
Nerolidol and Farnesol Inhibit Some Cytochrome P450 Activities but Did Not Affect Other Xenobiotic-Metabolizing Enzymes in Rat and Human Hepatic Subcellular Fractions

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Screening for Enzyme Inhibition
2.2. Determination of IC50
2.3. Mechanism of Enzyme Inhibition
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of Liver Microsomal and Cytosolic Fractions
4.3. Enzyme Assays
4.4. Screening for Enzyme Inhibition
4.5. Determination of IC50
4.6. Mechanism of Enzyme Inhibition
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartikova, H.; Hanusova, V.; Skalova, L.; Ambroz, M.; Bousova, I. Antioxidant, Pro-Oxidant and Other Biological Activities of Sesquiterpenes. Curr. Top. Med. Chem. 2014, 14, 2478–2494. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.M.; Lin, J.Y. Farnesol, a sesquiterpene alcohol in essential oils, ameliorates serum allergic antibody titres and lipid profiles in ovalbumin-challenged mice. Allergol. Immunopath. 2016, 44, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.M.; Lin, J.Y. Farnesol, a Sesquiterpene Alcohol in Herbal Plants, Exerts Anti-Inflammatory and Antiallergic Effects on Ovalbumin-Sensitized and -Challenged Asthmatic Mice. Evid.-Based Complement. Altern. 2015. [Google Scholar] [CrossRef] [PubMed]
- Santhanasabapathy, R.; Sudhandiran, G. Farnesol attenuates lipopolysaccharide-induced neurodegeneration in Swiss albino mice by regulating intrinsic apoptotic cascade. Brain Res. 2015, 1620, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.H.; Jetten, A.M. Molecular mechanisms involved in farnesol-induced apoptosis. Cancer Lett. 2010, 287, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Kim, S.H.; Sethi, G.; Ahn, K.S. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 2015, 360, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Szucs, G.; Murlasits, Z.; Torok, S.; Kocsis, G.F.; Paloczi, J.; Gorbe, A.; Csont, T.; Csonka, C.; Ferdinandy, P. Cardioprotection by Farnesol: Role of the Mevalonate Pathway. Cardiovasc. Drug Ther. 2013, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Tan, L.T.H.; Chan, K.G.; Lee, L.H.; Goh, B.H. Nerolidol: A Sesquiterpene Alcohol with Multi-Faceted Pharmacological and Biological Activities. Molecules 2016, 21, 529. [Google Scholar] [CrossRef] [PubMed]
