
Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Stable β-Arrestin-G-Protein-G Protein-Coupled Receptor Megaplexes
3. Sphingosine 1-Phosphate Receptor 1 Signaling
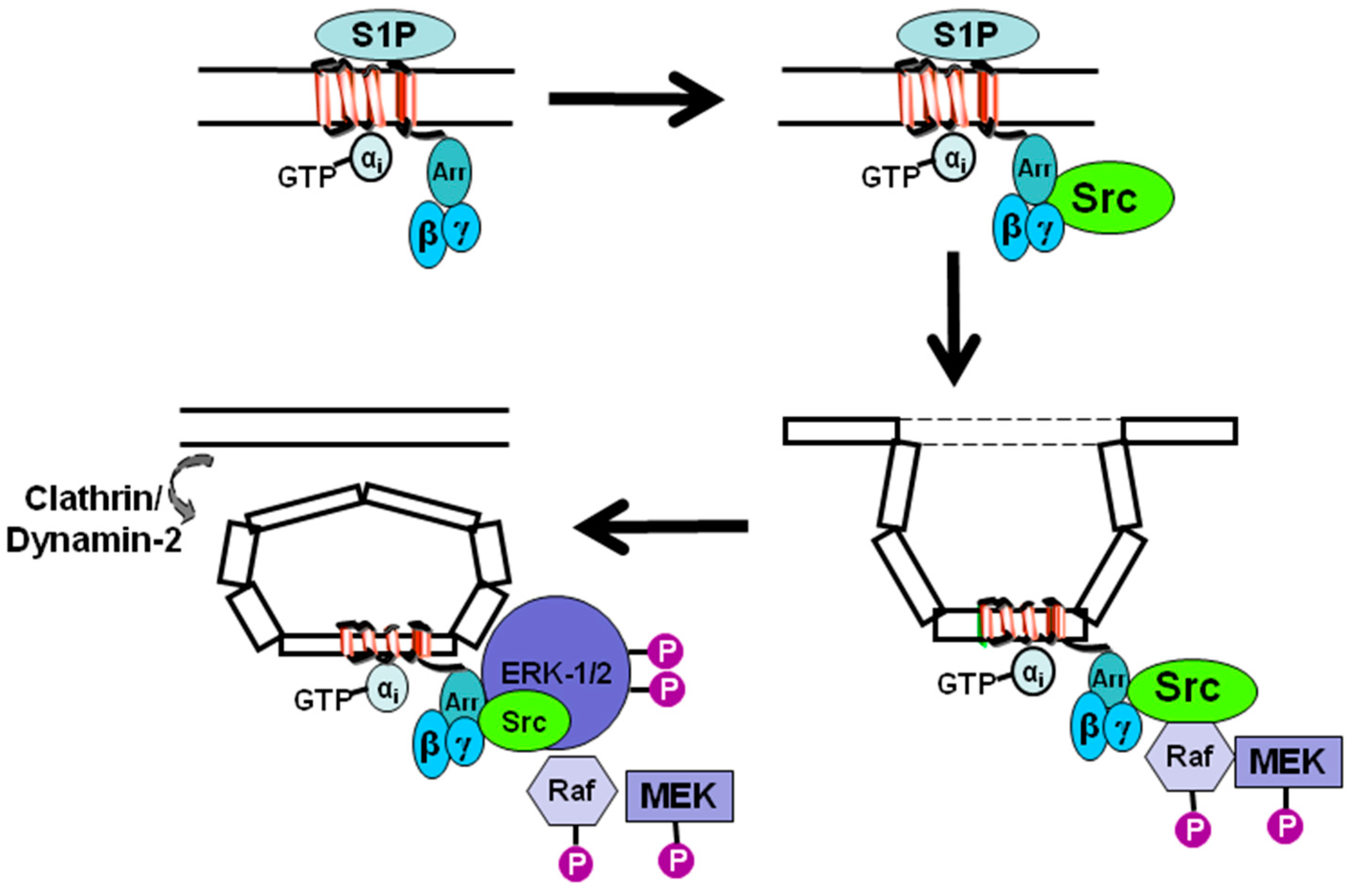
4. Sphingosine 1-Phosphate Receptor 1 Tyrosine Kinase Signaling Complexes
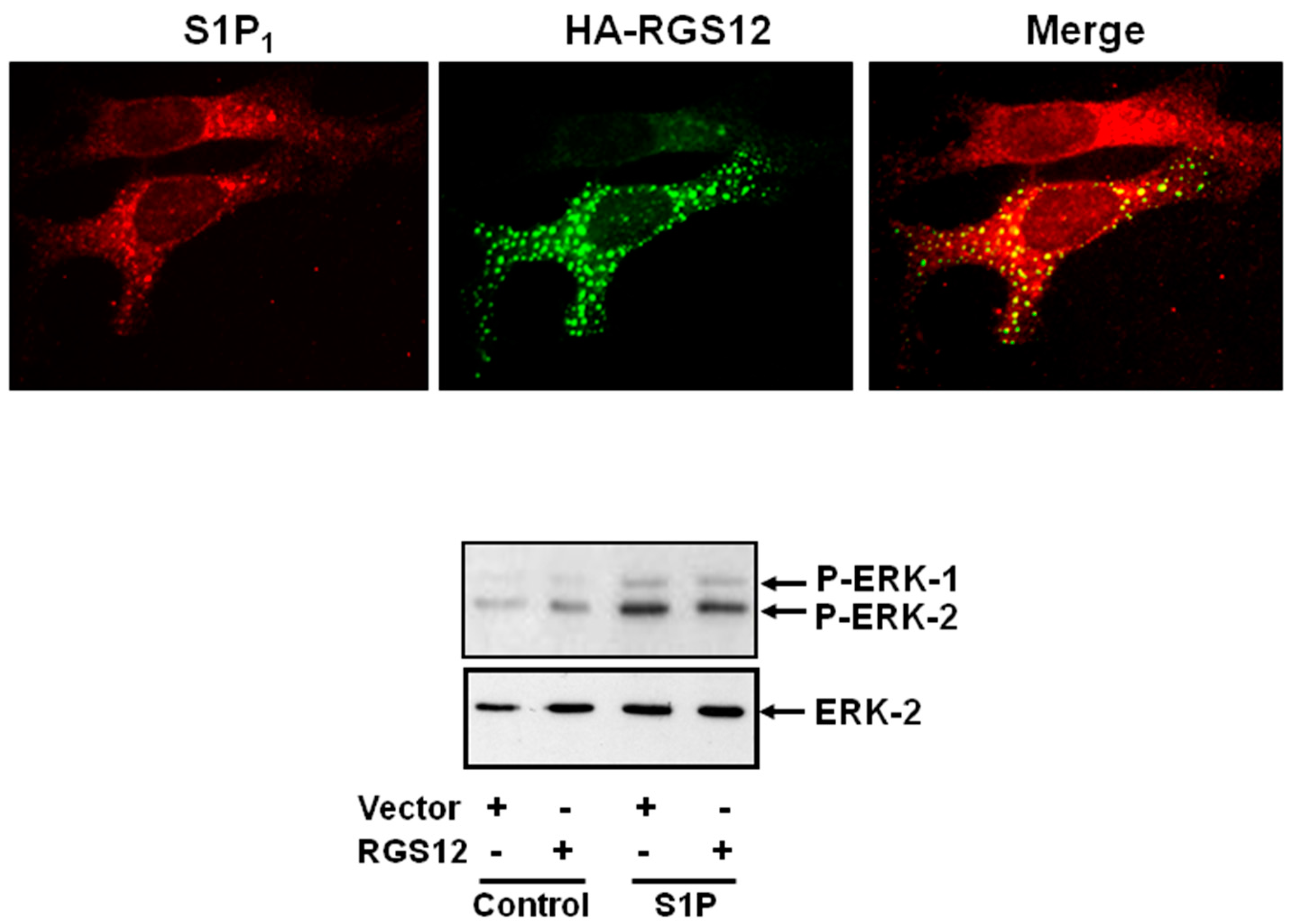
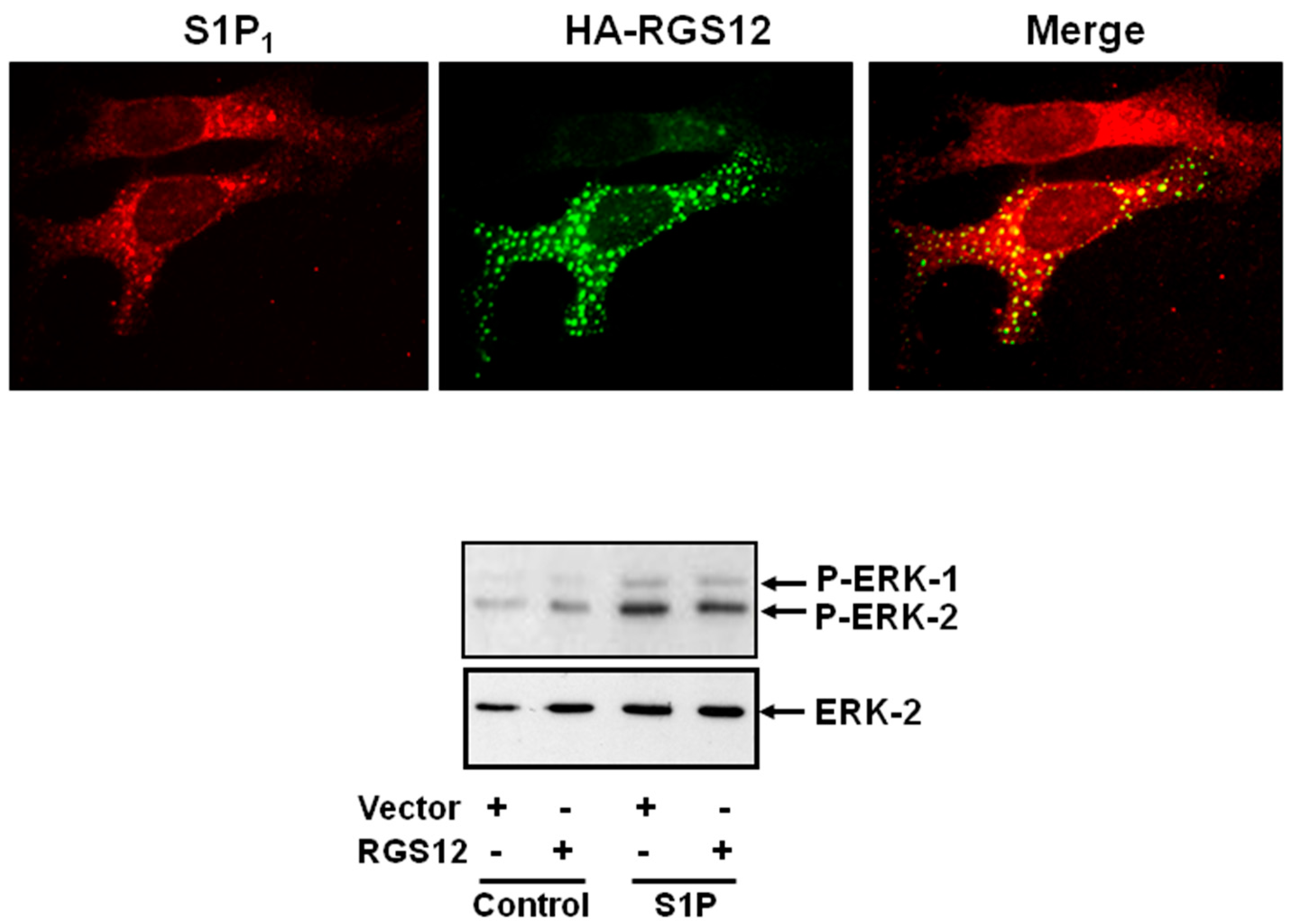
5. Sphingosine 1-Phosphate Receptor 1 and Regulator of G-Protein Signaling 12
6. Sphingosine 1-Phosphate Receptor 1 and Sphingosine 1-Phosphate Carriers/Chaperones
7. Sphingosine 1-Phosphate Receptor 1 in Health and Disease
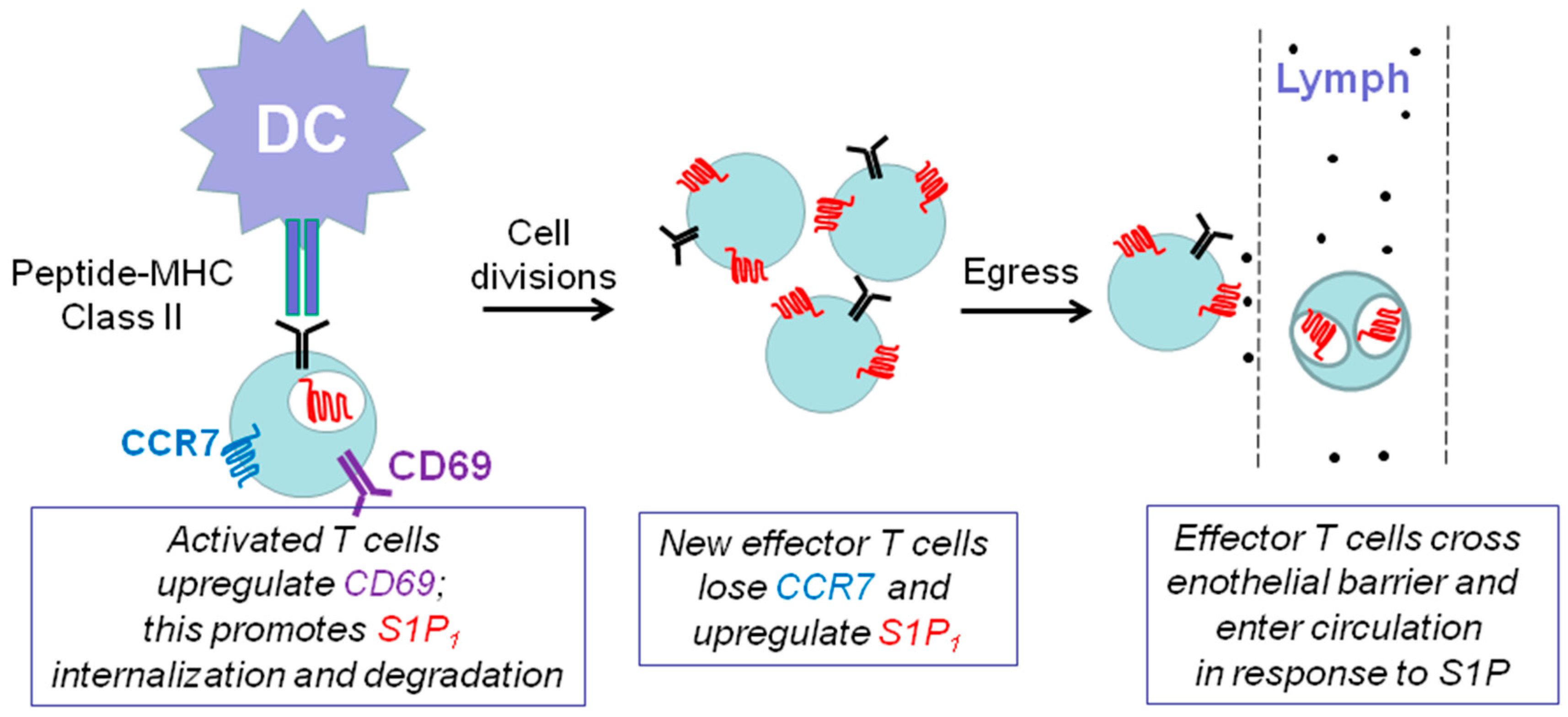
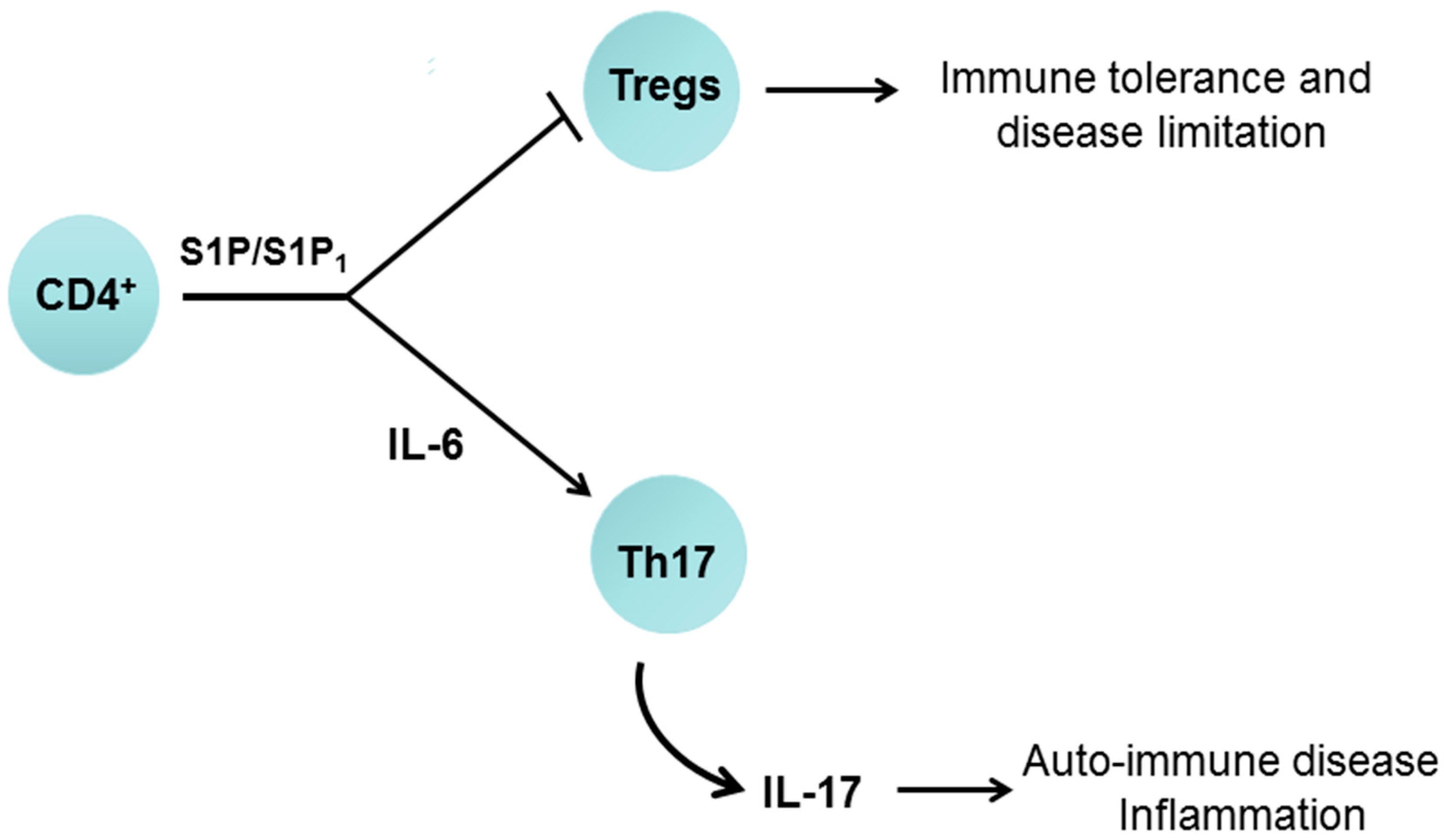
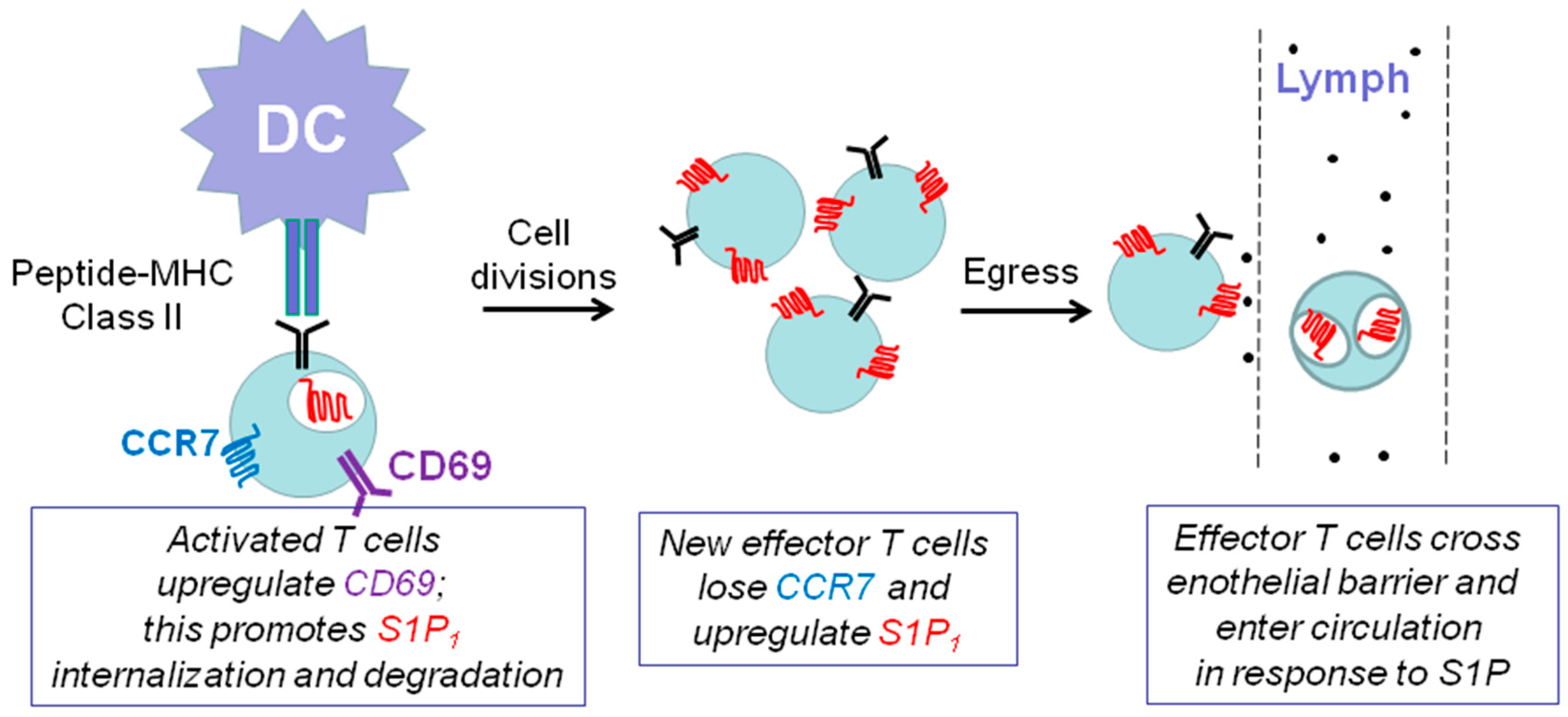
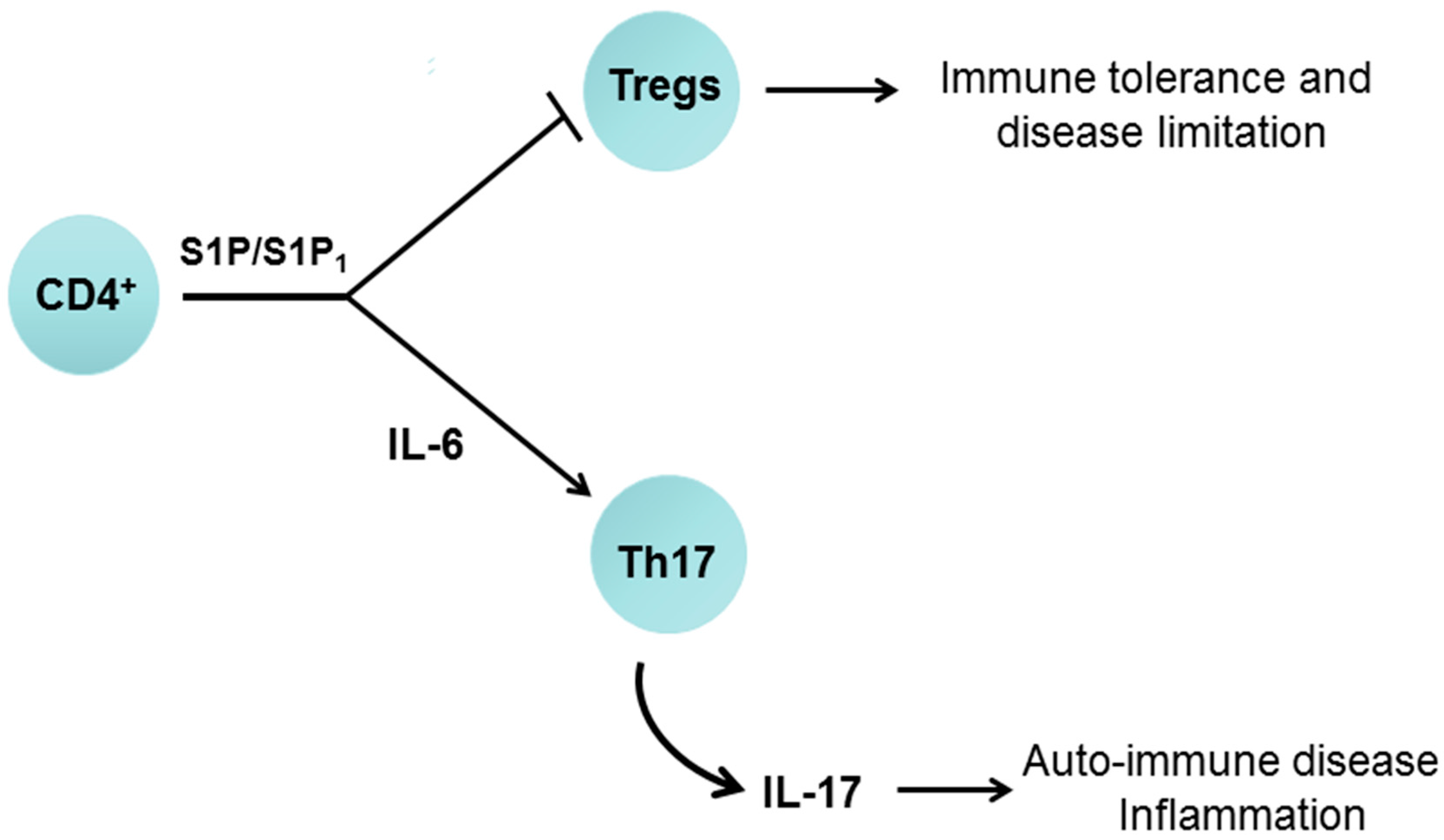
7.1. Sphingosine 1-Phosphate Receptor 1 and Immune Function
7.2. Sphingosine 1-Phosphate Receptor 1 and the Nervous System
7.3. Sphingosine 1-Phosphate Receptor 1 and Neovascularisation
7.4. Sphingosine 1-Phosphate Receptor 1 and the Heart
7.5. Sphingosine 1-Phosphate Receptor 1 and Cancer
8. Clinical Evidence for Sphingosine 1-Phosphate Receptor 1 Modulators
9. Conclusions
Author Contributions
Conflicts of Interest
References
- Pyne, S.; Pyne, N.J. Translational aspects of sphingosine 1-phosphate biology. Trends Mol. Med. 2011, 17, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Characterisation of the ATP dependent sphingosine 1-phosphate transporter in rat erythrocytes. J. Biol. Chem. 2009, 284, 21192–21200. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS ONE 2012, 7, e38941. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Kobayashi, N.; Hisano, Y.; Kawahara, A.; Yamaguchi, A. Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochim. Biophys. Acta 2014, 1841, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Donoviel, M.S.; Hait, N.C.; Ramachandran, S.; Maceyka, M.; Takabe, K.; Milstien, S.; Oravecz, T.; Spiegel, S. Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J. 2015, 29, 5018–5028. [Google Scholar] [CrossRef] [PubMed]
- Weyden, L.V.; Arends, M.J.; Campbell, A.D.; Bald, T.; Wardle-Jones, H.; Griggs, N.; Velasco-Herrera, M.D.; Tüting, T.; Sansom, O.J.; Karp, N.A.; et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonisation. Nature 2017, 541, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Goetzl, E.J.; Hla, T.; Igarashi, Y.; Lynch, K.R.; Moolenaar, W.; Pyne, S.; Tigyi, G. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2002, 54, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Roth, C.B.; Jo, E.; Griffith, M.T.; Scott, F.L.