
Ginsenoside PPD’s Antitumor Effect via Down-Regulation of mTOR Revealed by Super-Resolution Imaging

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
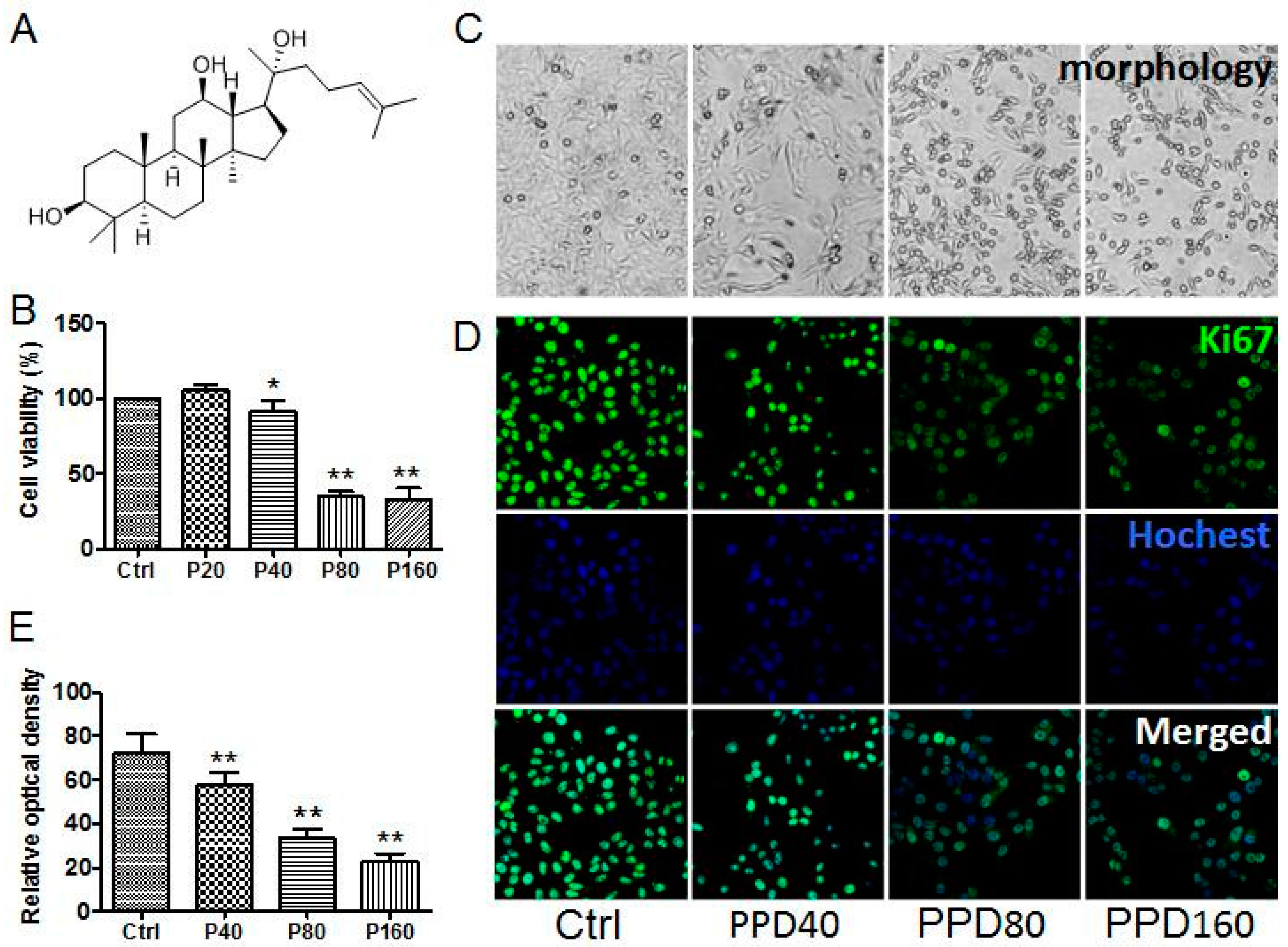
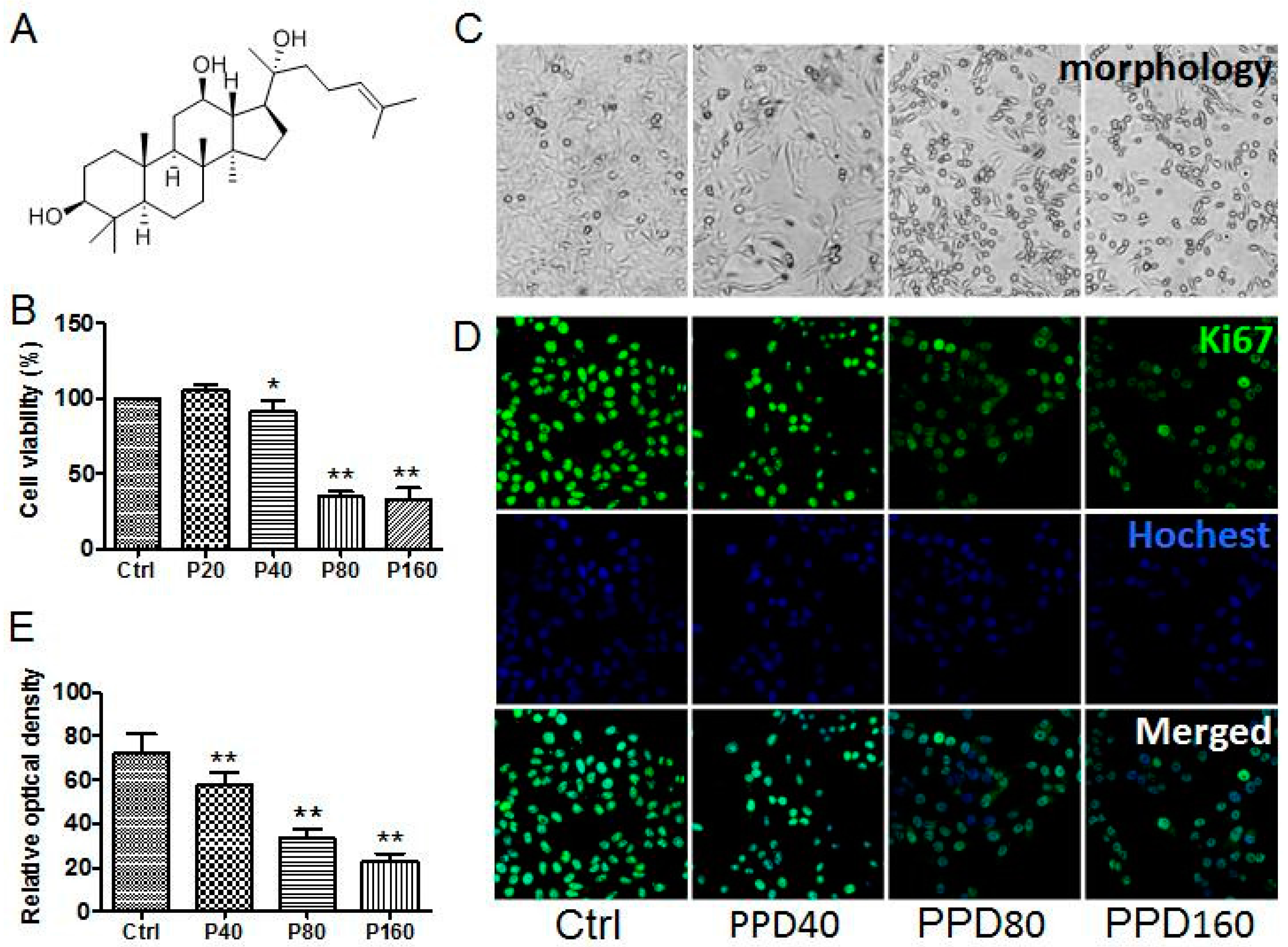
2.1. PPD Suppresses the Proliferation of Hep-2 Cells
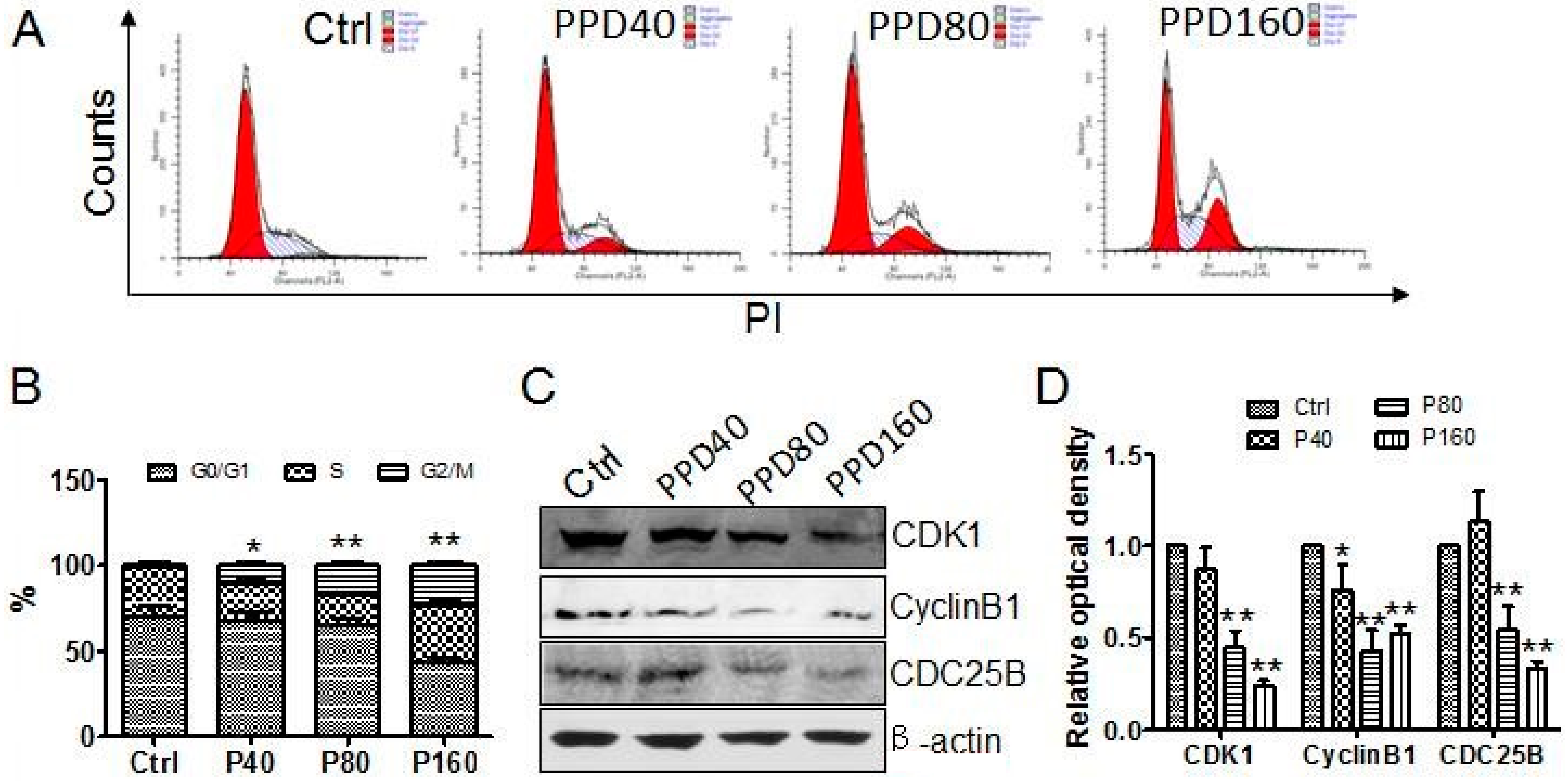
2.2. PPD Induces G2/M Cell Cycle Arrest
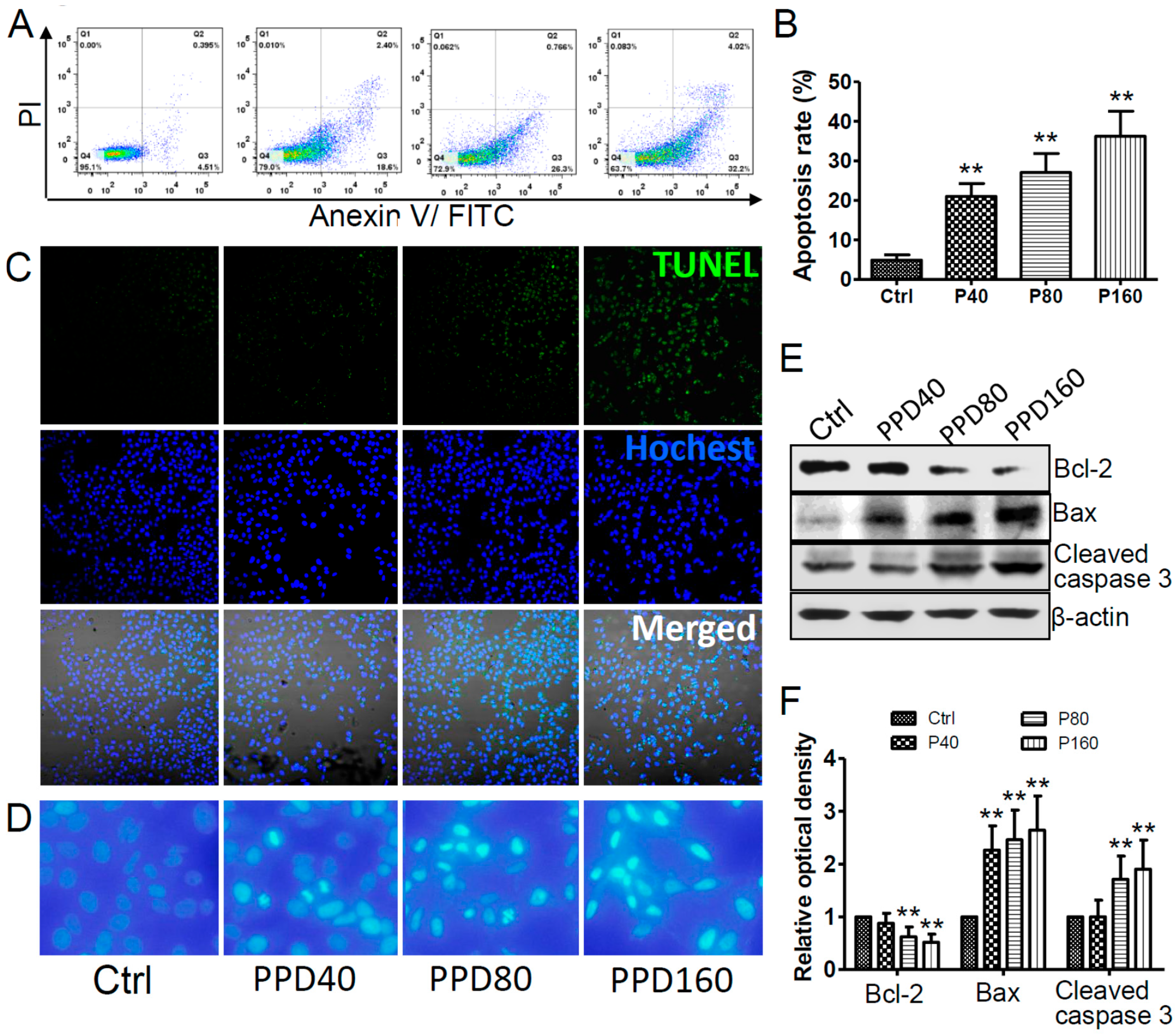
2.3. PPD Promotes the Apoptosis of Hep-2 Cells
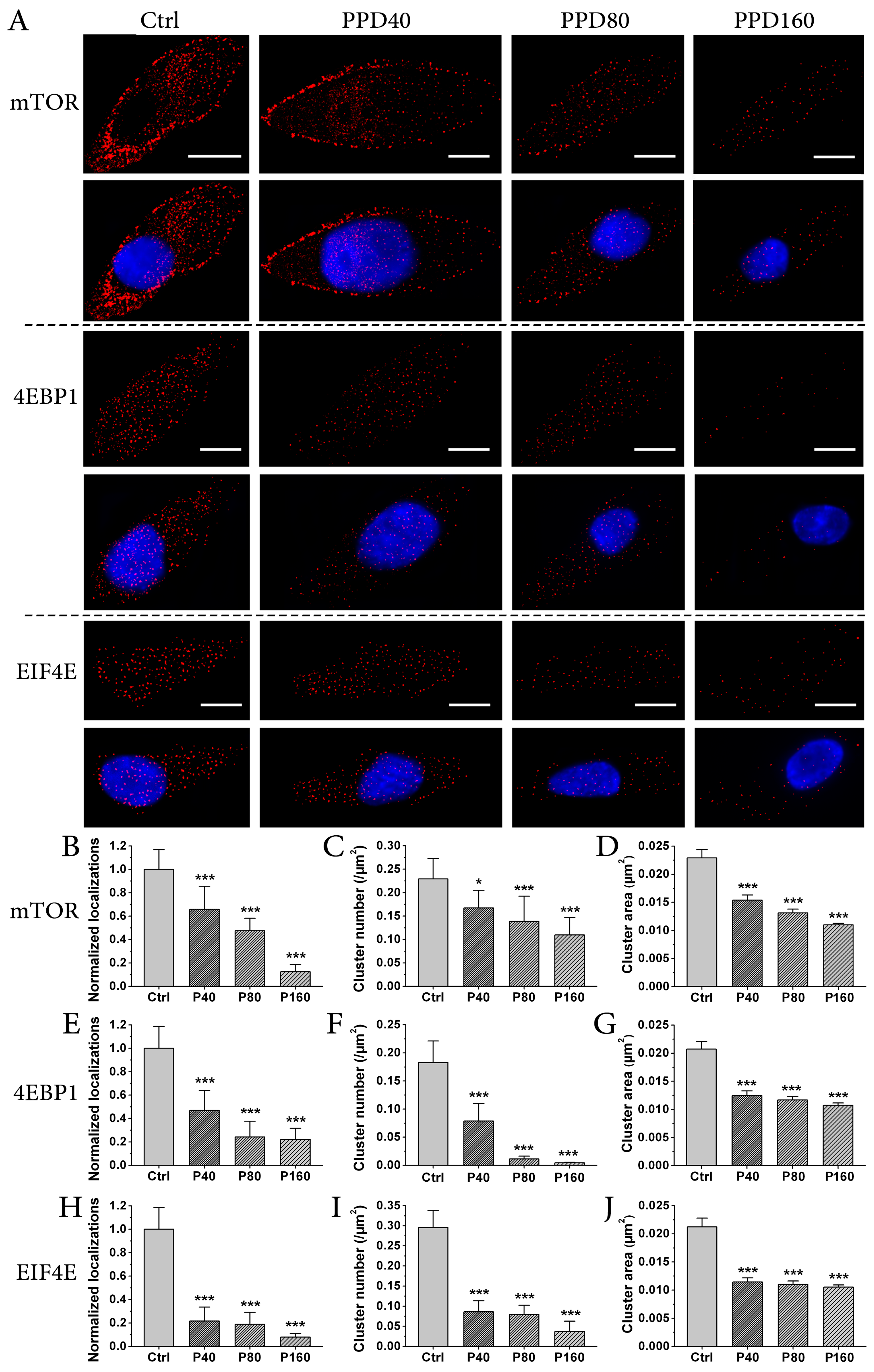
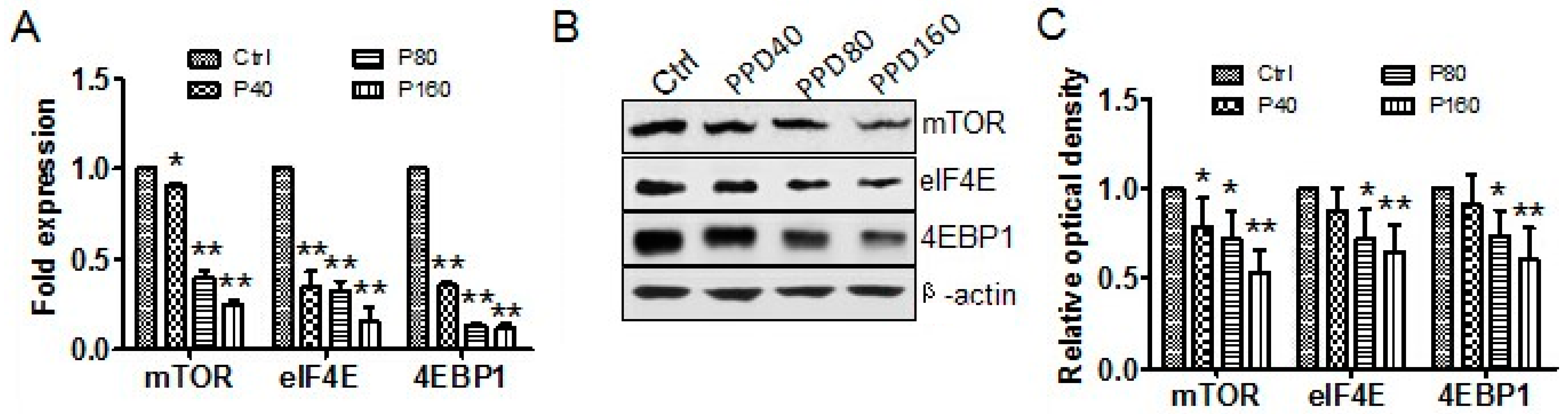
2.4. PPD Down-Regulates the Expression of mTOR Signaling Pathway
3. Materials and Methods
3.1. Cell Culture
3.2. Cytotoxicity Assay
3.3. Immunofluorescence
3.4. Flow Cytometry Analysis of Apoptosis and Cell Cycle
3.5. Western Blotting
3.6. dSTORM Imaging and Data Analysis
3.7. Quantitative Real-Time Reverse Transcription (RT) PCR
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Souza Viana, L.; de Aguiar Silva, F.C.; Andrade Dos Anjos Jacome, A.; Calheiros Campelo Maia, D.; Duarte de Mattos, M.; Arthur Jacinto, A.; Elias Mamere, A.; Boldrini Junior, D.; de Castro Capuzzo, R.; Roberto Santos, C.; et al. Efficacy and safety of a cisplatin and paclitaxel induction regimen followed by chemoradiotherapy for patients with locally advanced head and neck squamous cell carcinoma. Head Neck 2016, 38, E970–E980. [Google Scholar] [CrossRef] [PubMed]
