 48.5, 38.6, 37.2, 37.0, 30.9, 28.3, 28.1, 27.8, 27.5, 27.2, 16.5, 16.4, 16.3, 16.24, 16.21, 16.19, 16.1, 13.3, 13.1, 13.0, 12.6, 12.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C53H76F12N4NaO21Si2 1411,4247, found 1411.4190.
6,3′,4′-Tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin (17). Compound 15 (0.48 g, 0.35 mmol) was dissolved in a mixture of triethylamine trihydrofluoride and acetonitrile (1:10, v/v, 2.0 mL). The reaction was stirred overnight at ambient temperature, 10% aqueous KH2PO4 was added and the mixture was extracted with ethyl acetate. The combined organic layers were washed with saturated NaHCO3 and saturated NaCl, dried with Na2SO4, filtered and evaporated to dryness. The crude product was purified by a silica gel chromatography (10 % MeOH in CH2Cl2) to yield 0.35 g (73 %) of 17 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.11 (d, 1H, J = 3.5 Hz), 5.21 (dd, 1H, J = 10.6 Hz & 9.6 Hz), 5.16 (d, 1H, J = 4.3 Hz), 4.99 (dd, 1H, J = 10.2 Hz & 9.6 Hz), 4.95 (dd, 1H, J = 9.8 Hz, both), 4.38 (dd, 1H, J = 10.8 Hz & 3.9 Hz), 4.24 (m, 1H), 4.15 (m, 1H), 4.12 (dd, 1H, J = 4.1 Hz, both), 4.04–4.01 (m, 2H), 3.85 (dd, 1H, J = 10.0 Hz & 8.8 Hz), 3.78 (m, 1H), 3.65 (dd, 1H, J = 14.4 Hz & 4.0 Hz), 3.56–3.52 (m, 3H), 3.47 (dd, 1H, J = 14.5 Hz & 2.4 Hz), 3.39 (s, 3H), 2.84–2.72 (m, 6H), 2.70–2.41 (m, 6H), 2.19 (s, 3H), 2.17 (s, 3H), 2.14 (s, 3H), 2.01–1.97 (m, 2H); 13C (125 MHz, CD3OH): δ 208.0, 207.7, 207.6, 172.6, 171.98, 171.96, 158.5–156.9 (m), 119.5–112.5 (m), 108.1, 95.1, 84.7, 85.6, 82.9, 75.5, 75.1, 70.1, 69.0, 68.6, 67.5, 61.6, 57.5, 48.7, 48.5, 39.0, 37.3, 37.1, 30.8, 28.3, 28.2, 27.8, 27.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C41H50F12N4NaO20 1169.2724, found 1169.2744.
6,3′,4′-Tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α(1′′−5)-ribostamycin (18). The α-anomer (18) was synthesized as described for 17 above. 0.35 g (0.25 mmol) of 16 gave 0.28 g (80%) of 18 as white foam. 1H NMR (500 MHz, CD3OH): δ 6.09 (d, 1H, J = 2.6 Hz), 5.22 (dd, 1H, J = 10.7 Hz & 9.6 Hz), 5.08–5.04 (m, 2H), 4.94 (dd, 1H, J = 9.9 Hz & 9.8 Hz), 4.36 (dd, 1H, J = 10.9 Hz & 3.9 Hz), 4.22 (m, 1H), 4.17–4.12 (m, 2H), 4.04–3.98 (m, 3H), 3.89 (dd, 1H, J = 10.1 Hz & 8.8 Hz), 3.65 (dd, 1H, J = 14.8 Hz & 3.5 Hz), 3.58–3.54 (m, 3H), 3.49 (dd, 1H, J = 14.4 Hz & 2.0 Hz), 3.35 (s, 3H), 2.86–2.41 (m, 12H), 2.17 (s, 2 × 3H), 2.15 (s, 3H), 2.04–1.94 (m, 2H); 13C (125 MHz, CD3OH): δ 207.9, 207.8, 207.5, 172.9, 172.0, 158.4–156.9 (m), 119.4–112.5 (m), 103.8, 96.1, 86.9, 84.1, 80.9, 77.2, 74.3, 70.2, 68.5, 67.7, 67.6, 61.8, 57.2, 53.5, 48.7, 48.6, 38.6, 37.3, 37.2, 37.1, 30.9, 28.4, 28.3, 28.0, 27.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C41H50F12N4NaO20 1169.2724, found 1169.2716.