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.M.; Schuerer, S.C.; et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012, 335, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.A.; Milano, S.K.; Benovic, J.L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 2007, 69, 451–482. [Google Scholar] [CrossRef] [PubMed]
- Goodman, O.B.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.; Caron, M.G.; Barak, L.S. The beta2-adrenergic receptor/beta arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Barak, L.S.; Caron, M.G. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis. J. Biol. Chem. 2001, 276, 19452–19460. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de Filippis, T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent cAMP-signals triggered by internalised G-protein-coupled receptors. PLoS Biol. 2009, 7, e1000172. [Google Scholar] [CrossRef]
- Feinstein, T.N.; Yui, N.; Webber, M.J.; Wehbi, V.L.; Stevenson, H.P.; King, J.D.; Hallows, K.R.; Brown, D.; Bouley, R.; Vilardaga, J.P. Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J. Biol. Chem. 2013, 288, 27849–27860. [Google Scholar] [CrossRef] [PubMed]
- Ferrandon, S.; Feinstein, T.N.; Castro, M.; Wang, B.; Bouley, R.; Potts, J.T.; Gardella, T.J.; Vilardaga, J.P. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat. Chem. Biol. 2009, 5, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; Chevalier, M.; Mahoney, J.P.; Steyaert, J.; Rasmussen, S.G.; Sunahara, R.K.; El-Samad, H.; Huang, B.; et al. Conformational biosensors reveal GPCR signaling from endosomes. Nature 2013, 495, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Zecri, F.; Cetin, C.; Billich, A.; Guerini, D.; Seuwen, K. Persistent signaling induced by FTY720-phosphate is mediated by internalised S1P1 receptors. Nat. Chem. Biol. 2009, 5, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; et al. Visualisation of arrestin recruitment by a G-protein-coupled receptor. Nature 2014, 512, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, A.R.; Plouffe, B.; Cahill, T.J., 3rd; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ahn, S.; Shenoy, S.K.; Karnik, S.S.; Hunyady, L.; Luttrell, L.M.; Lefkowitz, R.J. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA 2003, 100, 10782–10787. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.; Yee, D.K.; Faulconbridge, L.F.; Fluharty, S.J. Divergent behavioural roles of angiotensin receptor intracellular signaling cascades. Endocrinology 2005, 146, 5552–5560. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.; Connell, M.C.; Pyne, S.; Pyne, N.J. c-Src is involved in regulating signal transmission from PDGFβ receptor-GPCR signal complexes in mammalian cells. Cell Signal. 2005, 17, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.; Sambi, B.; Kong, K.C.; Thompson, D.; Pitson, S.M.; Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate and platelet-derived growth factor (PDGF) act via PDGF beta receptor-sphingosine 1-phosphate receptor complexes in airway smooth muscle cells. J. Biol. Chem. 2003, 278, 6282–6290. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase-G-protein-coupled receptor signaling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Alderton, F.; Rakhit, S.; Kong, K.C.; Palmer, T.; Sambi, B.; Pyne, S.; Pyne, N.J. Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J. Biol. Chem. 2001, 276, 28578–28585. [Google Scholar] [CrossRef] [PubMed]
- Watterson, K.R.; Johnston, E.; Chalmers, C.; Pronin, A.; Cook, S.J.; Benovic, J.L.; Palmer, T.M. Dual regulation of EDG1/S1P(1) receptor phosphorylation and internalisation by protein kinase C and G-protein-coupled receptor kinase 2. J. Biol. Chem. 2002, 277, 5767–5777. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.D.; Balius, A.M.; Yamaguchi, K.; Freilich, E.R.; Barak, L.S.; Kwatra, M.M. Human substance P receptor lacking the C-terminal domain remains competent to desensitise and internalise. J. Neurochem. 2003, 84, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Gurevich, V.V.; Preninger, A.; Hamm, H.E.; Bader, M.F.; Fazleabas, A.T.; Birnbaumer, L.; Hunzicker-Dunn, M. Aspartic acid 564 in the third cytoplasmic loop of the luteinising hormone/choriogonadotropin receptor is crucial for phosphorylation-independent interaction with arrestin 2. J. Biol. Chem. 2002, 277, 17916–17927. [Google Scholar] [CrossRef] [PubMed]
- Jala, V.R.; Shao, W.H.; Haribabu, B. Phosphorylation-independent beta-arrestin translocation and internalisation of leukotriene B4 receptors. J. Biol. Chem. 2005, 280, 4880–4887. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.M.; Kang, Y.L.; Kirchhausen, T. Endocytosis of Ligand-Activated Sphingosine 1-Phosphate Receptor 1 Mediated by the Clathrin-Pathway. Traffic 2016, 17, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Ferguson, S.M.; Pereira, J.P.; De Camilli, P.; Flavell, R.A. Dynamin-2-dependent endocytosis is required for sustained S1PR1 signaling. J. Exp. Med. 2014, 211, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.M.; Long, J.; Gorshkova, I.; Fujiwara, Y.; Connell, M.; Belmonte, K.E.; Tigyi, G.; Natarajan, V.; Pyne, S.; Pyne, N.J. Cell migration activated by platelet-derived growth factor receptor is blocked by an inverse agonist of the sphingosine 1-phosphate receptor-1. FASEB J. 2006, 20, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, C.; Childs, S.; Ohotski, J.; McGlynn, L.; Riddick, M.; MacFarlane, S.; Tasker, D.; Pyne, S.; Pyne, N.J.; Edwards, J.; et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013, 4, e927. [Google Scholar] [CrossRef] [PubMed]
- Gorshkova, I.; He, D.; Berdyshev, E.; Usatuyk, P.; Burns, M.; Kalari, S.; Zhao, Y.; Pendyala, S.; Garcia, J.G.; Pyne, N.J.; et al. Protein kinase C-epsilon regulates sphingosine 1-phosphate-mediated migration of human lung endothelial cells through activation of phospholipase D2, protein kinase C-zeta, and Rac1. J. Biol. Chem. 2008, 283, 11794–11806. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; He, D.; Bindokas, V.; Gorshkova, I.A.; Berdyshev, E.V.; Garcia, J.G.; Natarajan, V. Photolysis of caged sphingosine-1-phosphate induces barrier enhancement and intracellular activation of lung endothelial cell signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L840–L850. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Natarajan, V.; Tigyi, G.; Pyne, S.; Pyne, N.J. The functional PDGFβ receptor-S1P1 receptor signaling complex is involved in regulating migration of mouse embryonic fibroblasts in response to platelet derived growth factor. Prostaglandins Other Lipid Mediat. 2006, 80, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Schmidt, T.T.; Yazbeck, P.; Rajput, C.; Desai, B.; Sukriti, S.; Giantsos-Adams, K.; Knezevic, N.; Malik, A.B.; Mehta, D. S1PR1 Tyr143 phosphorylation downregulates endothelial cell surface S1PR1 expression and responsiveness. J. Cell Sci. 2015, 128, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Bergelin, N.; Löf, C.; Balthasar, S.; Kalhori, V.; Törnquist, K. S1P1 and VEGFR-2 form a signaling complex with extracellularly regulated kinase 1/2 and protein kinase C-alpha regulating ML-1 thyroid carcinoma cell migration. Endocrinology 2010, 151, 2994–3005. [Google Scholar] [PubMed]