- Marioni, G.; Marchese-Ragona, R.; Cartei, G.; Marchese, F.; Staffieri, A. Current opinion in diagnosis and treatment of laryngeal carcinoma. Cancer Treat. Rev. 2006, 32, 504–515. [Google Scholar] [CrossRef] [PubMed]
- S antoro, R.; Meccariello, G.; Mannelli, G.; Bini, B.; Paiar, F.; Gallo, O. Surgical options in radiotherapy-failed early glottic cancer. Eur. Arch. Oto Rhino Laryngol. 2014, 271, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Pellini, R.; Pichi, B.; Ruscito, P.; Ceroni, A.R.; Caliceti, U.; Rizzotto, G.; Pazzaia, A.; Laudadio, P.; Piazza, C.; Peretti, G.; et al. Supracricoid partial laryngectomies after radiation failure: A multi-institutional series. Head Neck 2008, 30, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ortiz, K.; Pasche, P.; Tamez-Velarde, M.; Villavicencio-Valencia, V. Supracricoid partial laryngectomy with cricohyoidoepiglottopexy in patients with radiation therapy failure. World J. Surg. Oncol. 2009, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Kouloulias, V.E.; Zygogianni, A.; Mosa, E.; Platoni, K.; Georgakopoulos, J.; Antypas, C.; Beli, I.; Tolia, M.; Maragoudakis, P.; Giotakis, I.; et al. Evaluation of acute/late toxicity and local recurrence in T1–T2 glottic carcinoma treated with accelerated hypofractionated 3D-conformal external beam radiotherapy (3D-CRT). Radiol. Oncol. 2013, 47, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Z.; Yuan, C.S. Potential role of ginseng in the treatment of colorectal cancer. Am. J. Chin. Med. 2008, 36, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Z.; Aung, H.H.; Zhang, B.; Sun, S.; Li, X.L.; He, H.; Xie, J.T.; He, T.C.; Du, W.; Yuan, C.S. Chemopreventive effects of heat-processed Panax quinquefolius root on human breast cancer cells. Anticancer Res. 2008, 28, 2545–2551. [Google Scholar] [PubMed]
- Gao, J.L.; Lv, G.Y.; He, B.C.; Zhang, B.Q.; Zhang, H.; Wang, N.; Wang, C.Z.; Du, W.; Yuan, C.S.; He, T.C. Ginseng saponin metabolite 20(S)-protopanaxadiol inhibits tumor growth by targeting multiple cancer signaling pathways. Oncol. Rep. 2013, 30, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Zhang, R.; Xu, H.L.; Yu, X.F.; Qu, S.C.; Sui, D.Y. 20(S)-protopanaxadiol triggers mitochondrial-mediated apoptosis in human lung adenocarcinoma