5′′-O-(4,4′-Dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin (19). 17 (0.35 g, 0.31 mmol) was dissolved in dry pyridine (2.0 mL) and 4′,4′-dimethoxytritylchloride (0.12 g, 0.34 mmol) was added. The mixture was stirred overnight at ambient temperature, poured to saturated aqueous NaHCO3 and the product was extracted with dichloromethane. The organic layers were combined, dried over Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel chromatography (10% MeOH in CH2Cl2) to give 0.37 g (83%) of the product (19) as white foam. 1H NMR (500 MHz, CD3OH): δ 7.48 (2H, d, J = 7.5 Hz), 7.35–7.32 (m, 6H), 7.25 (m, 1H), 6.92 (d, 4H, J = 8.8 Hz), 6.16 (d, 1H, J = 4.0 Hz), 5.18 (d, 1H, J = 4.9 Hz), 5.14 (dd, 1H, J = 10.4 Hz & 9.8 Hz), 5.07 (dd, 1H, J = 10.2 Hz & 9.7 Hz), 4.88 (dd, 1H, J = 9.9 Hz, both), 4.23 (m, 1H), 4.16 (m, 1H), 4.05–3.97 (m, 4H), 3.90 (dd, 1H, J = 10.2 Hz & 8.9 Hz), 3.85 (m, 1H), 3.82 (s, 2 × 3H), 3.63 (dd, 1H, J = 4.6 Hz, both), 3.54 (dd, 1H, J = 14.6 Hz & 4.3 Hz), 3.42 (dd, 1H, J = 14.6 Hz & 3.4 Hz), 3.31 (s, 3H), 3.21 (d, 2H, J = 4.0 Hz), 2.84–2.71 (m, 6H), 2.69–2.40 (m, 6H), 2,17 (s, 3H), 2.16 (s, 3H), 2.14 (s, 3H), 2.07 (ddd, 1H, J = 12.8 Hz, each), 2.00 (ddd, 1H, J = 12.8 Hz, 4.6 Hz & 4.6 Hz); 13C (125 MHz, CD3OH): δ 207.9, 207.6, 207.4, 172.6, 172.2, 171.8, 158.84, 158.80, 158.4–156.6 (m), 145.1, 135.7, 135.4, 130.2, 130.1, 128.0, 127.4, 126.4, 119.5–112.5 (m), 112.8, 112.7, 108.3, 94.9, 86.2, 84.8, 83.9, 83.2, 75.1, 75.0, 70.2, 69.2, 69.0, 66.9, 62.5, 57.3, 54.4, 51.2, 48.7, 48.5, 39.1, 37.3, 37.1, 30.8, 28.2, 28.15, 28.12, 27.9, 27.49, 27.46. HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C62H68F12N4NaO22 1471.4031, found 1471.4055.
5′′-O-(4,4′-Dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α1′′−5-ribostamycin (20). The α-anomer (20) was synthesized as described for 19 above. 0.28 g (0.24 mmol) of 18 gave 0.25 g (71%) of 20 as white foam. 1H NMR (500 MHz, CD3OH): δ 7.41 (d, 2H, J = 7.6 Hz), 7.31–7.28 (m, 6H), 7.22 (m, 1H), 6.86 (d, 4H, J = 8.7 Hz), 6.12 (b, 1H), 5.26 (dd, 1H, J = 10.4 Hz & 9.9 Hz), 5.16 (d, 1H, J = 4.3 Hz), 5.06 (dd, 1H, J = 9.9 Hz & 9.4 Hz), 4.95 (dd, 1H, J = 9.9Hz, both), 4.38 (dd, 1H, J = 10.9 Hz & 3.9 Hz), 4.27–4.14 (m, 3H), 4.08–4.04 (m, 3H), 3.91 (dd, 1H, J = 10.2 Hz & 9.1 Hz), 3.81 (m, 1H), 3.79 (s, 2 × 3H), 3.68 (dd, 1H, J = 14.8 Hz & 3.2 Hz), 3.50 (dd, 1H, J = 14.8 Hz & 2.0 Hz), 3.37 (s, 3H), 3.21 (dd, 1H, J = 10.5 Hz & 3.5 Hz), 3.09 (dd, 1H, J = 10.4 Hz & 3.2 Hz), 2.84–2.71 (m, 4H), 2.61–2.41 (m, 8H), 2.14 (s, 3H), 2.12 (s, 3H), 2.02 (s, 3H), 2.00–1.96 (m, 2H); 13C (125 MHz, CD3OH): δ 207.8, 207.4, 207.1, 172.7, 172.9, 171.9, 158.1, 158.4–156.8 (m), 144.9, 135.7, 135.6, 129.9, 127.8, 127.4, 126.5, 119.5–112.5 (m), 112.7, 104.0, 96.2, 86.1, 85.8, 81.1, 77.4, 74.3, 70.2, 68.6, 67.8, 67.7, 63.6, 57.2, 54.3, 51.7, 48.73, 48.70, 38.6, 37.13, 37.05, 31.0, 28.4, 28.17, 28.15, 28.0, 27.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C62H68F12N4NaO22 1471.4031, found 1471.3997.