- AbouLaila, M.; Sivakumar, T.; Yokoyama, N.; Igarashi, I. Inhibitory effect of terpene nerolidol on the growth of Babesia parasites. Parasitol. Int. 2010, 59, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.P.N.; Oliveira, G.L.S.; De Carvalho, R.B.F.; De Sousa, D.P.; Freitas, R.M.; Pinto, P.L.S.; De Moraes, J. Antischistosomal Activity of the Terpene Nerolidol. Molecules 2014, 19, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.Y.; Rodriguez, A.A.M.; Vega, D.S.M.; Sussmann, R.A.C.; Kimura, E.A.; Katzin, A.M. Antimalarial activity of the terpene nerolidol. Int. J. Antimicrob. Agent 2016, 48, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Azimullah, S.; Khair, S.B.A.; Ojha, S.; Haque, M.E. Neuroprotective effect of nerolidol against neuroinflammation and oxidative stress induced by rotenone. BMC Neurosci. 2016, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Ryabchenko, B.; Tulupova, E.; Schmidt, E.; Wlcek, K.; Buchbauer, G.; Jirovetz, L. Investigation of anticancer and antiviral properties of selected aroma samples. Nat. Prod. Commun. 2008, 3, 1085–1088. [Google Scholar]
- Malatkova, P.; Wsol, V. Carbonyl reduction pathways in drug metabolism. Drug Metab. Rev. 2014, 46, 96–123. [Google Scholar] [CrossRef] [PubMed]
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 2008, 40, 553–624. [Google Scholar] [CrossRef] [PubMed]
- Bousova, I.; Skalova, L.; Soucek, P.; Matouskova, P. The modulation of carbonyl reductase 1 by polyphenols. Drug Metab. Rev. 2015, 47, 520–533. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Maser, E. Carbonyl reductases and pluripotent hydroxysteroid dehydrogenases of the short-chain dehydrogenase/reductase superfamily. Drug Metab. Rev. 2007, 39, 87–144. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, U. Carbonyl reductases: The complex relationships of mammalian carbonyl- and quinone-reducing enzymes and their role in physiology. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.J.; Wang, X.D.; Morris, M.E. Dietary flavonoids: Effects on xenobiotic and carcinogen metabolism. Toxicol. In Vitro 2006, 20, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Taneja, I.; Raju, K.S.R.; Wahajuddin, M. Dietary Isoflavones as Modulators of Drug Metabolizing Enzymes and Transporters: Effect on Prescription Medicines. Crit. Rev. Food Sci. 2016, 56, S95–S109. [Google Scholar] [CrossRef] [PubMed]
- Bousova, I.; Skalova, L. Inhibition and induction of glutathione S-transferases by flavonoids: Possible pharmacological and toxicological consequences. Drug Metab. Rev. 2012, 44, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Hodek, P. Flavonoids. In Metabolism of Drugs and Other Xenobiotics; Anzenbacher, P., Zanger, U.M., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp. 543–582. [Google Scholar]
- Bamba, Y.; Yun, Y.S.; Kunugi, A.; Inoue, H. Compounds isolated from Curcuma aromatica Salisb. inhibit human P450 enzymes. J. Nat. Med. 2011, 65, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Pimkaew, P.; Kublbeck, J.; Petsalo, A.; Jukka, J.; Suksamrarn, A.; Juvonen, R.; Auriola, S.; Piyachaturawat, P.; Honkakoski, P. Interactions of sesquiterpenes zederone and germacrone with the human cytochrome P450 system. Toxicol. In Vitro 2013, 27, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.Z.; Lv, Q.L.; Wu, N.Y.Y.; Cheng, L.; Chu, Y.C.; Chu, T.Y.; Hu, L.; Cheng, Y.; Zhang, X.; Zhou, H.H. Mechanism-based inhibition of Alantolactone on human cytochrome P450 3A4 in vitro and activity of hepatic cytochrome P450 in mice. J. Ethnopharmacol. 2015, 168, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.U.; Kwon, S.S.; Kong, T.Y.; Kim, J.H.; Lee, H.S. Inhibitory Effects of Cedrol, beta-Cedrene, and Thujopsene on Cytochrome P450 Enzyme Activities in Human Liver Microsomes. J. Toxicol. Environ. Health Part A 2014, 77, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.D.; Thompson, S.; Weaver, R.J.; Wolf, C.R.; Mayer, R.T. Cytochrome-P450 specificities of alkoxyresorufin O-dealkylation in human and rat liver. Biochem. Pharmacol. 1994, 48, 923–936. [Google Scholar]