A549 cells via inhibiting the PI3K/Akt signaling pathway. Am. J. Chin. Med. 2013, 41, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Pacelli, R.; Della Vittoria Scarpati, G.; Cella, L.; Giuliano, M.; Caponigro, F.; Pepe, S. Radioresistance in head and neck squamous cell carcinoma: Biological bases and therapeutic implications. Head Neck 2015, 37, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Lei, C. Acquisition of epithelial-mesenchymal transition/cancer stem cell is associated with activation of PI3K pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 112, e875. [Google Scholar]
- Heavey, S.; O’Byrne, K.J.; Gately, K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat. Rev. 2014, 40, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Teng, B.; Wen, L.; Feng, Q.; Wang, H.; Li, N.; Wang, Y.; Liang, Z. mTOR inhibitor AZD8055 inhibits proliferation and induces apoptosis in laryngeal carcinoma. Int. J. Clin. Exp. Med. 2014, 7, 337–347. [Google Scholar]
- Chi, K.R. Super-resolution microscopy: Breaking the limits. Nat. Methods 2009, 6, 15–18. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- van de Linde, S.; Loschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, F.; Liu, Y.; Cai, M.; Xu, H.; Jiang, J.; Wang, H. Revealing the cellular localization of STAT1 during the cell cycle by super-resolution imaging. Sci. Rep. 2015, 5, 9045. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, D.; Huang, B. Development in the STORM. Dev. Cell 2012, 23, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, F.; Chen, J.; Wang, J.; Cai, M.; Xu, H.; Jiang, J.; Wang, H. Super-resolution imaging of STAT3 cellular clustering during nuclear transport. RSC Adv. 2016, 6, 54597–54607. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, Y.; Rayburn, E.R.; Hill, D.L.; Wang, H.; Zhang, R. In vitro anti-cancer activity and structure-activity relationships of natural products isolated from fruits of Panax ginseng. Cancer Chemother. Pharmacol. 2007, 59, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, H.; Rayburn, E.R.; Zhao, Y.; Hill, D.L.; Zhang, R. 20(S)-25-methoxyl-dammarane-3beta, 12beta, 20-triol. Br. J. Cancer 2008, 98, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.Y.; Li, Y.W.; Tse, K.W.; Hau, K.P.; Leung, C.H.; Yu, Z.L.; Fong, W.F. 20(S)-Protopanaxadiol, a metabolite of ginsenosides, induced cell apoptosis through endoplasmic reticulum stress in human hepatocarcinoma HepG2 cells. Eur. J.Pharmacol. 2011, 668, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [PubMed]
