
Structural Probing and Molecular Modeling of the A3 Adenosine Receptor: A Focus on Agonist Binding

Abstract
:
1. Introduction
2. Structural Probing of the A3 Adenosine Receptor
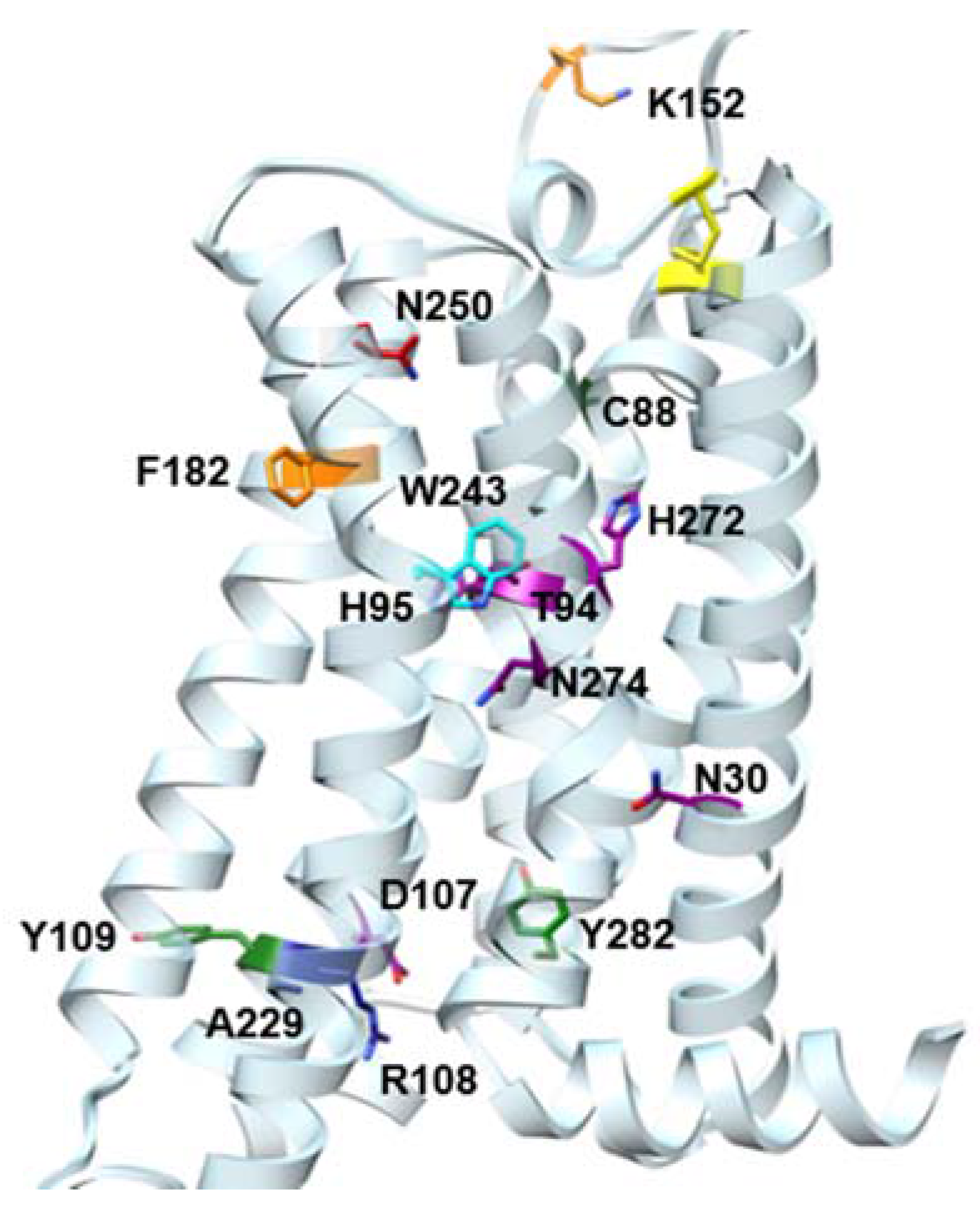
2.1. Site-Directed Mutagenesis
2.2. Homology Modeling
2.3. Receptor Reengineering
3. Molecular Modeling of the A3 Adenosine Receptor
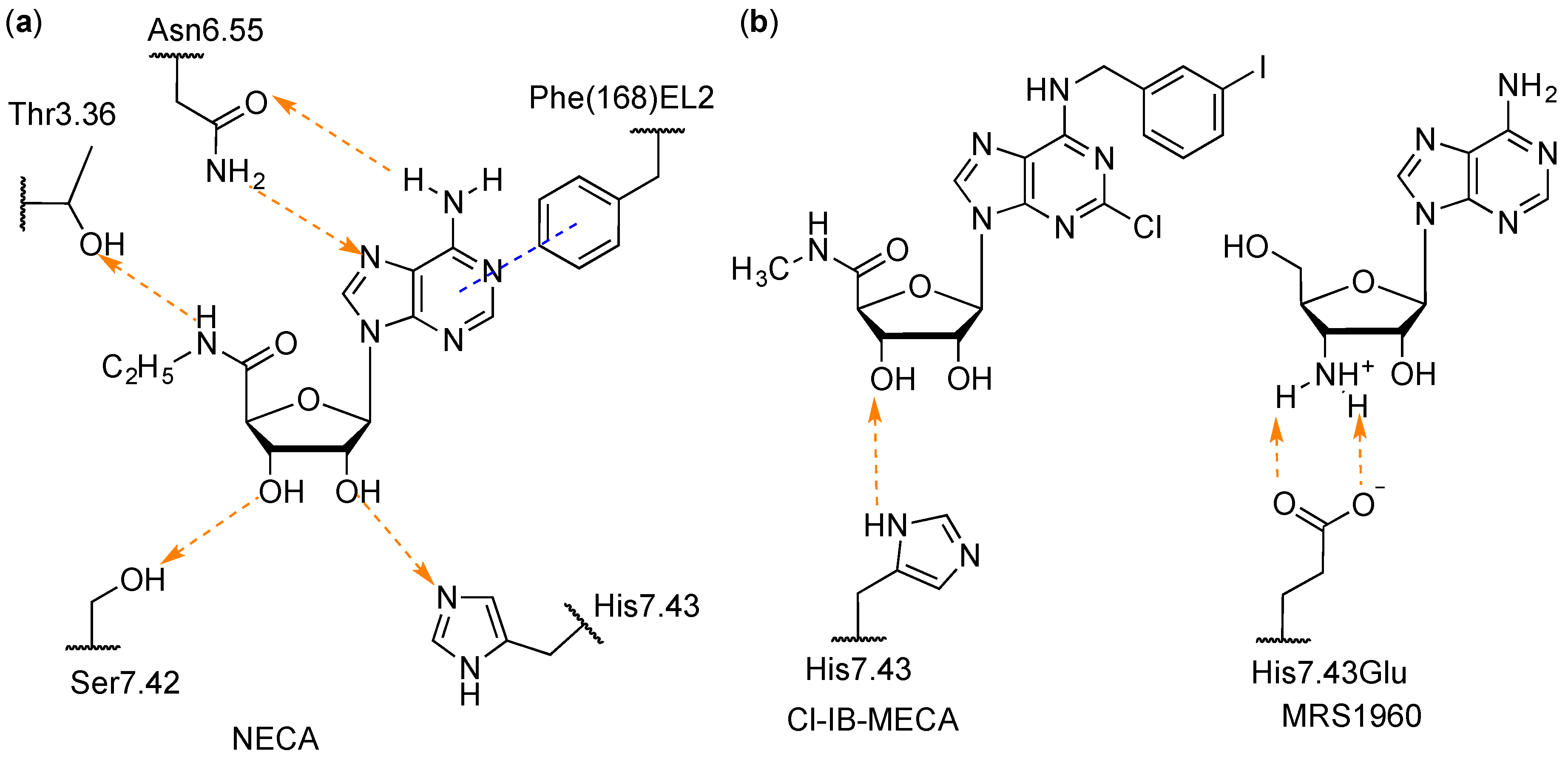
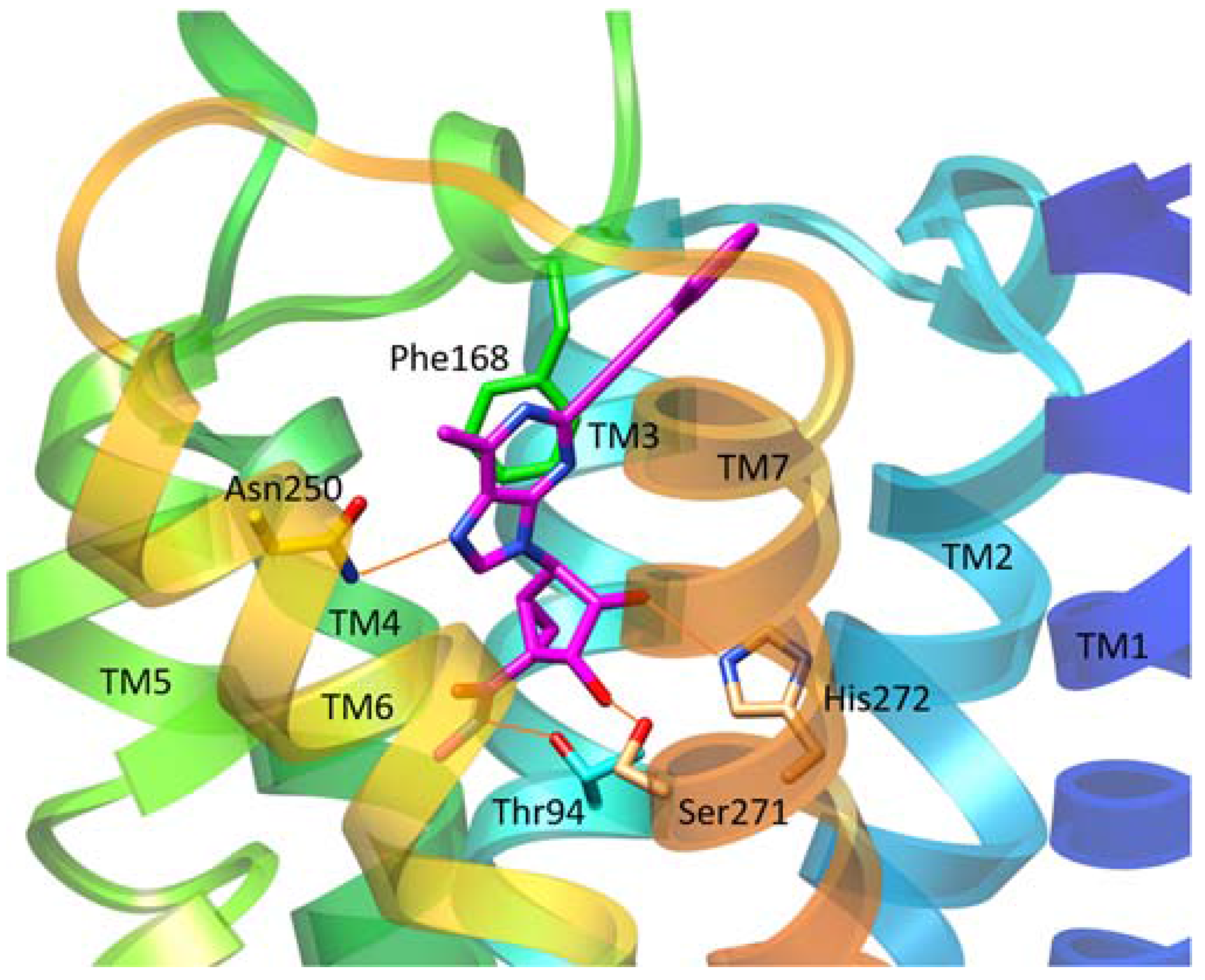
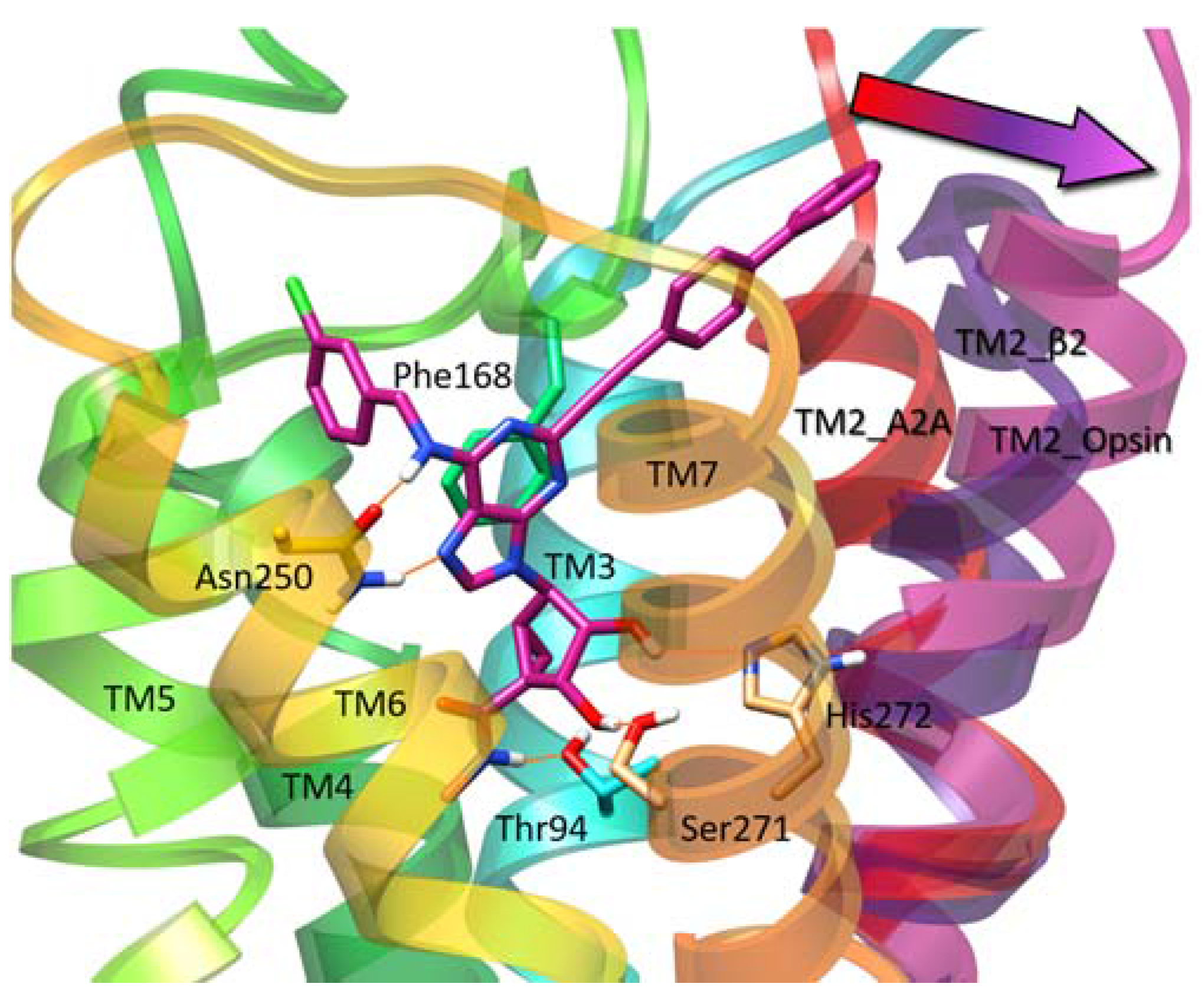
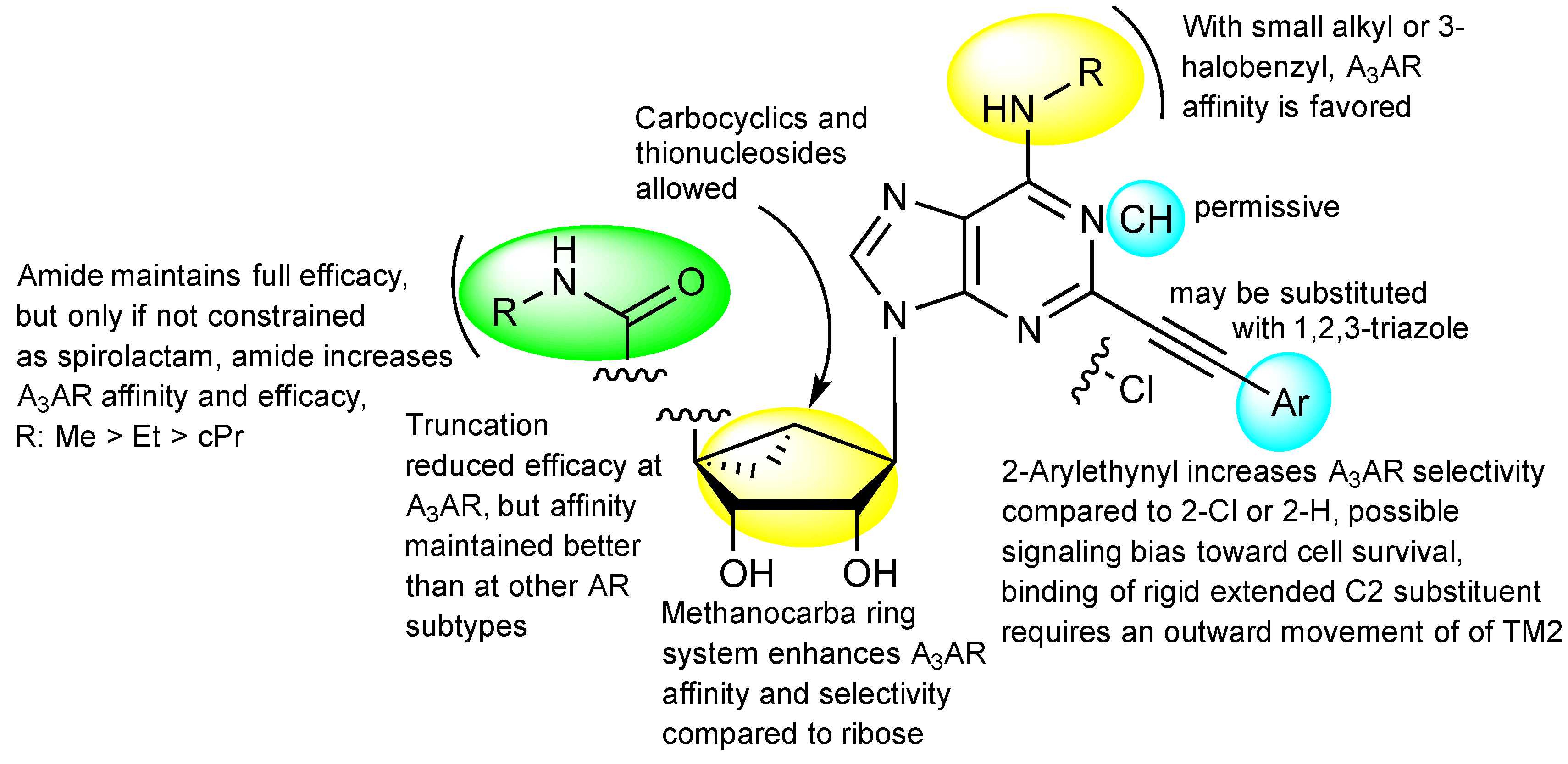
3.1. Docking
3.2. Molecular Dynamics (MD)
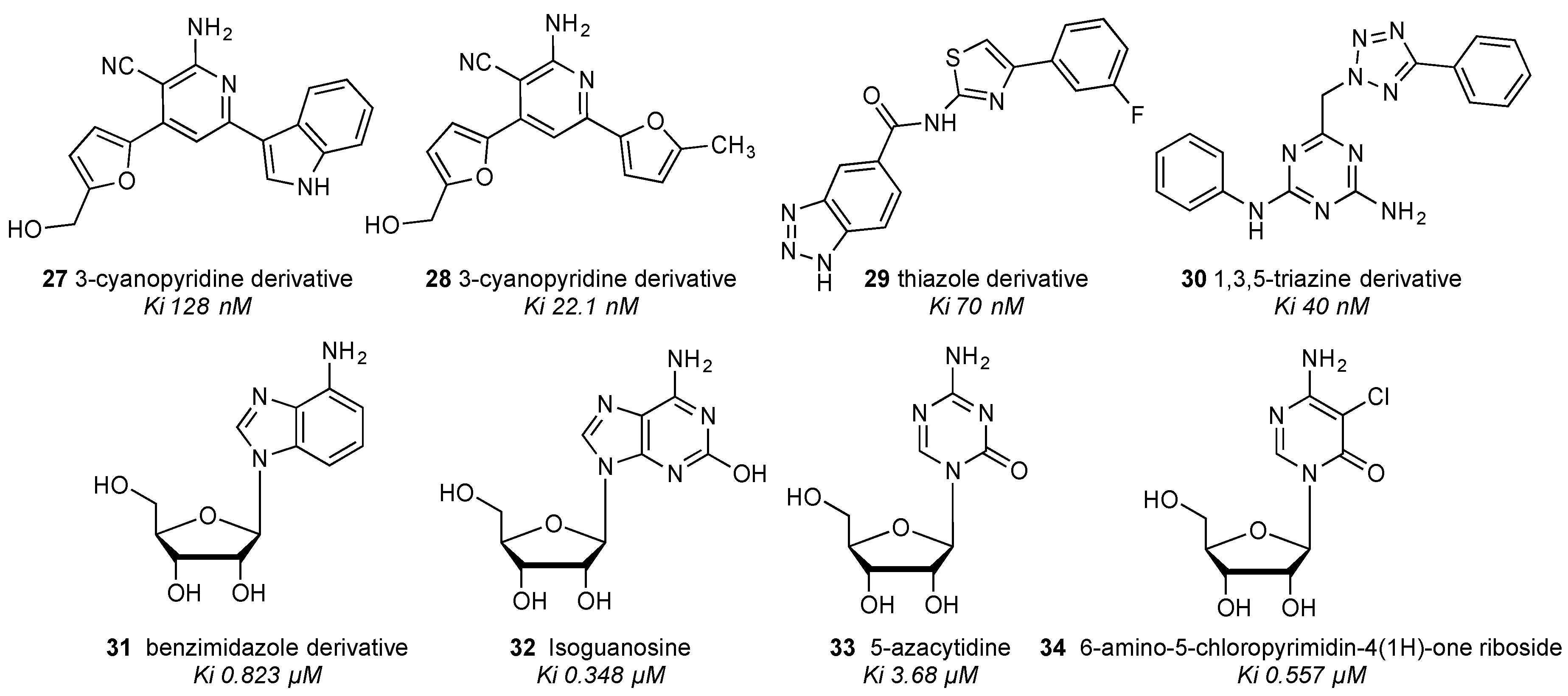
4. Library Screening to Discover Novel Ligands of the A3 Adenosine Receptor
5. Receptor Dimerization
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Linden, J. Cloned adenosine A3 receptors: Pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 1994, 15, 298–306. [Google Scholar] [CrossRef]
- Lopes, L.V.; Rebola, N.; Pinheiro, P.C.; Richardson, P.J.; Oliveira, C.R.; Cunha, R.A. Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport 2003, 14, 1645–1648. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.; Symons-Liguori, A.; Jacobson, K.A.; Salvemini, D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics: A3 receptor agonists as non-narcotic analgesics. Br. J. Pharmacol. 2016, 173, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Avila, M.Y.; Peterson-Yantorno, K.; Coca-Prados, M.; Stone, R.A.; Jacobson, K.A.; Civan, M.M. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr. Eye Res. 2005, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Do, C.W.; Avila, M.Y.; Peterson-Yantorno, K.; Stone, R.A.; Gao, Z.-G.; Joshi, B.; Besada, P.; Jeong, L.S.; Jacobson, K.A.; et al. Nucleoside-derived antagonists to A3 adenosine receptors lower mouse intraocular pressure and act across species. Exp. Eye Res. 2010, 90, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Spina, D.; Page, C.P. Adenosine receptors and asthma. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S446–S456. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Bar-Yehuda, S.; Fishman, P. A3 adenosine receptor: pharmacology and role in disease. Handb. Exp. Pharmacol. 2009, 297–327. [Google Scholar]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; IJzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.; Tuccinardi, T. Molecular modeling of adenosine receptors: new results and trends. Med. Res. Rev. 2008, 28, 247–277. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Morizzo, E.; Jacobson, K.A. Molecular modeling and reengineering of A3 adenosine receptors. In A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics; Borea, P.A., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 149–161. [Google Scholar]
- Cheong, S.L.; Federico, S.; Venkatesan, G.; Mandel, A.L.; Shao, Y.-M.; Moro, S.; Spalluto, G.; Pastorin, G. The A3 adenosine receptor as multifaceted therapeutic target: Pharmacology, medicinal chemistry, and in silico approaches. Med. Res. Rev. 2013, 33, 235–335. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Kim, S.-K.; Gross, A.S.; Chen, A.; Blaustein, J.B.; Jacobson, K.A. Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol. Pharmacol. 2003, 63, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Gao, Z.-G.; Barak, D.; Liang, B.T.; Jacobson, K.A. Constitutive Activation of A3 Adenosine Receptors by Site-Directed Mutagenesis. Biochem. Biophys. Res. Commun. 2001, 284, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Chen, A.; Barak, D.; Kim, S.-K.; Müller, C.E.; Jacobson, K.A. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J. Biol. Chem. 2002, 277, 19056–19063. [Google Scholar] [CrossRef] [PubMed]