- Krasulova, K.; Siller, M.; Holas, O.; Dvorak, Z.; Anzenbacher, P. Enantiospecific effects of chiral drugs on cytochrome P450 inhibition in vitro. Xenobiotica 2016, 46, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Raner, G.M.; Muir, A.Q.; Lowry, C.W.; Davis, B.A. Farnesol as an inhibitor and substrate for rabbit liver microsomal P450 enzymes. Biochem. Biophys. Res. Commun. 2002, 293, 1–6. [Google Scholar] [CrossRef]
- Gillette, J. Techniques for studying drug metabolism in vitro. In Fundamentals of Drug Metabolism and Drug Disposition; La Du, B.N., Mandel, H.G., Way, E.L., Eds.; The Williams and Wilkins Company: Baltimore, MA, USA, 1971; pp. 400–418. [Google Scholar]
- Phillips, I.R.; Shephard, E.A. Cytochrome P450 Protocols; Humana Press: Totowa, NJ, USA, 1998. [Google Scholar]
- Soucek, P. Novel sensitive high-performance liquid chromatographic method for assay of coumarin 7-hydroxylation. J. Chromatogr. B Biomed. Sci. Appl. 1999, 734, 23–29. [Google Scholar] [CrossRef]
- Morse, M.A.; Lu, J. High-performance liquid chromatographic method for measurement of cytochrome P450-mediated metabolism of 7-ethoxy-4-trifluoromethylcoumarin. J. Chromatogr. B Biomed. Sci. Appl. 1998, 708, 290–293. [Google Scholar] [CrossRef]
- Donato, M.T.; Jimenez, N.; Castell, J.V.; Gomez-Lechon, M.J. Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab. Dispos. 2004, 32, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Crespi, C.L.; Chang, T.K.; Waxman, D.J. Determination of CYP2C9-catalyzed diclofenac 4′-hydroxylation by high-performance liquid chromatography. Methods Mol. Biol. 1998, 107, 129–133. [Google Scholar] [PubMed]
- Crespi, C.L.; Chang, T.K.; Waxman, D.J. CYP2C19-mediated (S)-mephenytoin 4′-hydroxylation assayed by high-performance liquid chromatography with radiometric detection. Methods Mol. Biol. 1998, 107, 135–139. [Google Scholar] [PubMed]
- Crespi, C.L.; Chang, T.K.; Waxman, D.J. CYP2D6-dependent bufuralol 1′-hydroxylation assayed by reversed-phase ion-pair high-performance liquid chromatography with fluorescence detection. Methods Mol. Biol. 1998, 107, 141–145. [Google Scholar] [PubMed]
- Lucas, D.; Menez, J.F.; Berthou, F. Chlorzoxazone: An in vitro and in vivo substrate probe for liver CYP2E1. Methods Enzymol. 1996, 272, 115–123. [Google Scholar] [PubMed]