- Zhou, J.; Luo, Y.H.; Wang, J.R.; Lu, B.B.; Wang, K.M.; Tian, Y. Gambogenic acid induction of apoptosis in a breast cancer cell line. Asian Pac. J.Cancer Prev. 2013, 14, 7601–7605. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Double identity for proteins of the Bcl-2 family. Nature 1997, 387, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska-Izydorczyk, O.; Smolewski, P. mTOR kinase inhibitors as a treatment strategy in hematological malignancies. Future Med. Chem. 2012, 4, 487–504. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Duran, I.; Clarke, B.A.; Oza, A.M. Biologic rationale and clinical activity of mTOR inhibitors in gynecological cancer. Cancer Treat. Rev. 2012, 38, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, X.; Ke, J.; Duan, Y.; He, Y.; Zhang, D.; Cai, M.; Sun, G.; Sun, X. Procyanidins, from Castanea mollissima Bl. shell, induces autophagy following apoptosis associated with PI3K/AKT/mTOR inhibition in HepG2 cells. Biomed. Pharmacother. 2016, 81, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nogami, A.; Oshikawa, G.; Okada, K.; Fukutake, S.; Umezawa, Y.; Nagao, T.; Kurosu, T.; Miura, O. FLT3-ITD confers resistance to the PI3K/Akt pathway inhibitors by protecting the mTOR/4EBP1/Mcl-1 pathway through STAT5 activation in acute myeloid leukemia. Oncotarget 2015, 6, 9189–9205. [Google Scholar] [CrossRef] [PubMed]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 102, 15500–15505. [Google Scholar] [CrossRef] [PubMed]
- Lillemeier, B.F.; Mortelmaier, M.A.; Forstner, M.B.; Huppa, J.B.; Groves, J.T.; Davis, M.M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010, 11, 543. [Google Scholar] [CrossRef]
- Sengupta, P.; Jovanovic-Talisman, T.; Skoko, D.; Renz, M.; Veatch, S.L.; Lippincott-Schwartz, J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods 2011, 8, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Y.; Cai, M.; Pan, Y.; Xu, H.; Jiang, J.; Ji, H.; Wang, H. Mechanistic insights into EGFR membrane clustering revealed by super-resolution imaging. Nanoscale 2015, 7, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Henriques, R.; Lelek, M.; Fornasiero, E.F.; Valtorta, F.; Zimmer, C.; Mhlanga, M.M. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Methods 2010, 7, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.M.M.; Pascau, J. Image Processing with ImageJ. Biophotonics Int. 2003, 11, 36–42. [Google Scholar]