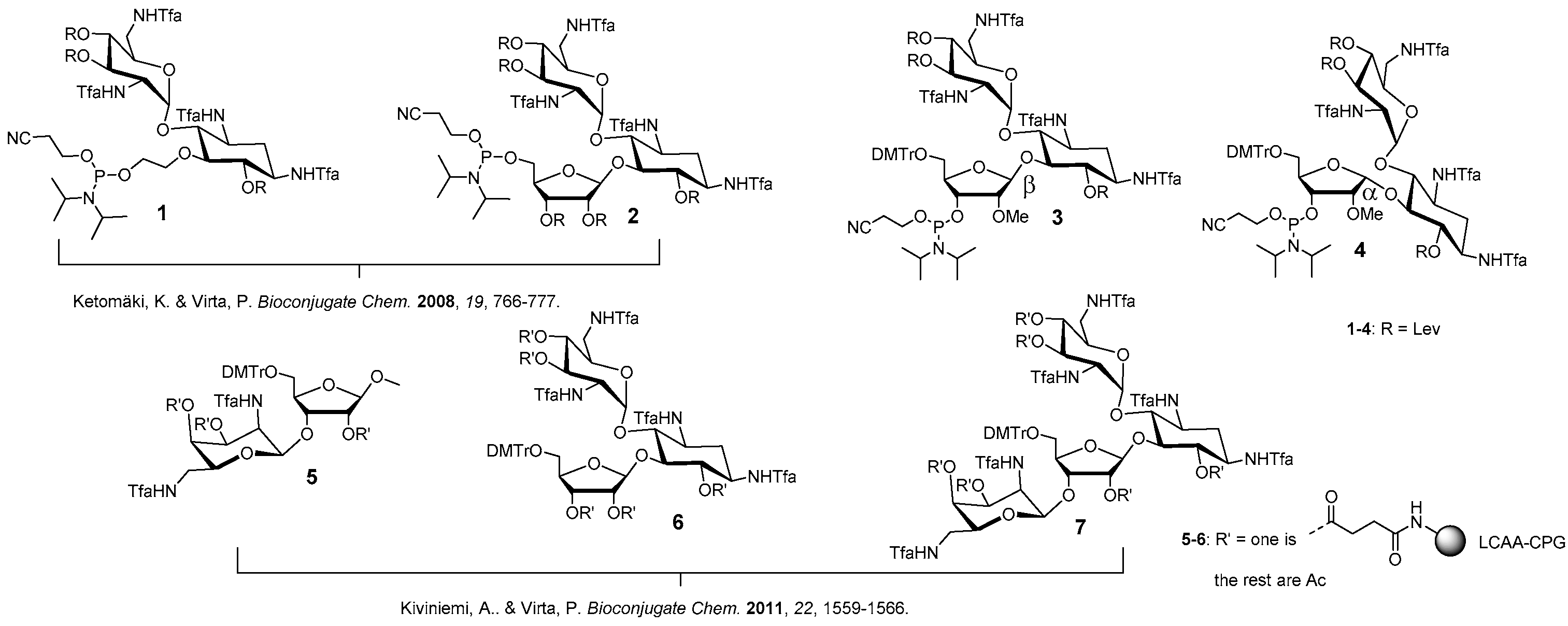
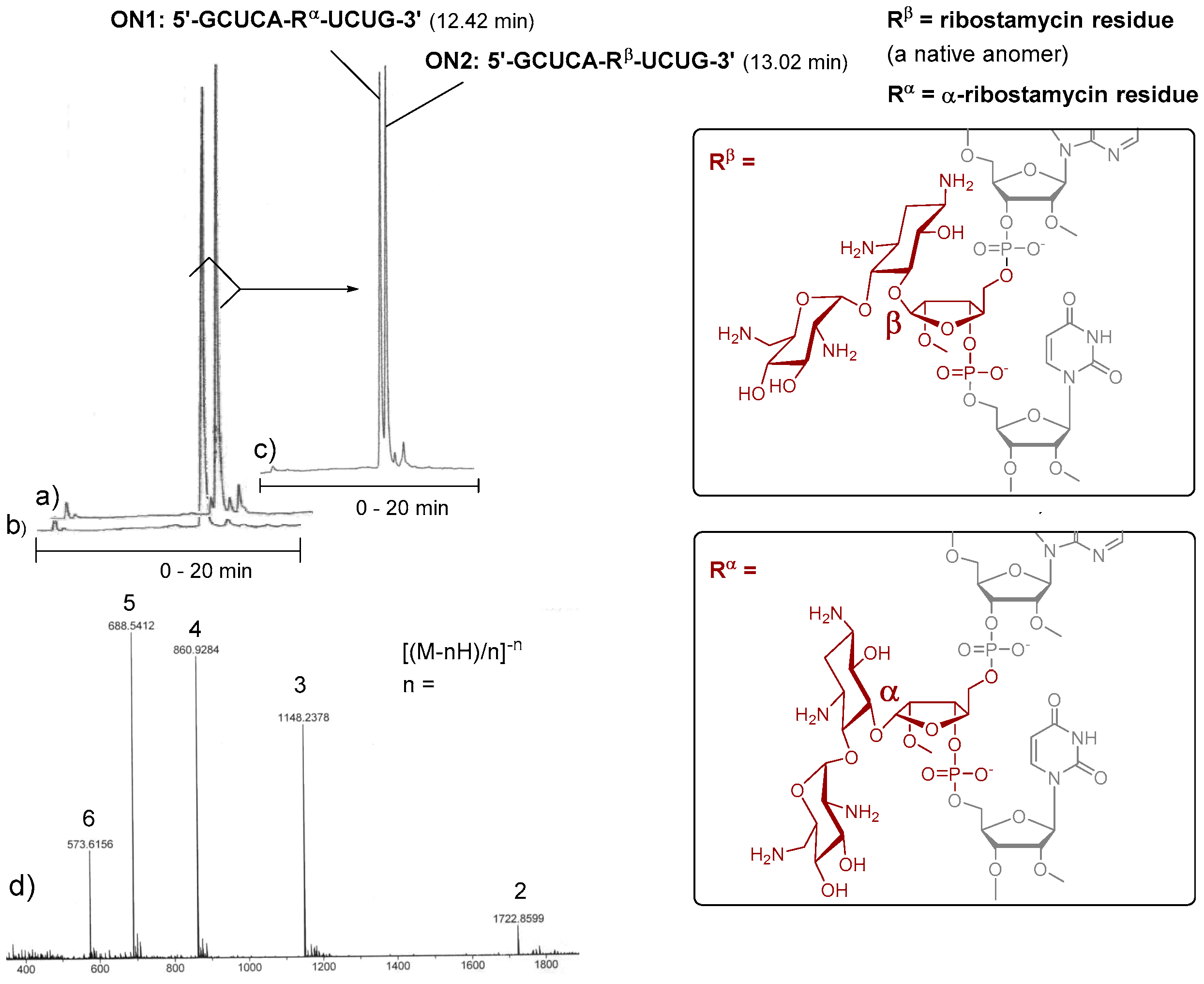
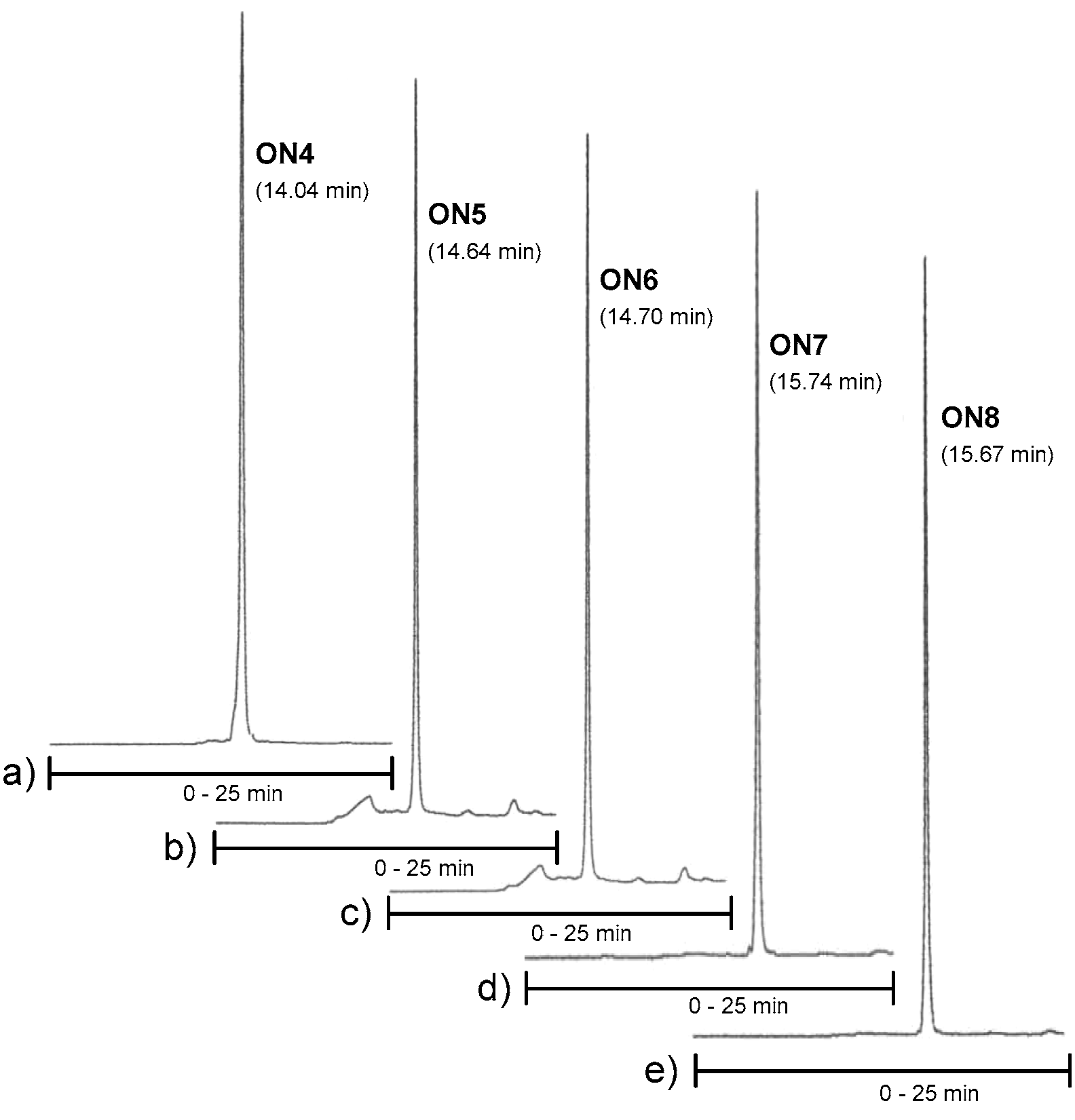
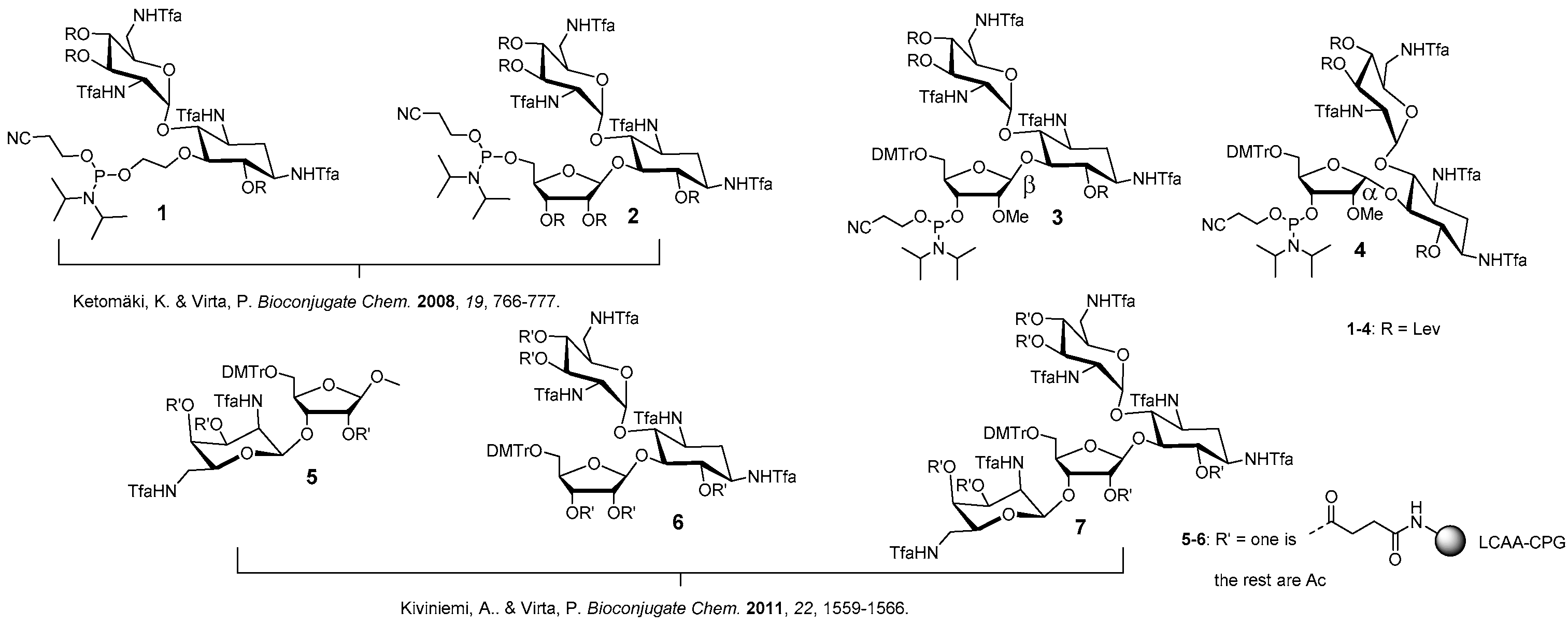
2-Cyanoethyl [5′′-O-(4,4′-dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl ribostamycin-3′′-O-yl] N,N-diisopropylphosphoramidite (3). Triethylamine (0.16 mL, 1.3 mmol) and 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (0.12 g, 0.52 mmol) were added to a mixture of 19 (0.37 g, 0.26 mmol) in dry dichloromethane (1.0 mL). The mixture was stirred at ambient temperature for 1 h under nitrogen and filtered through a short silica gel column (5% Et3N in EtOAc). The product fractions were evaporated to dryness to give 0.40 g (96%) of the product (3) as white foam. 1H NMR (500 MHz, CD3OH): δ 8.00 (d, 1H, J = 8.7 Hz), 7.48–7.44 (2H), 7.38–7.27 (m, 6H), 7.20–7.25 (m, 