- Tchilibon, S.; Kim, S.-K.; Gao, Z.-G.; Harris, B.A.; Blaustein, J.B.; Gross, A.S.; Duong, H.T.; Melman, N.; Jacobson, K.A. Exploring distal regions of the A3 adenosine receptor binding site: Sterically constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg. Med. Chem. 2004, 12, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, D.; Buccioni, M.; Lambertucci, C.; Kachler, S.; Falgner, N.; Marucci, G.; Thomas, A.; Cristalli, G.; Volpini, R.; Klotz, K.-N. Different efficacy of adenosine and NECA derivatives at the human A3 adenosine receptor: Insight into the receptor activation switch. Biochem. Pharmacol. 2014, 87, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hallmen, C.; Wiese, M. Molecular dynamics simulation of the human adenosine A3 receptor: Agonist induced conformational changes of Trp243. J. Comput. Aided Mol. Des. 2006, 20, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Deganutti, G.; Cuzzolin, A.; Ciancetta, A.; Moro, S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Kim, S.-K.; Biadatti, T.; Chen, W.; Lee, K.; Barak, D.; Kim, S.G.; Johnson, C.R.; Jacobson, K.A. Structural determinants of A3 adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J. Med. Chem. 2002, 45, 4471–4484. [Google Scholar] [CrossRef] [PubMed]
- Toti, K.S.; Moss, S.M.; Paoletta, S.; Gao, Z.-G.; Jacobson, K.A.; van Calenbergh, S. Synthesis and evaluation of N6-substituted apioadenosines as potential adenosine A3 receptor modulators. Bioorg. Med. Chem. 2014, 22, 4257–4268. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Ciancetta, A.; Warnick, E.; O’Connor, R.; Chen, Z.; Gizewski, E.; Crane, S.; Gao, Z.-G.; Auchampach, J.A.; Salvemini, D.; et al. Purine (N)-methanocarba nucleoside derivatives lacking an exocyclic amine as selective A3 adenosine receptor agonists. J. Med. Chem. 2016, 59, 3249–3263. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.-G.; Chen, A.; Barak, D.; Kim, S.-A.; Lee, K.; Link, A.; Rompaey, P.V.; van Calenbergh, S.; Liang, B.T. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J. Med. Chem. 2001, 44, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-G.; Duong, H.T.; Sonina, T.; Kim, S.