- Guengerich, F.P.; Martin, M.V.; Beaune, P.H.; Kremers, P.; Wolff, T.; Waxman, D.J. Characterization of rat and human liver microsomal cytochrome P-450 forms involved in nifedipine oxidation, a prototype for genetic polymorphism in oxidative drug metabolism. J. Biol. Chem. 1986, 261, 5051–5060. [Google Scholar] [PubMed]
- Ghosal, A.; Satoh, H.; Thomas, P.E.; Bush, E.; Moore, D. Inhibition and kinetics of cytochrome P4503A activity in microsomes from rat, human, and cdna-expressed human cytochrome P450. Drug Metab. Dispos. 1996, 24, 940–947. [Google Scholar] [PubMed]
- Kronbach, T.; Mathys, D.; Umeno, M.; Gonzalez, F.J.; Meyer, U.A. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol. 1989, 36, 89–96. [Google Scholar] [PubMed]
- Veinlichova, A.; Jancova, P.; Siller, M.; Anzenbacher, P.; Kuca, K.; Jun, D.; Fusek, J.; Anzenbacherova, E. Effect of acetylcholinesterase oxime-type reactivators K-48 and HI-6 on human liver microsomal cytochromes P450 in vitro. Chem. Biol. Int. 2009, 180, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Novotna, A.; Krasulova, K.; Bartonkova, I.; Korhonova, M.; Bachleda, P.; Anzenbacher, P.; Dvorak, Z. Dual effects of ketoconazole cis-enantiomers on CYP3A4 in human hepatocytes and HepG2 Cells. PLoS ONE 2014, 9, e111286. [Google Scholar] [CrossRef] [PubMed]
- Kopecna-Zapletalova, M.; Krasulova, K.; Anzenbacher, P.; Hodek, P.; Anzenbacherova, E. Interaction of isoflavonoids with human liver microsomal cytochromes P450: Inhibition of CYP enzyme activities. Xenobiotica 2017, 47, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Mate, L.; Virkel, G.; Lifschitz, A.; Ballent, M.; Lanusse, C. Hepatic and extra-hepatic metabolic pathways involved in flubendazole biotransformation in sheep. Biochem. Pharmacol. 2008, 76, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.J.; Hinkhouse, M.M.; Grady, M.; Gaut, A.W.; Liu, J.R.; Zhang, Y.P.; Weydert, C.J.D.; Domann, F.E.; Oberley, L.W. Dicumarol inhibition of NADPH: Quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003, 63, 5513–5520. [Google Scholar] [PubMed]