2H), 6.95–6.92 (m, 4H), 6.13 and 6.03 (2 × d, 1H, J = 3.8 Hz and 3.9 Hz), 5.15 (d, 1H, J = 6.2 Hz), 5.12–5.08 (m, 1H), 5.01–4.97 (m, 1H), 4.92–4.83 (m, 1H), 4.35–3.05 (m, 13H), 3.82 (s, 6H), 3.26 and 3.23 (2 × s, 3H), 2.81–2.31 (m, 14H), 2.13, 2.11 and 2.10 (3 × s, 9H), 2.15–1.96 (m, 2H), 1.32–1.08 (m, 12H); 13C (125 MHz, CD3OH): δ 207.01, 206.99, 206.45, 206.42, 206.1, 172.9, 172.8, 172.3, 172.2, 171.91, 171.88, 158.8, 157.4–156.6 (m), 145.2, 145.0, 135.61, 135.59, 135.43, 135.37, 130.31, 130.28, 128.1, 127.9, 126.94, 126.90, 118.6, 118.2, 119.5–112.5 (m), 113.1, 113.0, 108.4, 108.3, 94.5, 94.4, 86.5, 86.3, 84.3, 84.2, 83.8, 83.7, 83.3, 83.2, 75.0, 74.8, 74.7, 71.9, 71.8, 70.9, 70.8, 70.1, 70.0, 68.9, 68.8, 67.2, 67.0, 63.1, 62.9, 58.9, 58.8, 58.2, 58.1, 58.0, 57.9, 57.8, 57.6, 57.5, 55.0, 51.6, 51.5, 48.9, 48.8, 48.7, 48.6, 43.2, 