- Ryu, J.M.; Baek, Y.B.; Shin, M.S.; Park, J.H.; Park, S.H.; Lee, J.H.; Han, H.J. Sphingosine-1-phosphate-induced Flk-1 transactivation stimulates mouse embryonic stem cell proliferation through S1P1/S1P3-dependent β-arrestin/c-Src pathways. Stem Cell Res. 2014, 12, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Akekawatchai, C.; Holland, J.D.; Kochetkova, M.; Wallace, J.C.; McColl, S.R. Transactivation of CXCR4 by IGF-IR in human MDA-MB-231 breast cancer epithelial cells. J. Biol. Chem. 2005, 280, 39701–39708. [Google Scholar] [CrossRef] [PubMed]
- Delcourt, N.; Thouvenot, E.; Chanrion, B.; Galéotti, N.; Jouin, P.; Bockaert, J.; Marin, P. PACAP type 1 receptor transactivation is essential for IGF-1 receptor signaling and anti-apoptotic activity in neurons. EMBO J. 2007, 26, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.M.; Canals, D.; Jeong, N.; Kelkar, A.D.; Hernandez-Corbacho, M.; Pulkoski-Gross, M.J.; Donaldson, J.C.; Hannun, Y.A.; Obeid, L.M. Intracellular sphingosine kinase 2-derived sphingosine-1-phosphate mediates epidermal growth factor-induced ezrin-radixin-moesin phosphorylation and cancer cell invasion. FASEB J. 2015, 29, 4654–4669. [Google Scholar] [CrossRef] [PubMed]
- Sambi, B.S.; Hains, M.D.; Waters, C.M.; Connell, M.C.; Willard, F.S.; Kimple, A.J.; Pyne, S.; Siderovski, D.P.; Pyne, N.J. The effect of RGS12 on platelet derived growth factor beta receptor signaling to p42/p44 MAPK in mammalian cells. Cell. Signal. 2006, 18, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, B.A.; Grass, G.D.; Wing, S.B.; Argraves, W.S.; Argraves, K.M. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: High density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem. 2012, 287, 44645–44653. [Google Scholar] [CrossRef] [PubMed]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Conger, H.; Dahlbäck, B.; Kono, M.; Proia, R.L.; Smith, J.D.; Hla, T. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 2015, 8, ra79. [Google Scholar] [CrossRef] [PubMed]
- Blaho, V.A.; Galvani, S.; Engelbrecht, E.; Liu, C.; Swendeman, S.L.; Kono, M.; Proia, R.L.; Steinman, L.; Han, M.H.; Hla, T. HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 2015, 523, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.S.; Liu, C.H.; Sun, Y.; Chen, Y.; Swendeman, S.L.; Jung, B.; Chavez, D.; Cao, Z.; Christoffersen, C.; Nielsen, L.B.; et al. HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P(1)) promotes regeneration and suppresses fibrosis in the liver. JCI Insight 2016, 1, e87058. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Tucker, A.E.; Tran, J.; Bergner, J.B.; Turner, E.M.; Proia, R.L. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J. Clin. Investig. 