-K.; van Rompaey, P.; van Calenbergh, S.; Mamedova, L.; Kim, H.O.; Kim, M.J.; Kim, A.Y.; et al. Orthogonal activation of the reengineered A3 adenosine receptor (neoceptor) using tailored nucleoside agonists. J. Med. Chem. 2006, 49, 2689–2702. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Deflorian, F.; Phan, K.; Gao, Z.-G.; Wan, T.C.; Gizewski, E.; Auchampach, J.A.; Jacobson, K.A. Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J. Med. Chem. 2012, 55, 4847–4860. [Google Scholar] [CrossRef]
- Baltos, J.-A.; Paoletta, S.; Nguyen, A.T.N.; Gregory, K.J.; Tosh, D.K.; Christopoulos, A.; Jacobson, K.A.; May, L.T. Structure-activity analysis of biased agonism at the human adenosine A3 receptor. Mol. Pharmacol. 2016, 90, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Auchampach, J.A.; Gizewski, E.; Wan, T.C.; de Castro, S.; Brown, G.G.; Jacobson, K.A. Synthesis and pharmacological characterization of [125I]MRS5127, a high affinity, selective agonist radioligand for the A3 adenosine receptor. Biochem. Pharmacol. 2010, 79, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Jacobson, K.A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- Yaziji, V.; Rodríguez, D.; Gutiérrez-de-Terán, H.; Coelho, A.; Caamaño, O.; García-Mera, X.; Brea, J.; Loza, M.I.; Cadavid, M.I.; Sotelo, E. Pyrimidine derivatives as potent and selective A3 adenosine receptor antagonists. J. Med. Chem. 2011, 54, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.G. Allosteric modulators of adenosine, P2Y and P2X receptors. In Allosterism in Drug Discovery; Doller, D., Ed.; RSC Drug Discovery Series No. 56; Royal Society of Chemistry: London, UK, 2017; Chapter 11; pp. 247–270. [Google Scholar]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
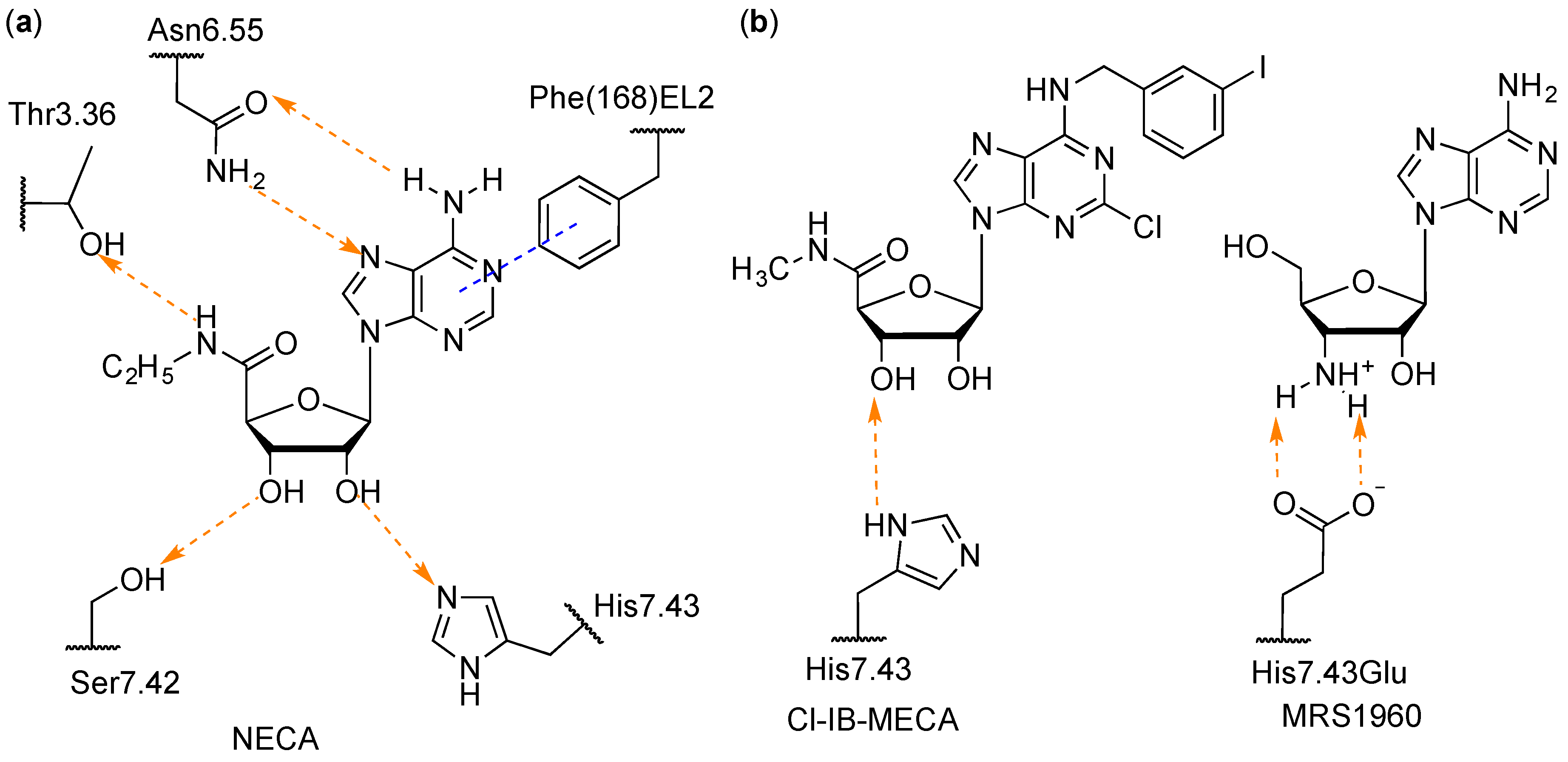
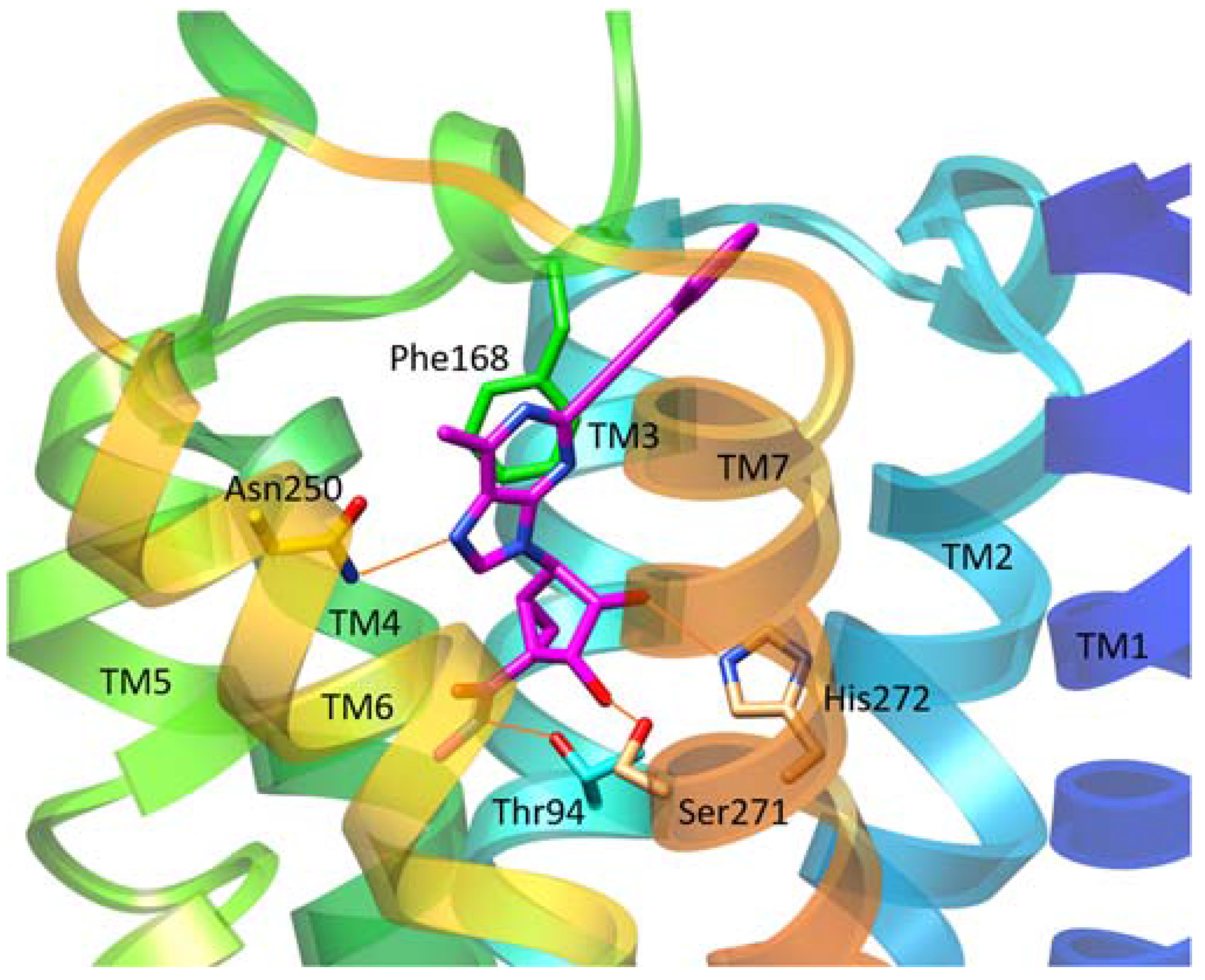