43.1, 42.9, 42.8, 39.5, 39.4, 37.6, 37.5, 37.3, 37.2, 31.0, 28.9, 28.8, 28.8, 28.7, 28.0, 27.6, 24.1, 24.0, 23.9, 23.9, 23.8, 23,7, 20.0, 19.9, 19.9, 19.8; 31P (200 MHz, CD3CN): δ 150.4, 148.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C71H86F12N6O23P 1649.5290, found 1649.5232.
2-Cyanoethyl [5′′-O-(4,4′-dimethoxytrityl)-6,3′,4′-tri-O-levulinoyl-tetra-N1,N3,N2′,N6′-trifluoroacetyl-2′′-O-methyl-α1′′−5-ribostamycin-3′′-O-yl] N,N-diisopropylphosphoramidite (4). The α-anomer (4) was synthesized as described for 3 above. 0.25 g (0.17 mmol) of 20 gave 0.28 g (98%) of 4 as white foam. 1H NMR (500 MHz, CD3OH): δ 8.00–7.90 (m, 1H), 7.55–7.37 (m, 4H), 7.37–7.16 (m, 7H), 6.91–6.87 (m, 4H), 6.18 and 6.87 (2 × b, 1H), 5.30–4.94 (m, 4H), 4.45–3.00 (m, 13H), 3.80 and 3.79 (2 × s, 6H), 3.38 and 3.28 (2 × s, 3H), 2.80–2.28 (m, 14H), 2.08–1.88 (m, 2H), 2.11, 2.10, 2.09, 2.02, 2.00 and 1.99 (6 × s, 9H), 1.33–1.14 (m, 12H); 13C (125 MHz, CD3OH): δ 207.0, 206.9, 206.5, 206.2, 206.0, 172.8, 172.6, 172.5, 172.4, 172.1, 172.0, 158.7, 157.5–156.4 (m), 145.2, 145.0, 136.0, 135.7, 135.7, 135.5, 130.1, 130.0, 129.9, 129.9, 127.9, 127.9, 127.8, 127.8, 126.9, 119.4, 118.4, 116.9–110.4 (m), 113.1, 113.1, 113.0, 104.3, 104.0, 96.2, 95.8, 86.1, 85.9, 84.7, 81.8, 80.9, 74.2, 73.8, 70.0, 69.7, 68.9, 68.3, 68.3, 67.8, 63.6, 63.1, 60.0, 59.1, 58.6, 57.7, 57.5, 57.5, 57.4, 54.9, 54.9, 52.0, 51.7, 49.6, 49.2, 48.7, 48.7, 43.2, 42.8, 39.2, 38.7, 37.4, 37.3, 37.3, 37.2, 31.1, 31.0, 28.8, 28.7, 28.2, 28.0, 27.6, 24.0, 23.9, 23.8, 23.7, 20.2, 20.1, 19.8, 19.7; 31P (200 MHz, CD3CN): δ 150.95, 149.60; HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C71H86F12N6O23P 1649.5290, found 1649.5221.