- Sample Availability: Samples of the compounds PPD are not available from the authors.


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, B.; Jiang, J.; Zhao, L.; Gao, J.; Chen, J.; Liu, Z.; Wang, H.; Lu, B. Ginsenoside PPD’s Antitumor Effect via Down-Regulation of mTOR Revealed by Super-Resolution Imaging. Molecules 2017, 22, 486. https://doi.org/10.3390/molecules22030486
Teng B, Jiang J, Zhao L, Gao J, Chen J, Liu Z, Wang H, Lu B. Ginsenoside PPD’s Antitumor Effect via Down-Regulation of mTOR Revealed by Super-Resolution Imaging. Molecules. 2017; 22(3):486. https://doi.org/10.3390/molecules22030486
Chicago/Turabian StyleTeng, Bo, Junguang Jiang, Lijing Zhao, Jing Gao, Junyu Chen, Zhe Liu, Hongda Wang, and Binfeng Lu. 2017. "Ginsenoside PPD’s Antitumor Effect via Down-Regulation of mTOR Revealed by Super-Resolution Imaging" Molecules 22, no. 3: 486. https://doi.org/10.3390/molecules22030486
APA StyleTeng, B., Jiang, J., Zhao, L., Gao, J., Chen, J., Liu, Z., Wang, H., & Lu, B. (2017). Ginsenoside PPD’s Antitumor Effect via Down-Regulation of mTOR Revealed by Super-Resolution Imaging. Molecules, 22(3), 486. https://doi.org/10.3390/molecules22030486