2014, 124, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Perez, W.D.; Fang, V.; Escalante-Alcalde, D.; Cammer, M.; Schwab, S.R. A map of the distribution of sphingosine 1-phosphate in the spleen. Nat. Immunol. 2015, 16, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Cyster, J.G.; Hla, T. FTY720: Sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am. J. Transplant. 2004, 4, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.; Lindberg, R.; Raulf, F.; Kuhle, J.; Hess, C.; Kappos, L.; Brinkmann, V. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology 2010, 75, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.J.; Huang, M.C.; Goetzl, E.J. Cutting edge: Alternative signaling of Th17 cell development by sphingosine 1-phosphate. J. Immunol. 2007, 178, 5425–5428. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Seki, N.; Kataoka, H.; Takemoto, K.; Utsumi, H.; Fukunari, A.; Sugahara, K.; Chiba, K. IL-17-Producing Vγ4+ γδ T Cells Require Sphingosine 1-Phosphate Receptor 1 for Their Egress from the Lymph Nodes under Homeostatic and Inflammatory Conditions. J. Immunol. 2015, 195, 1408–1416. [Google Scholar] [CrossRef]
- Liu, G.; Burns, S.; Huang, G.; Boyd, K.; Proia, R.L.; Flavell, R.A.; Chi, H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat. Immunol. 2009, 10, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Gutkind, S.; Stitham, J.; Okuno, T.; Yokomizo, T.; Hwa, J.; Hla, T. Individual variation of human S1P₁ coding sequence leads to heterogeneity in receptor function and drug interactions. J. Lipid Res. 2014, 55, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Wu, L.; Acharya, S.; Arac, A.; Blaho, V.A.; Huang, Y.; Moon, B.S.; Axtell, R.C.; Ho, P.P.; Steinberg, G.K.; et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat. Immunol. 2013, 14, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Tuymetova, G.; Lee, B.G.; Bonifacino, E.; Wu, Y.P.; Proia, R.L. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J. Exp. Med. 2010, 207, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Patrussi, L.; Capitani, N.; Martini, V.; Pizzi, M.; Trimarco, V.; Frezzato, F.; Marino, F.; Semenzato, G.; Trentin, L.; Baldari, C.T. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res. 2015, 75, 4153–4163. [Google Scholar] [CrossRef] [PubMed]