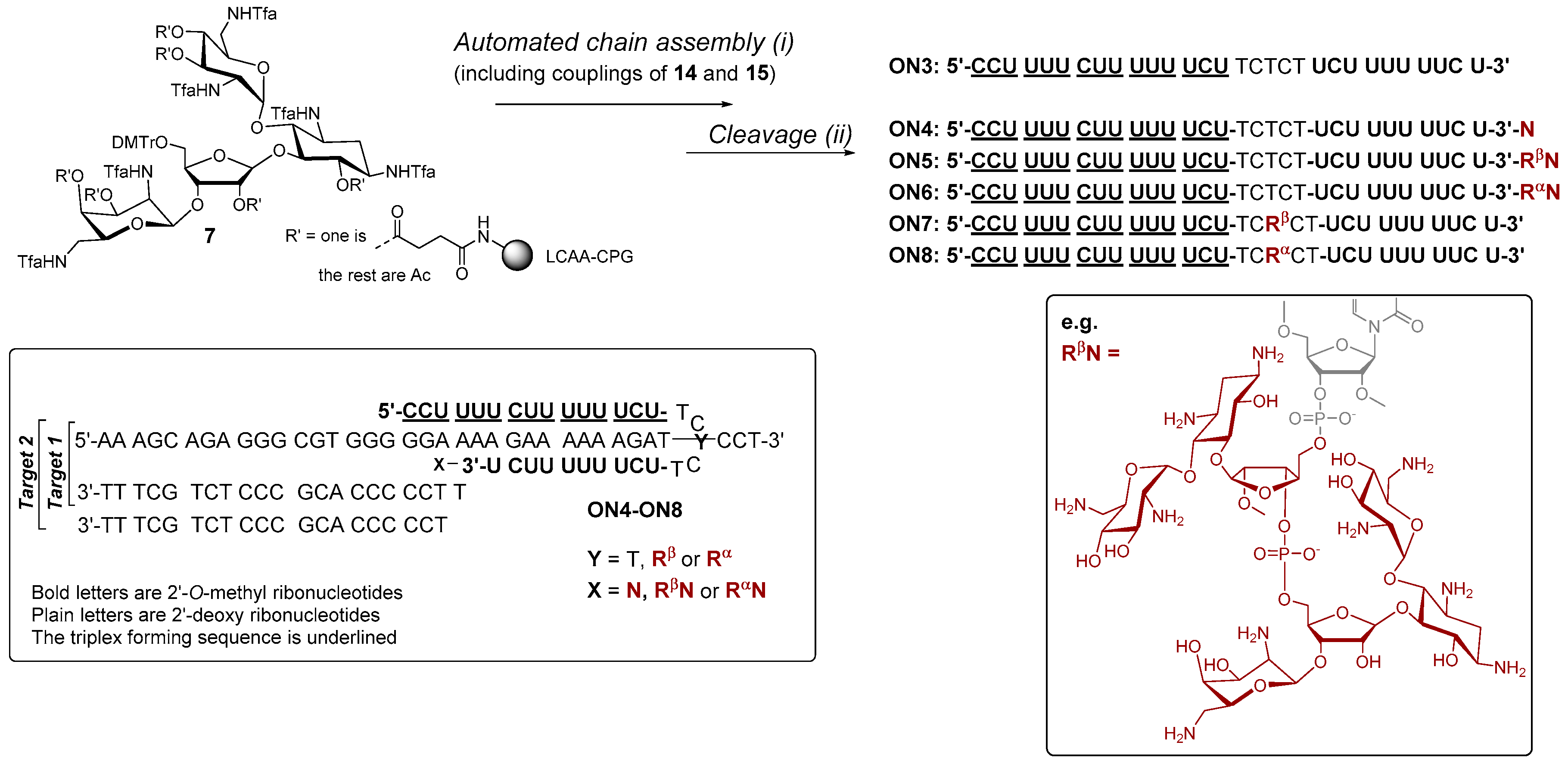
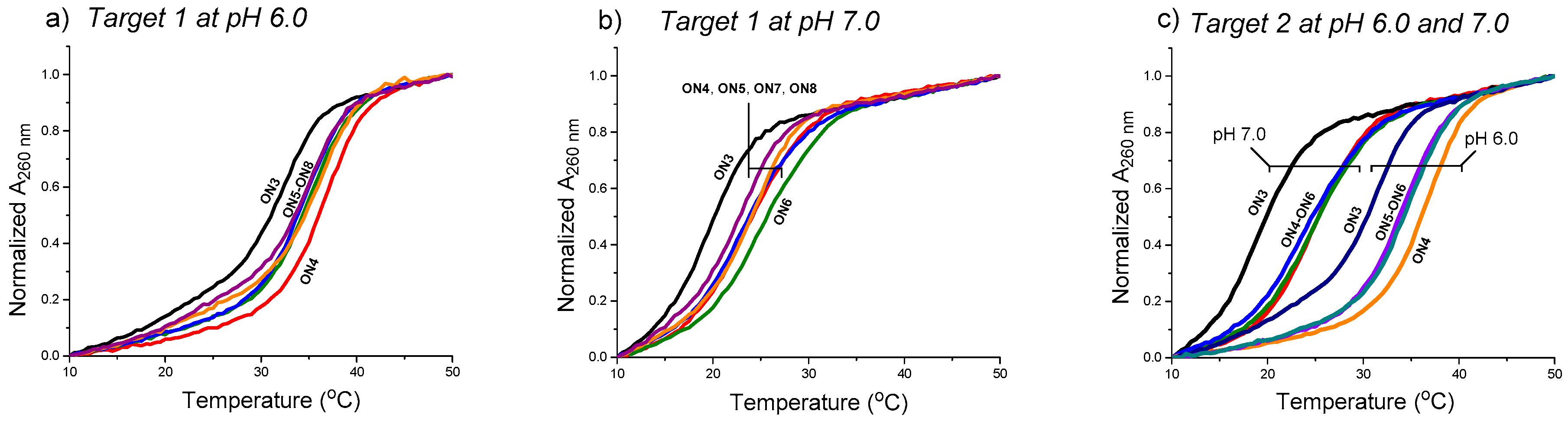

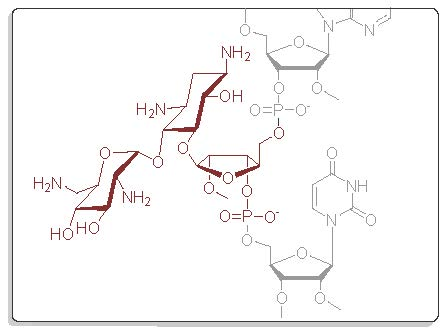