- Mizuma, T.; Machida, M.; Hayashi, M.; Awazu, S. Correlation of drug conjugate metabolism rates between in vivo and in vitro glucuronidation and sufatation of para-nitrophenol as a model comound in rat. J. Pharmacobio-Dyn. 1982, 5, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Frame, L.T.; Ozawa, S.; Nowell, S.A.; Chou, H.C.; Delongchamp, R.R.; Doerge, D.R.; Lang, N.P.; Kadlubar, F.F. Simple colorimetric assay for phenotyping the major human thermostable phenol sulfotransferase (SULT1A1) using platelet cytosols. Drug Metabol. Dispos. 2000, 28, 1063–1068. [Google Scholar]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferase A from rat liver. Arch. Biochem. Biophys. 1976, 175, 710–716. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available from the authors. |
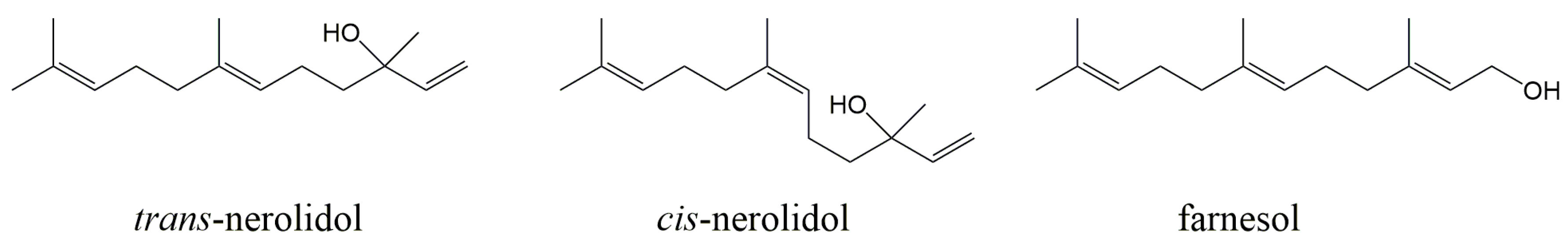

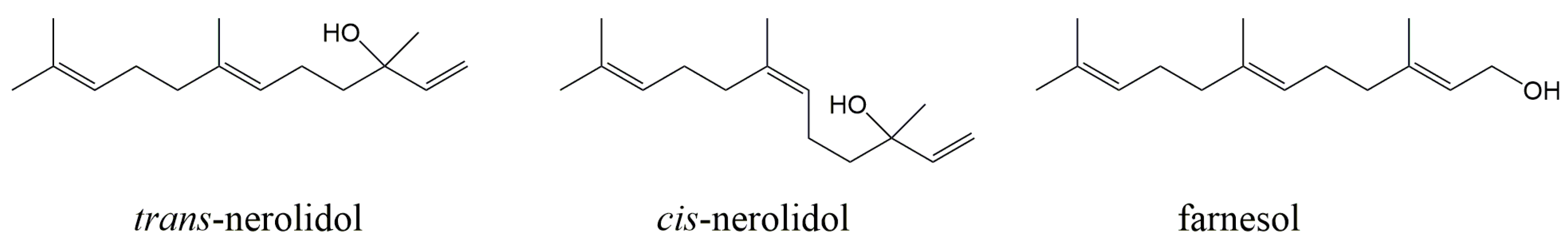{kind=link}
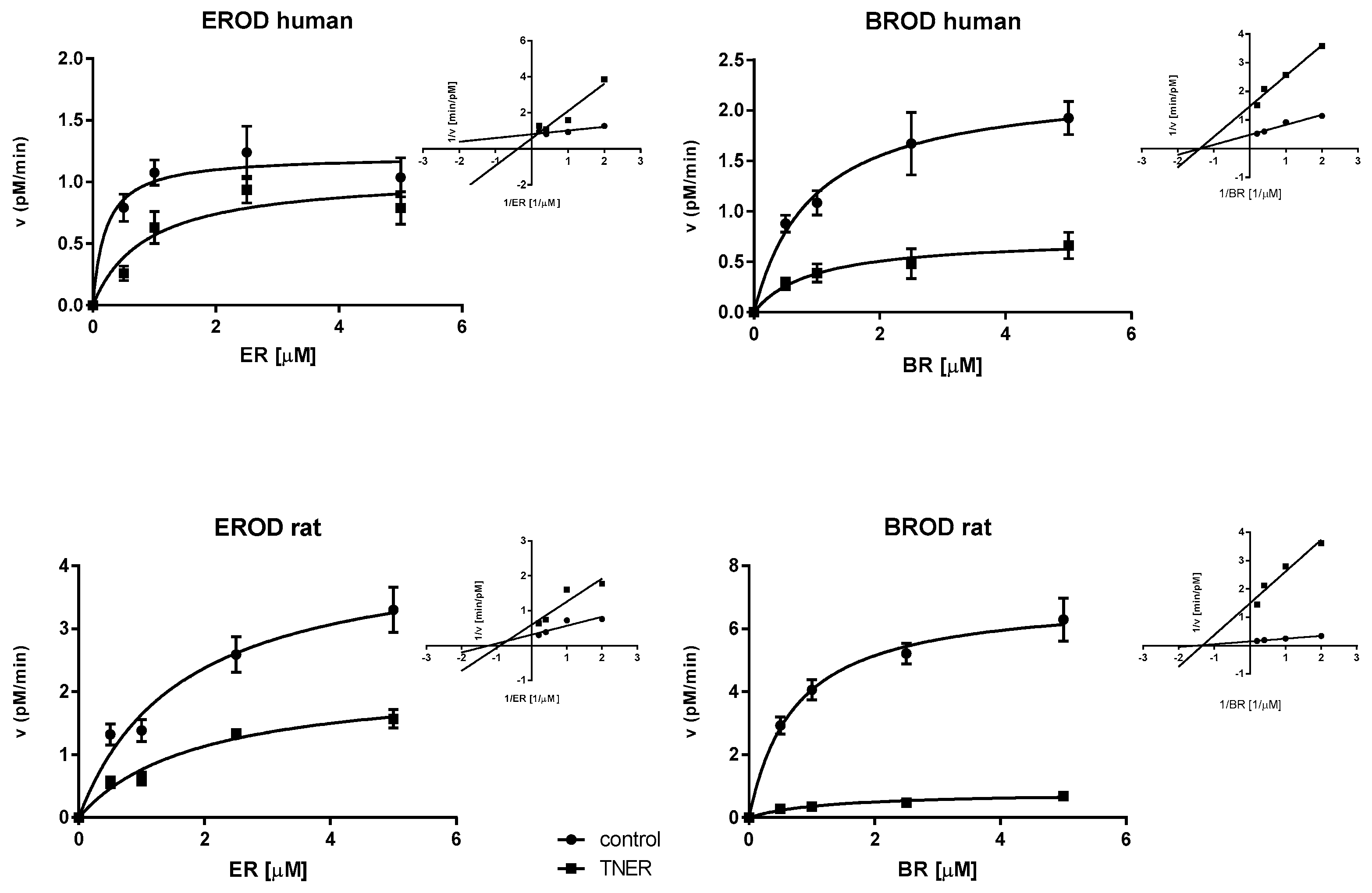
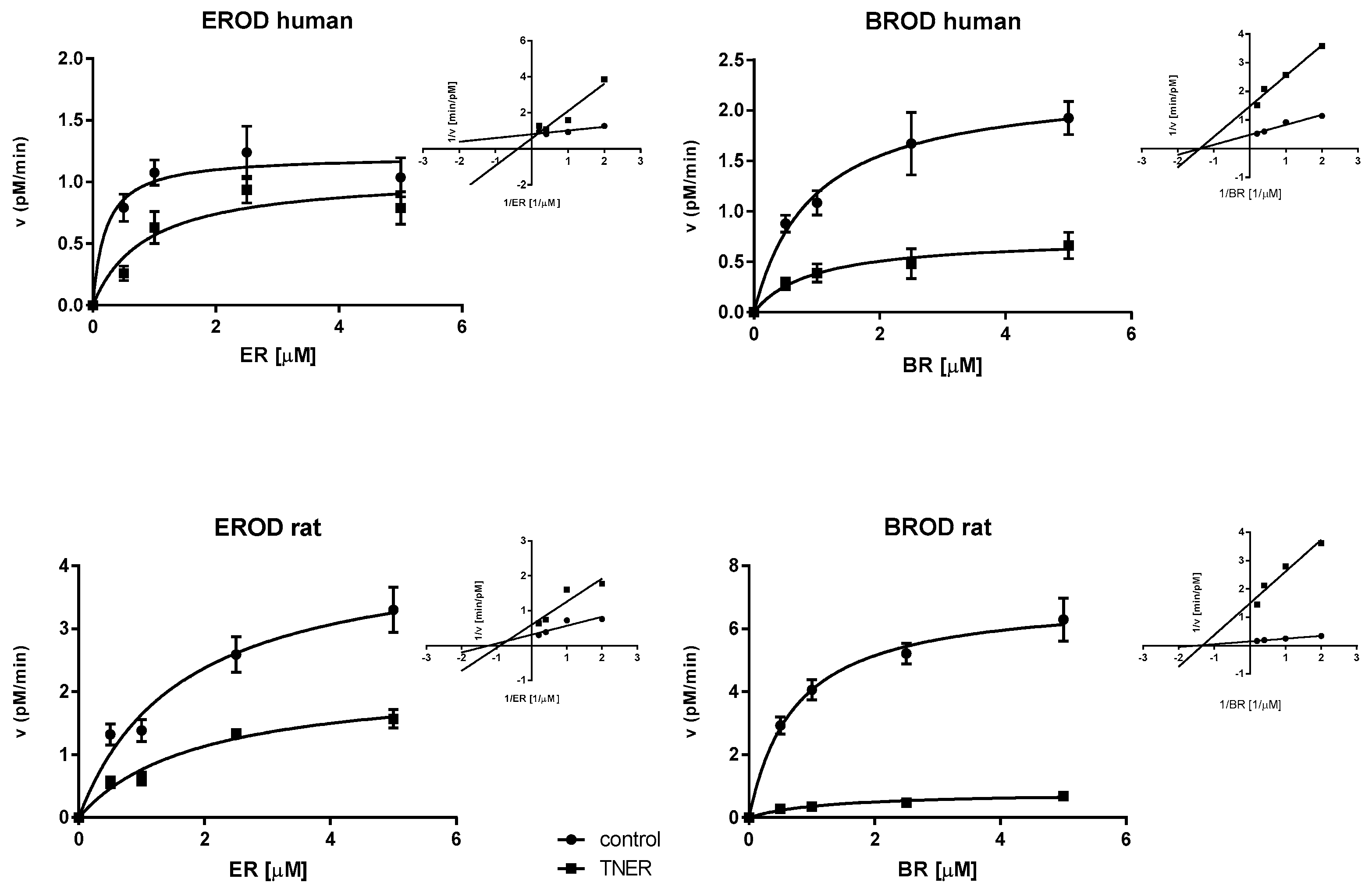{kind=link}
| Specific Activity (pmol/mg/min) | |||||
|---|---|---|---|---|---|
| Control | CNER | TNER | FAR | ||
| Human | CYP1A2 | 1.88 ± 0.19 | 0.163 ± 0.005 * | 0.287 ± 0.013 * | 0.083 ± 0.005 * |
| CYP2B/3A | 1.22 ± 0.05 | 0.168 ± 0.007 * | 0.188 ± 0.005 * | 0.033 ± 0.004 * | |
| Rat | CYP1A2 | 22.8 ± 0.1 | 2.16 ± 0.04 * | 1.53 ± 0.03 * | 1.55 ± 0.02 * |
| CYP2B/3A | 1.74 ± 0.02 | 0.331 ± 0.009 * | 0.153 ± 0.008 * | 0.212 ± 0.003 * | |
| CNER | TNER | FAR | ANF/KET | |||
|---|---|---|---|---|---|---|
| Human | CYP1A2 | IC50 (µM) | 2.49 | 8.69 | 1.83 | 0.41 |
| 95% CI | 1.38 to 4.48 | 5.96 to 12.7 | 1.39 to 2.41 | 0.22 to 0.75 | ||
| CYP2B/3A | IC50 (µM) | 1.32 | 2.40 | 1.78 | 2.68 | |
| 95% CI | 1.02 to 1.71 | 2.02 to 2.83 | 1.59 to 2.01 | 2.07 to 3.48 | ||
| Rat | CYP1A2 | IC50 (µM) | 5.70 | 4.38 | 16.1 | - |
| 95% CI | 5.30 to 6.14 | 3.91 to 4.92 | 14.0 to 18.5 | - | ||
| CYP2B/3A | IC50 (µM) | 6.47 | 5.11 | 4.43 | - | |
| 95% CI | 6.07 to 6.89 | 4.34 to 6.02 | 3.82 to 5.14 | - |
| CYP | Substrate | IC50 (µM) | ||
|---|---|---|---|---|
| CNER | TNER | FAR | ||
| CYP2A6 | coumarin | - | - | 85.9 ± 22.9 |
| CYP2B6 | 7-ethoxy-4-(trifluoromethyl)coumarin | - | - | - |
| CYP2C9 | diclofenac | - | - | - |
| CYP2C19 | S-mephenytion | - | 76.4 ± 52.1 | - |
| CYP2D6 | bufuralol | - | - | - |
| CYP2E1 | chlorzoxazone | - | - | - |