- Kim, S.-K.; Gao, Z.-G.; Jeong, L.S.; Jacobson, K.A. Docking studies of agonists and antagonists suggest an activation pathway of the A3 adenosine receptor. J. Mol. Graph. Model. 2006, 25, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.-G.; Cherezov, V.; Stevens, R.C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Edwards, P.C.; Leslie, A.G.W.; Tate, C.G. Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol. Pharmacol. 2015, 87, 907–915. [Google Scholar] [CrossRef] [PubMed]
- van Galen, P.J.; van Bergen, A.H.; Gallo-Rodriguez, C.; Melman, N.; Olah, M.E.; IJzerman, A.P.; Stiles, G.L.; Jacobson, K.A. A binding site model and structure-activity relationships for the rat A3 adenosine receptor. Mol. Pharmacol. 1994, 45, 1101–1111. [Google Scholar] [PubMed]
- Higgs, C.; Beuming, T.; Sherman, W. Hydration site thermodynamics explain SARs for triazolylpurines analogues binding to the A2A receptor. ACS Med. Chem. Lett. 2010, 1, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.-G.; Paoletta, S.; Kiselev, E.; Chakraborty, S.; Jayasekara, P.S.; Balasubramanian, R.; Tosh, D.K. John Daly Lecture: Structure-guided drug design for adenosine and P2Y receptors. Comput. Struct. Biotechnol. J. 2015, 13, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Schwyzer, R. ACTH: A short introductory review. Ann. New York Acad. Sci. 1977, 297, 3–26. [Google Scholar] [CrossRef]
- Ohno, M.; Gao, Z.-G.; van Rompaey, P.; Tchilibon, S.; Kim, S.-K.; Harris, B.A.; Gross, A.S.; Duong, H.T.; van Calenbergh, S.; Jacobson, K.A. Modulation of adenosine receptor affinity and intrinsic efficacy in adenine nucleosides substituted at the 2-position. Bioorg. Med. Chem. 2004, 12, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Morizzo, E.; Federico, S.; Spalluto, G.; Moro, S. Human A3 adenosine receptor as versatile G protein-coupled receptor example to validate the receptor homology modeling technology. Curr. Pharm. Des. 2009, 15, 4069–4084. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.-G.; Liang, B.T. Neoceptors: Reengineering GPCRs to recognize tailored ligands. Trends Pharmacol. Sci. 2007, 28, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Riley, L.; Abrol, R.; Jacobson, K.A.; Goddard, W.A. Predicted structures of agonist and antagonist bound complexes of adenosine A3 receptor. Proteins 2011, 79, 1878–1897. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Paoletta, S.; Phan, K.; Gao, Z.G.; Jacobson, K.A. Truncated nucleosides as A3 adenosine receptor ligands: Combined 2-arylethynyl and bicyclohexane substitutions. ACS Med. Chem. Lett. 2012, 3, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867.e13–877.e13. [Google Scholar] [CrossRef] [PubMed]
- Tosh, D.K.; Paoletta, S.; Chen, Z.; Crane, S.; Lloyd, J.; Gao, Z.-G.; Gizewski, E.T.; Auchampach, J.A.; Salvemini, D.; Jacobson, K.A. Structure-based design, synthesis by click chemistry and in vivo activity of highly selective A3 adenosine receptor agonists. Med. Chem. Commun. 2015, 6, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jonsson, A.L.; Beuming, T.; Shelley, J.C.; Voth, G.A. Ligand-dependent activation and deactivation of the human adenosine A2A receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the complexity of ligand-protein recognition pathways using Supervised Molecular Dynamics (SuMS) simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Gao, Z.-G.; Moss, S.M.; Jacobson, K.A.; Carlsson, J. Molecular docking screening using agonist-bound GPCR structures: Probing the A2A adenosine receptor. J. Chem. Inf. Model. 2015, 55, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Phan, K.; Gao, Z.-G.; Marko, A.C.; Sali, A.; Jacobson, K.A. Limits of ligand selectivity from docking to models: In silico screening for A1 adenosine receptor antagonists. PLoS ONE 2012, 7, e49910. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.; Chakraborty, S.; Warnick, E.; Crane, S.; Gao, Z.-G.; O’Connor, R.; Jacobson, K.A.; Carlsson, J. Structure-based screening of uncharted chemical space for atypical adenosine receptor agonists. ACS Chem. Biol. 2016, 11, 2763–2772. [Google Scholar] [CrossRef] [PubMed]