{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
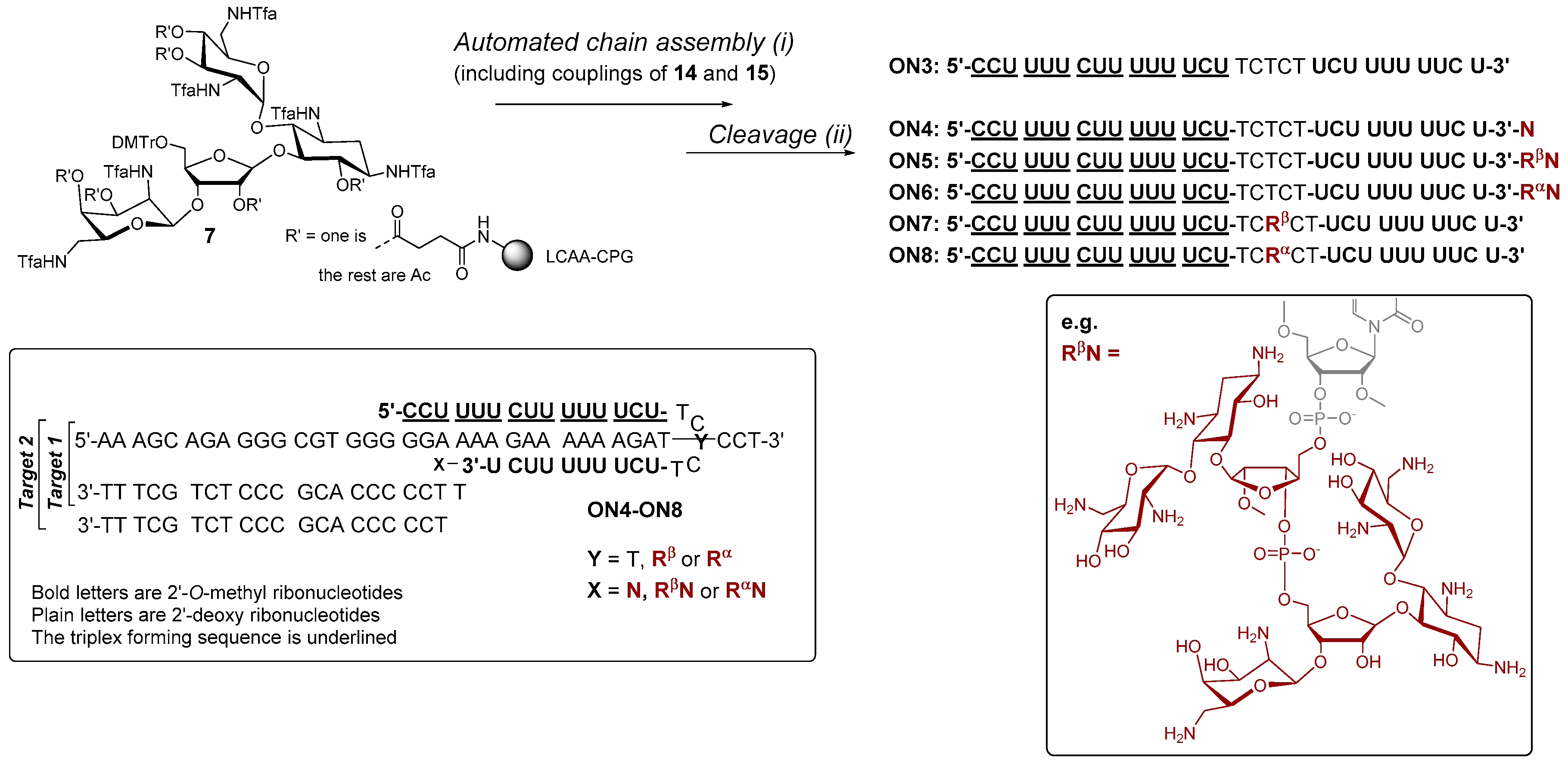{kind=link}