| CYP3A4/5 | testosterone | 186 ± 125 | 50.5 ± 13.5 | - |
| CYP3A4/5 | midazolam | 66.1 ± 30.5 | - | - |
| Enzyme | KM (µM) | K’M (µM) | VMAX (nM/min) | V’MAX (nM/min) | Ki (µM) | |
|---|---|---|---|---|---|---|
| Human | CYP1A2 | 0.21 ± 0.11 | 0.87 ± 0.41 | 1.21 ± 0.10 | 1.06 ± 0.16 | 0.92 ± 0.52 |
| CYP2B/3A | 0.93 ± 0.22 | 0.94 ± 0.39 | 2.27 ± 0.17 | 0.74 ± 0.10 | 2.41 ± 0.33 | |
| Rat | CYP1A2 | 1.64 ± 0.45 | 1.89 ± 0.49 | 4.32 ± 0.47 | 2.21 ± 0.24 | 4.68 ± 0.66 |
| CYP2B/3A | 0.74 ± 0.12 | 1.26 ± 0.39 | 7.04 ± 0.35 | 0.81 ± 0.09 | 0.55 ± 0.11 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Špičáková, A.; Szotáková, B.; Dimunová, D.; Myslivečková, Z.; Kubíček, V.; Ambrož, M.; Lněničková, K.; Krasulová, K.; Anzenbacher, P.; Skálová, L. Nerolidol and Farnesol Inhibit Some Cytochrome P450 Activities but Did Not Affect Other Xenobiotic-Metabolizing Enzymes in Rat and Human Hepatic Subcellular Fractions. Molecules 2017, 22, 509. https://doi.org/10.3390/molecules22040509
Špičáková A, Szotáková B, Dimunová D, Myslivečková Z, Kubíček V, Ambrož M, Lněničková K, Krasulová K, Anzenbacher P, Skálová L. Nerolidol and Farnesol Inhibit Some Cytochrome P450 Activities but Did Not Affect Other Xenobiotic-Metabolizing Enzymes in Rat and Human Hepatic Subcellular Fractions. Molecules. 2017; 22(4):509. https://doi.org/10.3390/molecules22040509
Chicago/Turabian StyleŠpičáková, Alena, Barbora Szotáková, Diana Dimunová, Zuzana Myslivečková, Vladimír Kubíček, Martin Ambrož, Kateřina Lněničková, Kristýna Krasulová, Pavel Anzenbacher, and Lenka Skálová. 2017. "Nerolidol and Farnesol Inhibit Some Cytochrome P450 Activities but Did Not Affect Other Xenobiotic-Metabolizing Enzymes in Rat and Human Hepatic Subcellular Fractions" Molecules 22, no. 4: 509. https://doi.org/10.3390/molecules22040509
APA StyleŠpičáková, A., Szotáková, B., Dimunová, D., Myslivečková, Z., Kubíček, V., Ambrož, M., Lněničková, K., Krasulová, K., Anzenbacher, P., & Skálová, L. (2017). Nerolidol and Farnesol Inhibit Some Cytochrome P450 Activities but Did Not Affect Other Xenobiotic-Metabolizing Enzymes in Rat and Human Hepatic Subcellular Fractions. Molecules, 22(4), 509. https://doi.org/10.3390/molecules22040509