- Hiller, C.; Kühorn, J.; Gmeiner, P. Class A G-Protein-Coupled Receptor (GPCR) Dimers and Bivalent Ligands. J. Med. Chem. 2013, 56, 6542–6559. [Google Scholar] [CrossRef] [PubMed]
- May, L.T.; Bridge, L.J.; Stoddart, L.A.; Briddon, S.J.; Hill, S.J. Allosteric interactions across native adenosine-A3 receptor homodimers: Quantification using single-cell ligand-binding kinetics. FASEB J. 2011, 25, 3465–3476. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Jacobson, K.A. Computational prediction of homodimerization of the A3 adenosine receptor. J. Mol. Graph. Model. 2006, 25, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Xie, R.; Young, L.; Chang, L.; Liang, B.T. A novel pharmacological approach to treating cardiac ischemia: Binary conjugates of A1 and A3 adenosine receptor agonists. J. Biol. Chem. 2000, 275, 30272–30279. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Heitman, L.H.; IJzerman, A.P. Kinetic aspects of the interaction between ligand and G protein-coupled receptor: The case of the adenosine receptors. Chem. Rev. 2017, 117, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]

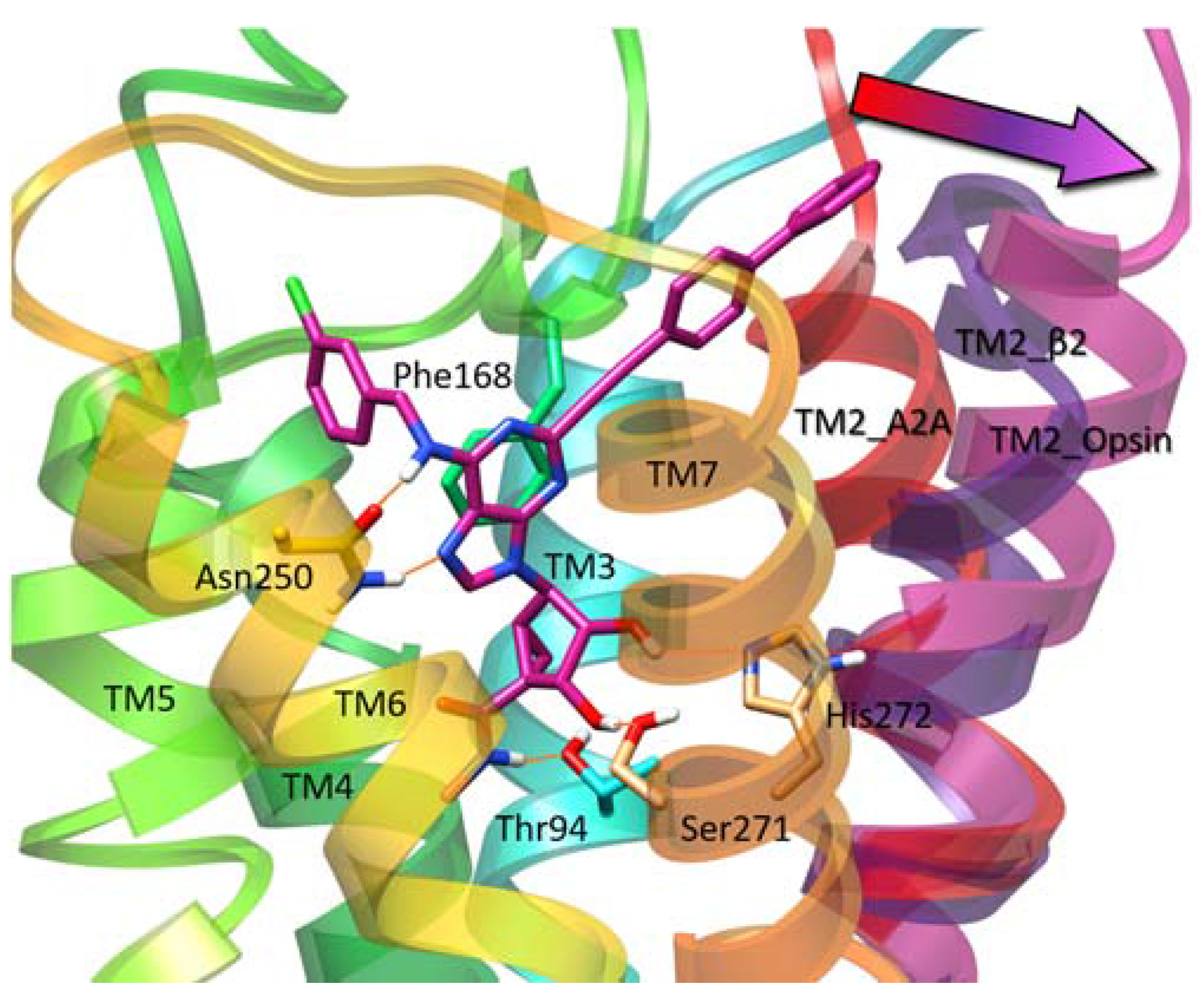
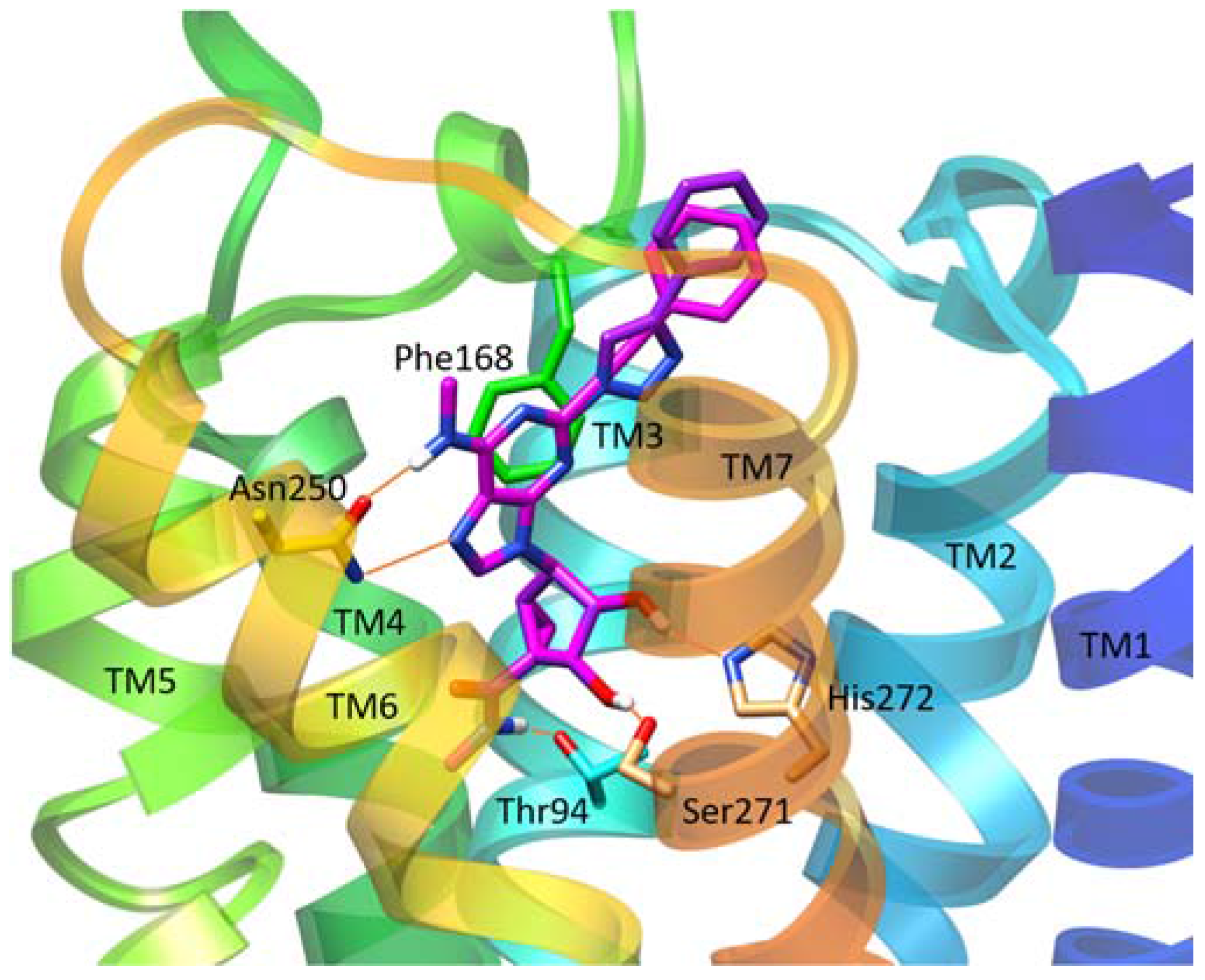
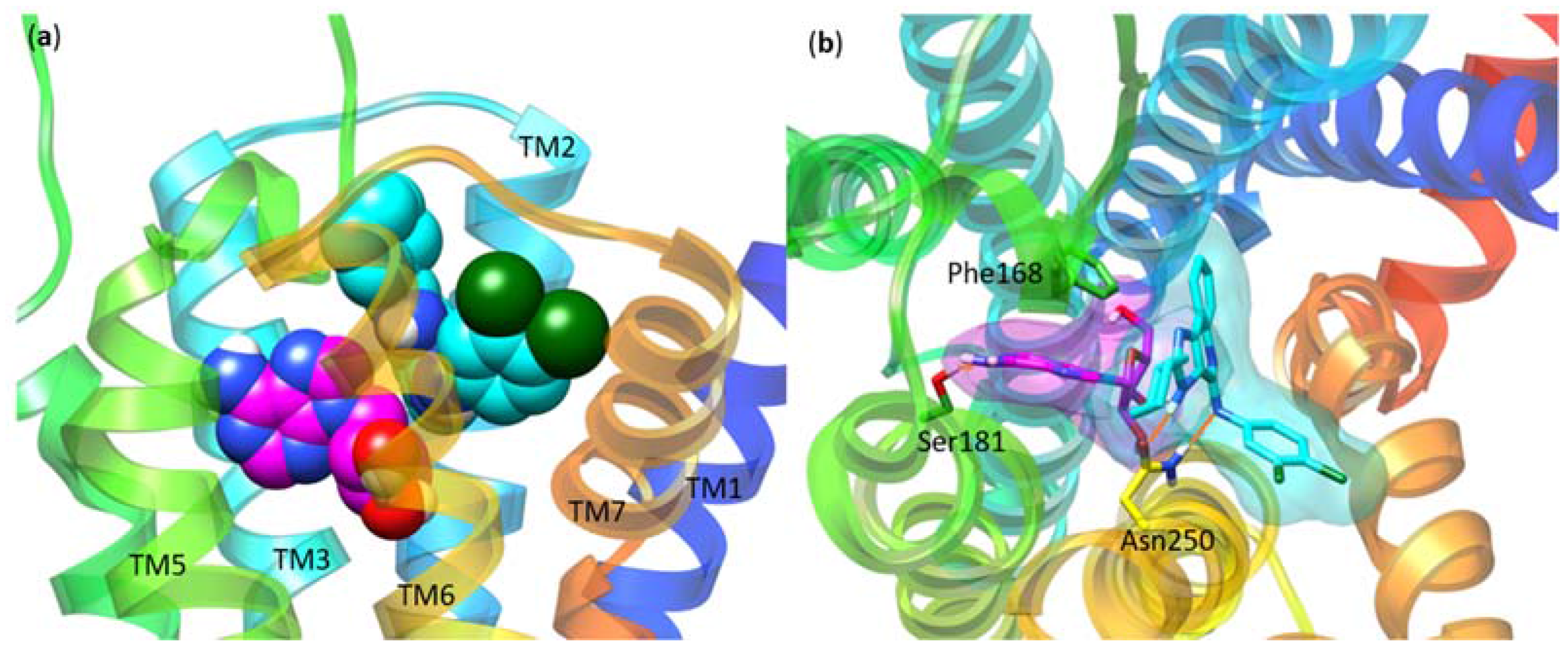
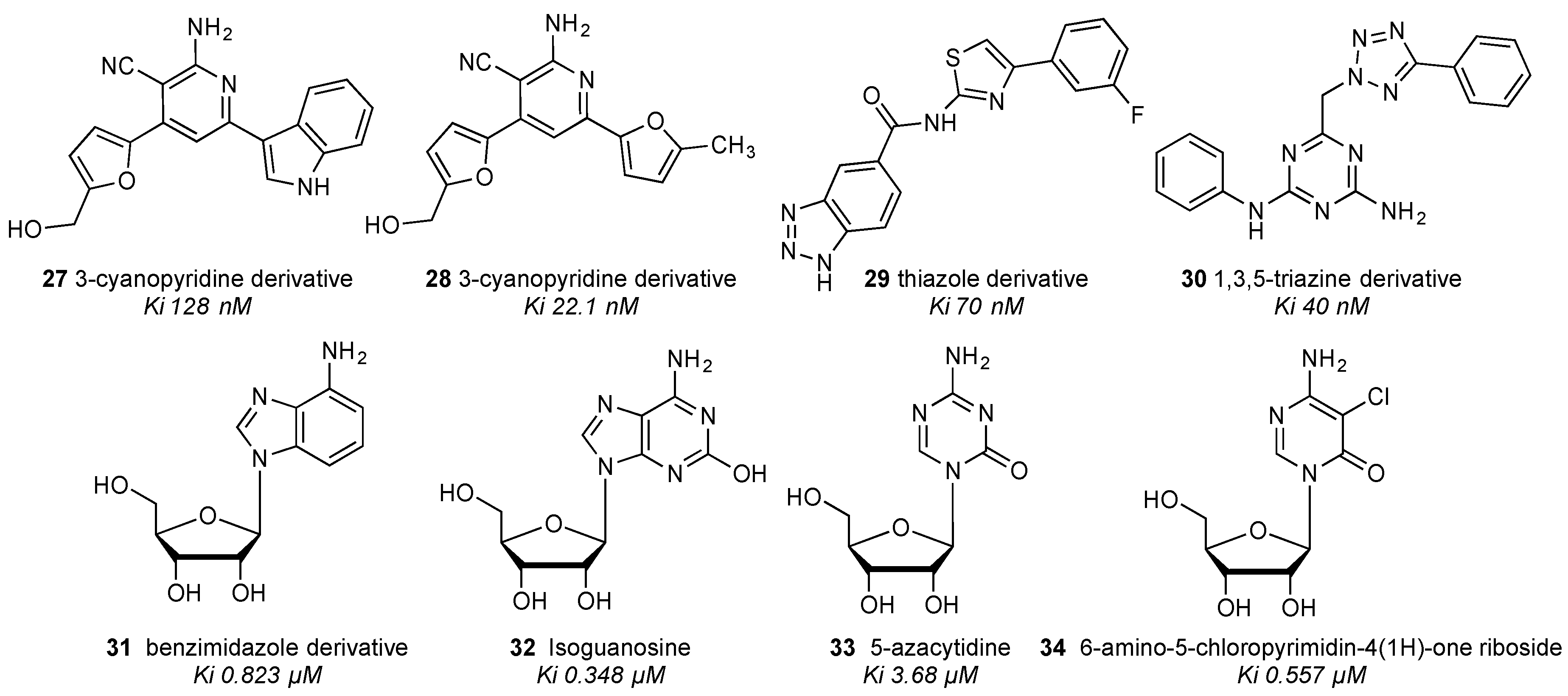
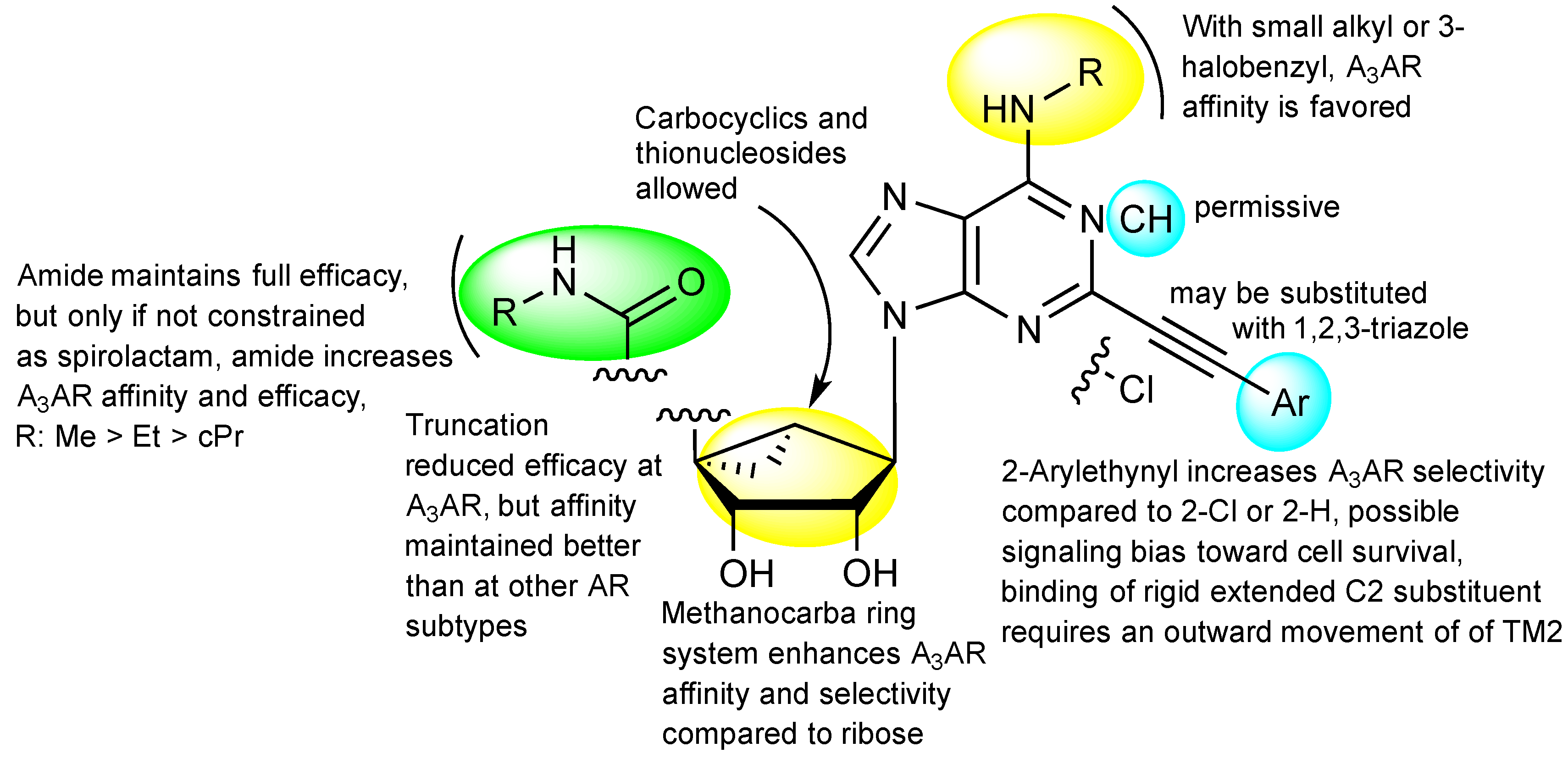

{kind=link}
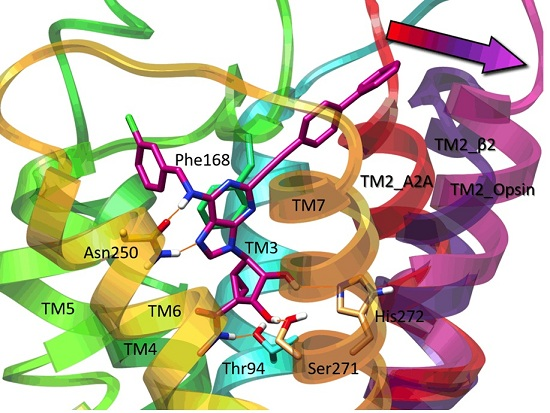
{kind=link}
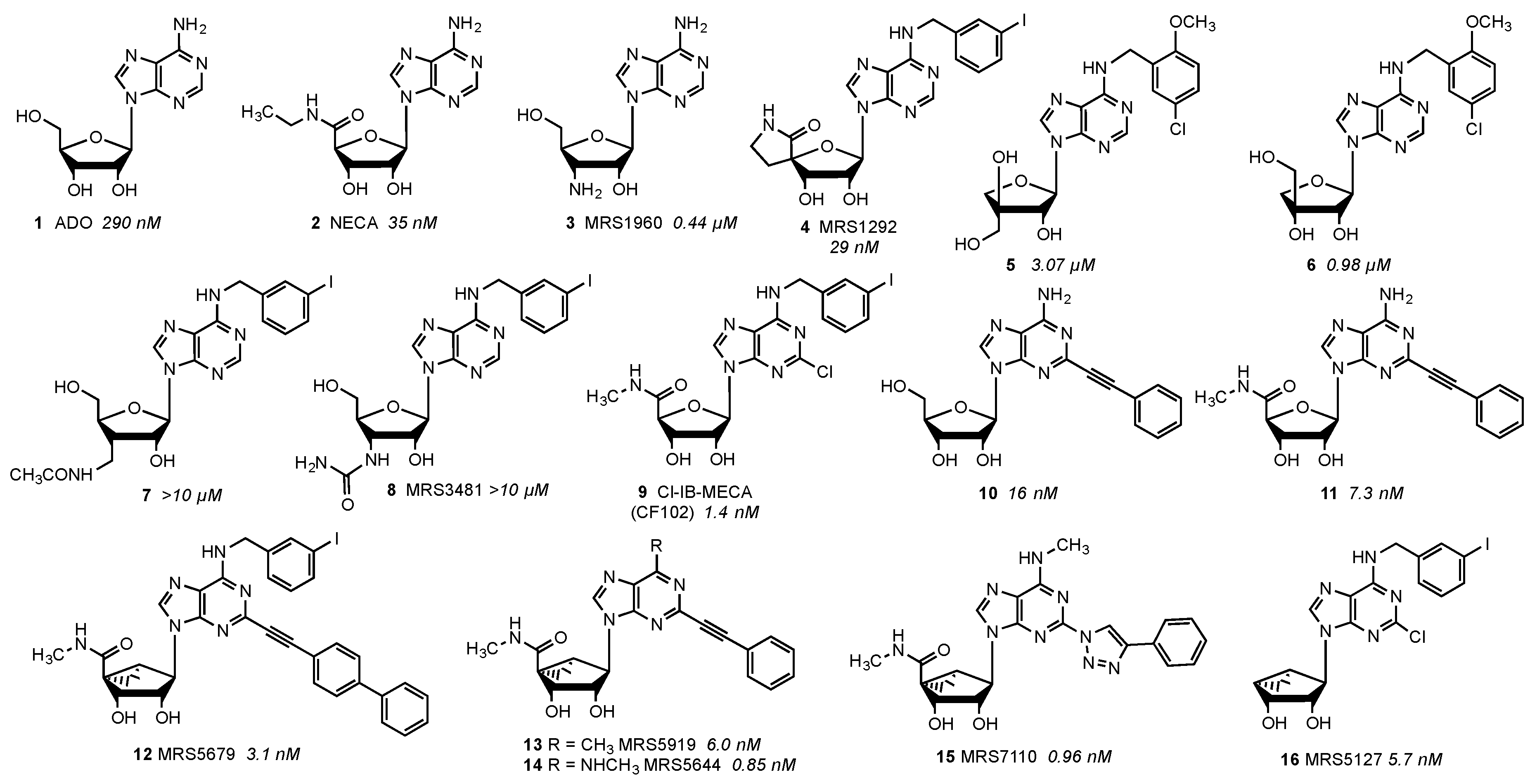
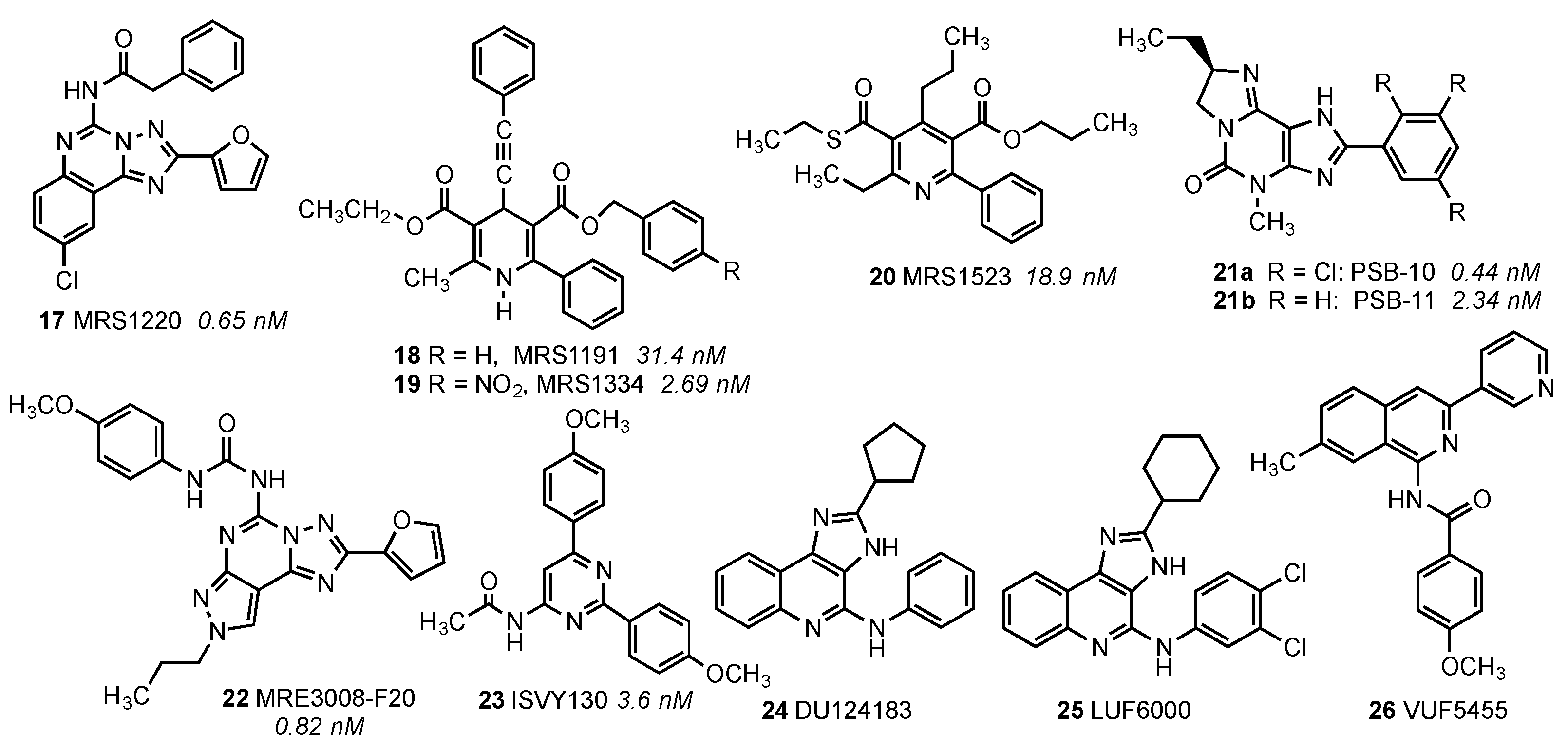{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
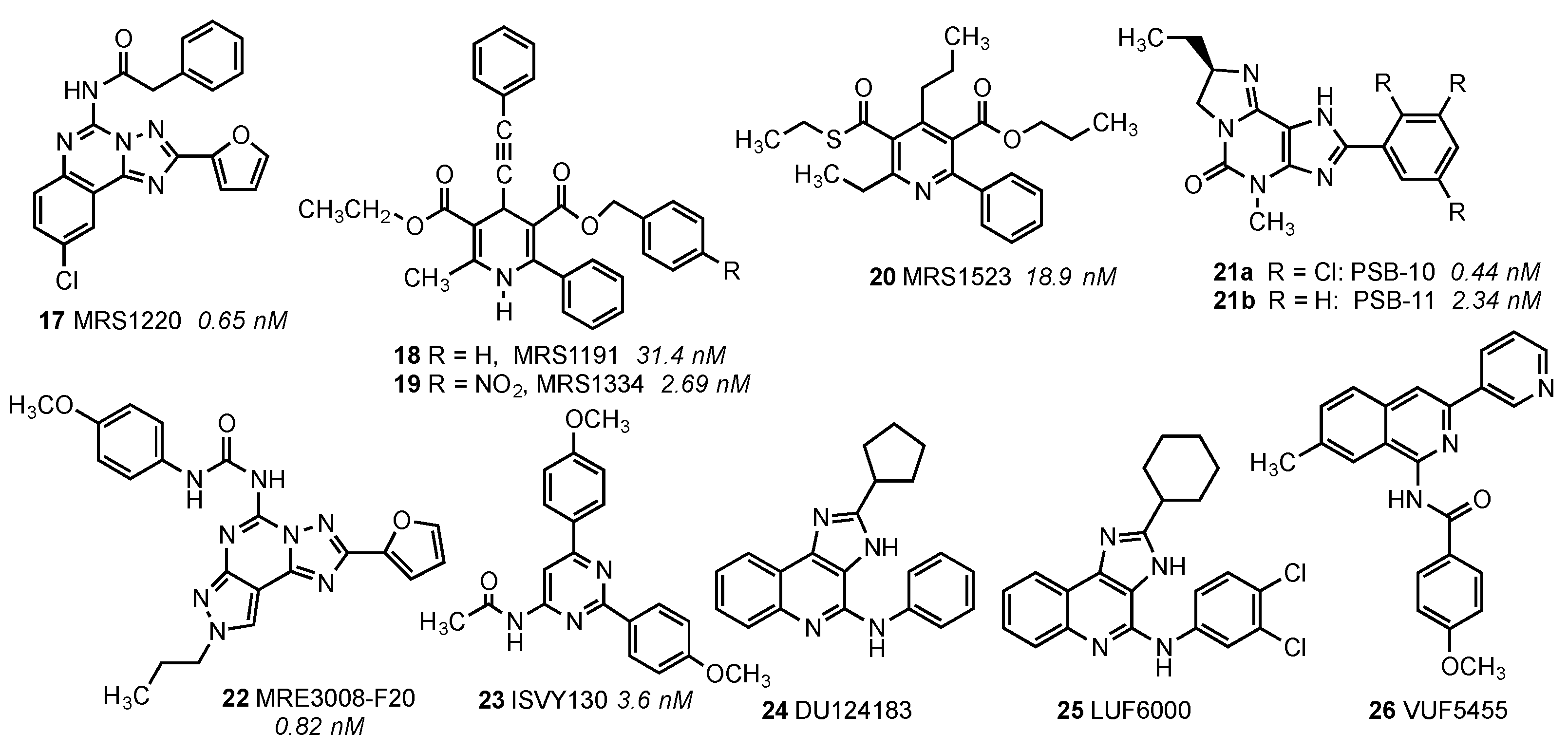
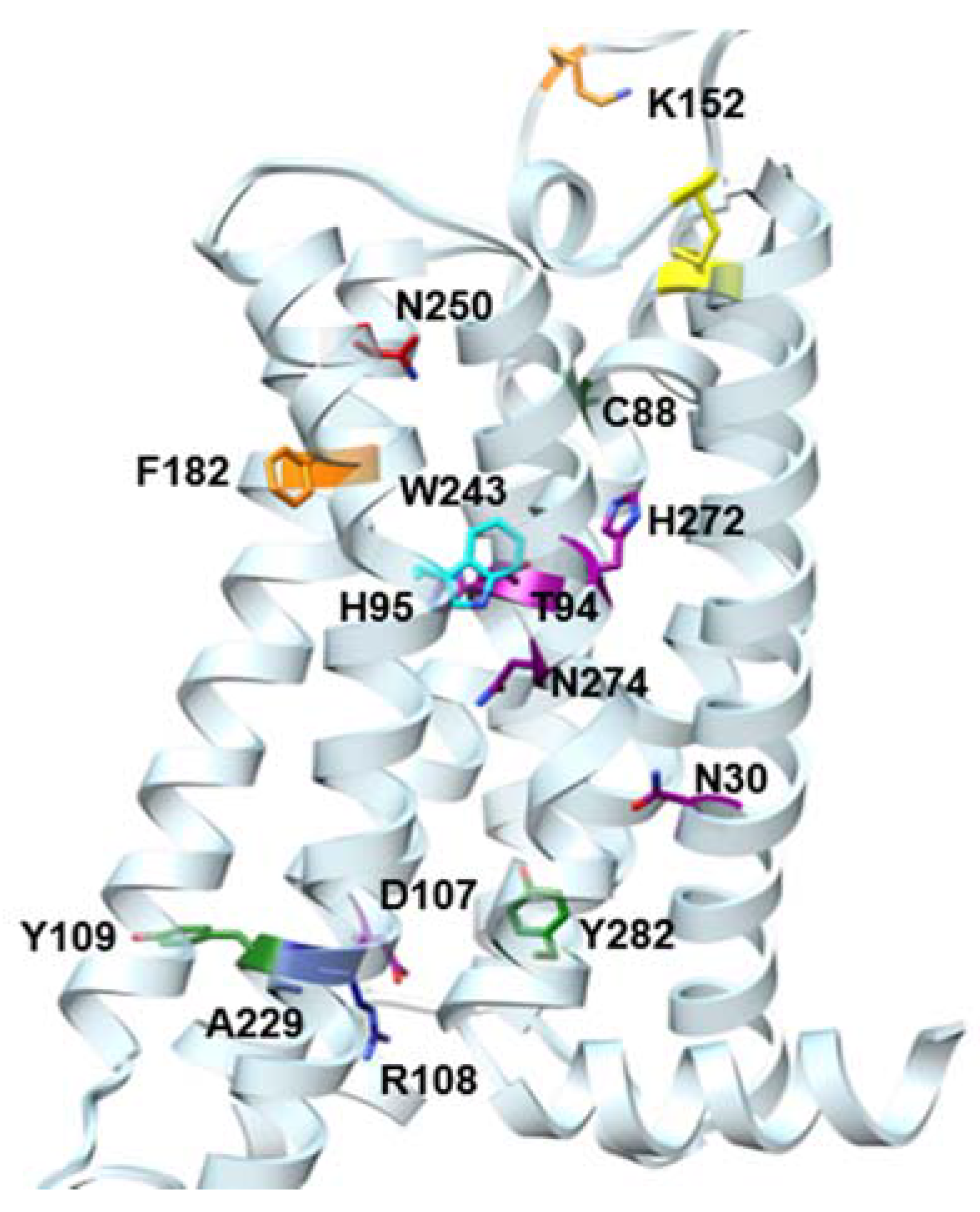
| Residue | Residue Number 1 | Mutation | Effect on Binding of 2 | Reference | |