- Bigaud, M.; Guerini, D.; Billich, A.; Bassilana, F.; Brinkmann, V. Second generation S1P pathway modulators: Research strategies and clinical developments. Biochim. Biophys. Acta 2014, 1841, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Rosas, G.; Pikor, N.; Schwartz, J.A.; Rojas, O.; Jian, A.; Summers-Deluca, L.; Ostrowski, M.; Nuesslein-Hildesheim, B.; Gommerman, J.L. A sphingosine-1-phosphate receptor 1-directed agonist reduces central nervous system inflammation in a plasmacytoid dendritic cell-dependent manner. J. Immunol. 2012, 189, 3700–3706. [Google Scholar] [CrossRef] [PubMed]
- Dukala, D.E.; Soliven, B. S1P1 deletion in oligodendroglial lineage cells: Effect on differentiation and myelination. Glia 2016, 64, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Laviña, B.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nyström, S.; Rymo, S.; Chen, L.L.; et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Ben Shoham, A.; Malkinson, G.; Krief, S.; Shwartz, Y.; Ely, Y.; Ferrara, N.; Yaniv, K.; Zelzer, E. S1P1 inhibits sprouting angiogenesis during vascular development. Development 2012, 139, 3859–3869. [Google Scholar] [CrossRef] [PubMed]
- Sabbadini, R.A. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br. J. Pharmacol. 2011, 162, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Tufail, A.; Patel, P.J.; Egan, C.; Hykin, P.; da Cruz, L.; Gregor, Z.; Dowler, J.; Majid, M.A.; Bailey, C.; Mohamed, Q.; et al. ABC Trial Investigators. Bevacizumab for neovascular age related macular degeneration (ABC Trial): Multicentre randomised double masked study. Br. Med. J. 2010, 340, c2459. [Google Scholar] [CrossRef] [PubMed]
- Caballero, S.; Swaney, J.; Moreno, K.; Afzal, A.; Kielczewski, J.; Stoller, G.; Cavalli, A.; Garland, W.; Hansen, G.; Sabbadini, R.; et al. Anti-sphingosine-1-phosphate monoclonal antibodies inhibit angiogenesis and sub-retinal fibrosis in a murine model of laser-induced choroidal neovascularization. Exp. Eye Res. 2009, 88, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H250–H261. [Google Scholar] [CrossRef] [PubMed]
- Clay, H.; Wilsbacher, L.D.; Wilson, S.J.; Duong, D.N.; McDonald, M.; Lam, I.; Park, K.E.; Chun, J.; Coughlin, S.R. Sphingosine 1-phosphate receptor-1 in cardiomyocytes is required for normal cardiac development. Dev. Biol. 2016, 418, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative role in heart failure. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Alvarez, S.E.; Milstien, S.; Spiegel, S. Filamin A links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol. Cell. Biol. 2008, 28, 5687–5697. [Google Scholar] [CrossRef] [PubMed]