|---|---|---|---|---|---|
| Agonist | Antagonist | ||||
| Asn30 | 1.50 | Ala | Decreased | Decreased | [14] |
| Asn58 | 2.50 | Asn | No changes | No Changes | [14] |
| Cys88 | 3.30 | Phe | Decreased | No effect | [15] |
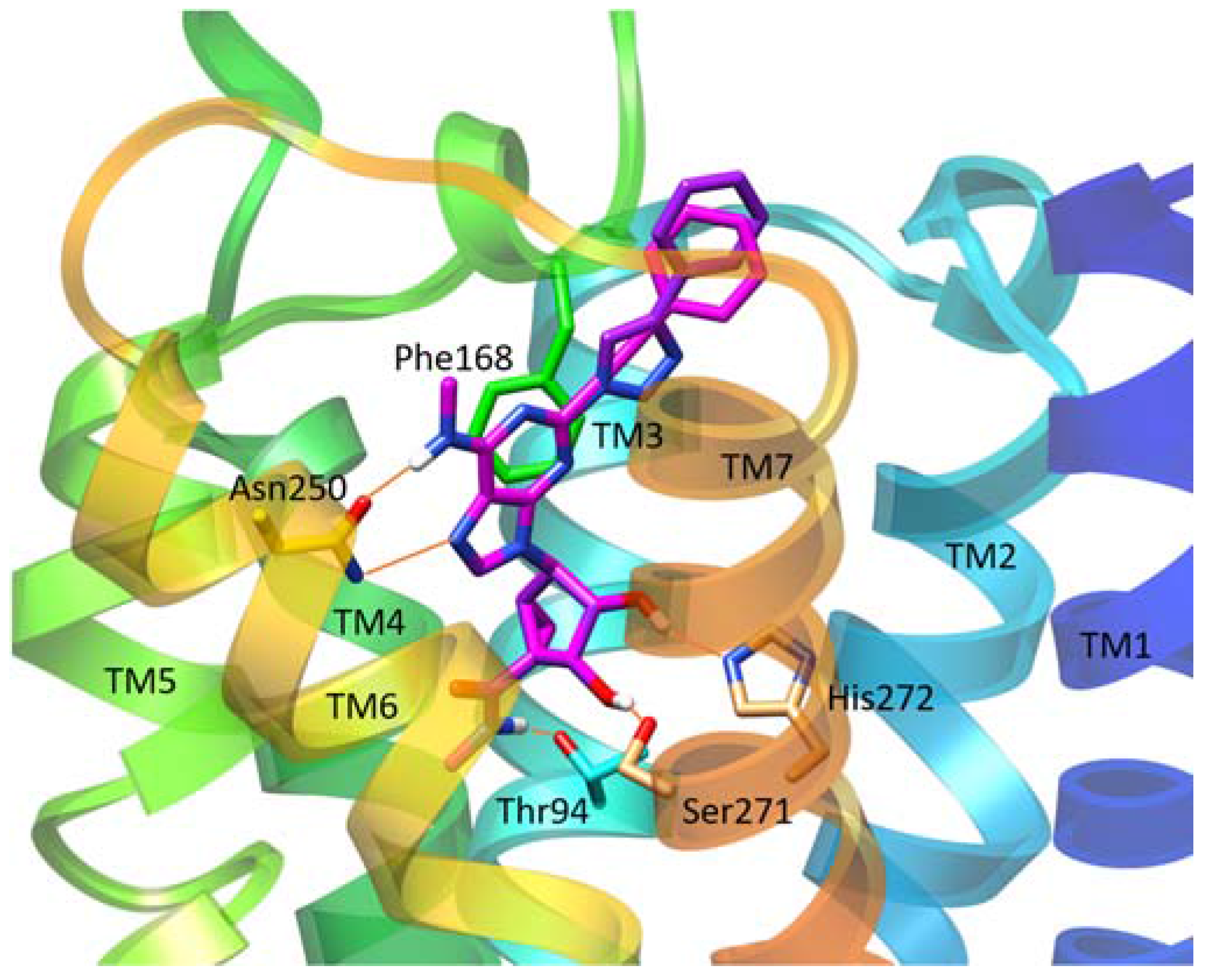
| Thr94 | 3.36 | Ala | Decreased | Decreased | [14] |
| His95 | 3.37 | Ala | Decreased | Decreased | [14] |
| Asp107 | 3.49 | Asn | Minor changes | Decreased | [14] |
| Arg108 | 3.50 | Arg,Lys,Asn, Glu,His | Minor changes | No effect | [15] |
| Tyr109 | 3.51 | Phe | Decreased | No effect | [15] |
| Lys152 | (EL2) | Ala | No effect | Decrease | [16] |
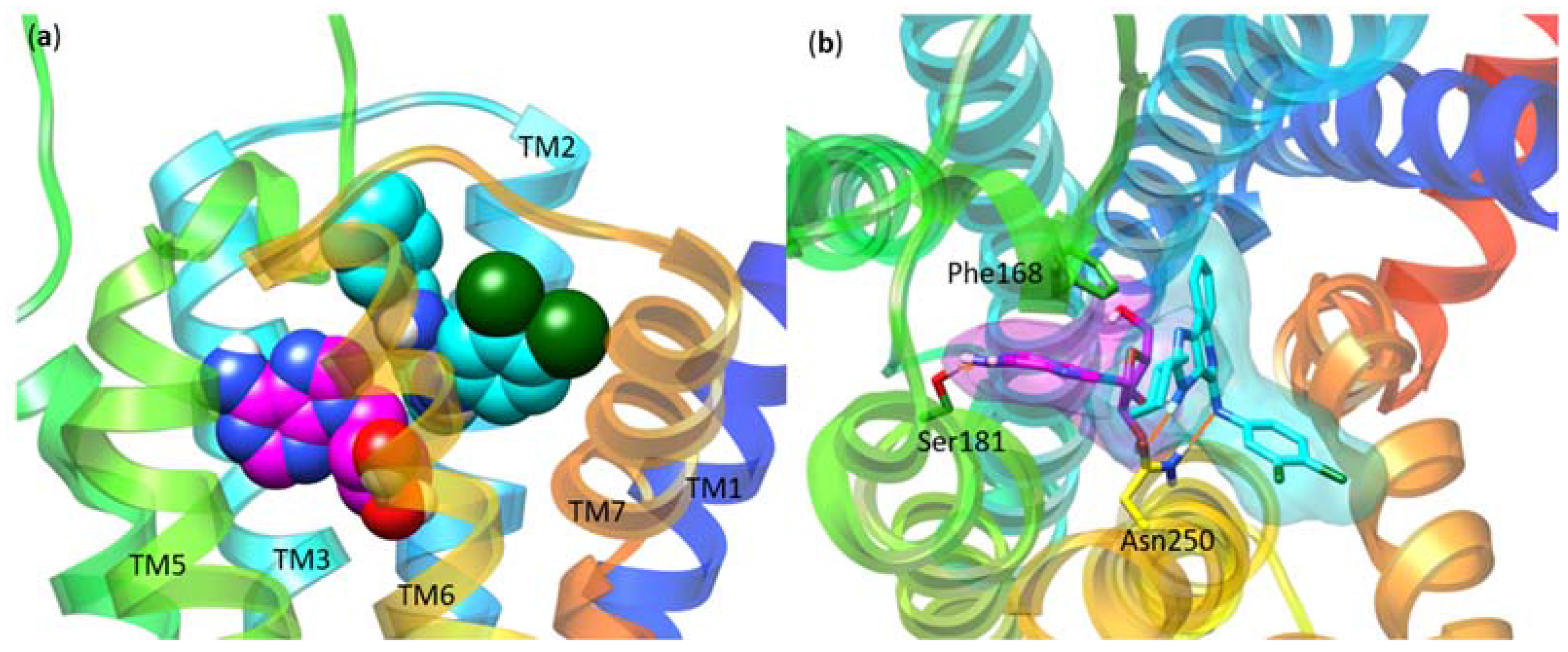
| Phe182 | 5.43 | Ala | No effect | Decreased | [14] |
| Trp243 | 6.48 | Ala,Phe | No effect 3 | Decreased | [16] |
| Asn250 | 6.55 | Ala | No binding | No binding | [16] |
| His272 | 7.43 | Glu | Decreased | Decreased | [16,17] |
| Asn274 | 7.45 | Ala | Decreased | Decreased | [14] |
| Tyr282 | 7.53 | Phe | Decreased | No effect | [14] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciancetta, A.; Jacobson, K.A. Structural Probing and Molecular Modeling of the A3 Adenosine Receptor: A Focus on Agonist Binding. Molecules 2017, 22, 449. https://doi.org/10.3390/molecules22030449
Ciancetta A, Jacobson KA. Structural Probing and Molecular Modeling of the A3 Adenosine Receptor: A Focus on Agonist Binding. Molecules. 2017; 22(3):449. https://doi.org/10.3390/molecules22030449
Chicago/Turabian StyleCiancetta, Antonella, and Kenneth A. Jacobson. 2017. "Structural Probing and Molecular Modeling of the A3 Adenosine Receptor: A Focus on Agonist Binding" Molecules 22, no. 3: 449. https://doi.org/10.3390/molecules22030449
APA StyleCiancetta, A., & Jacobson, K. A. (2017). Structural Probing and Molecular Modeling of the A3 Adenosine Receptor: A Focus on Agonist Binding. Molecules, 22(3), 449. https://doi.org/10.3390/molecules22030449