- LaMontagne, K.; Littlewood-Evans, A.; Schnell, C.; O’Reilly, T.; Wyder, L.; Sanchez, T.; Probst, B.; Butler, J.; Wood, A.; Liau, G.; et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularisation. Cancer Res. 2006, 66, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Yao, X.; Chen, L.; Yan, Z.; Liu, J.; Zhang, Y.; Feng, T.; Wu, J.; Liu, X. Sphingosine-1-phosphate induced epithelial-mesenchymal transition of hepatocellular carcinoma via an MMP-7/ syndecan-1/TGF-β autocrine loop. Oncotarget 2016, 7, 63324–63337. [Google Scholar] [CrossRef] [PubMed]
- Bouquerel, P.; Gstalder, C.; Müller, D.; Laurent, J.; Brizuela, L.; Sabbadini, R.A.; Malavaud, B.; Pyronnet, S.; Martineau, Y.; Ader, I.; et al. Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2α expression and activity in cancer. Oncogenesis 2016, 5, e209. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.; Wadham, C.; Gamble, J.; Xia, P. Sphingosine-1-phosphate receptor 1 transmits estrogens’ effects in endothelial cells. Steroids 2015, 104, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Tamashiro, P.M.; Furuya, H.; Shimizu, Y.; Kawamori, T. Sphingosine kinase 1 mediates head & neck squamous cell carcinoma invasion through sphingosine 1-phosphate receptor 1. Cancer Cell Int. 2014, 14, 76. [Google Scholar] [PubMed]
- Sekine, Y.; Suzuki, K.; Remaley, A.T. HDL and sphingosine-1-phosphate activate stat3 in prostate cancer DU145 cells via ERK1/2 and S1P receptors, and promote cell migration and invasion. Prostate 2011, 71, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate is a missing link between chronic inflammation and colon cancer. Cancer Cell 2013, 23, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Arnold, D.L.; Comi, G.; Bar-Or, A.; Gujrathi, S.; Hartung, J.P.; Cravets, M.; Olson, A.; Frohna, P.A.; Selmaj, K.W. Phase 2 results of the RADIANCE trial: A randomised, double-blind, placebo-controlled trial of oral RPC1063 in relapsing multiple sclerosis (LB1.1). Mult. Scler. 2014, 20 (Suppl. 1), 497. [Google Scholar]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cabrera, P.J.; Brown, S.; Studer, S.M.; Rosen, H. S1P signaling: New therapies and opportunities. F1000Prime Rep. 2014, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Vaclavkova, A.; Chimenti, S.; Arenberger, P.; Holló, P.; Sator, P.G.; Burcklen, M.; Stefani, M.; D’Ambrosio, D. Oral ponesimod in patients with chronic plaque psoriasis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 384, 2036–2045. [Google Scholar] [CrossRef]
- Bar-Or, A.; Zipp, F.; Scaramozza, M.; Vollmer, T.; Due, B.; Thangavelu, K.; Fischer, T.; Selmaj, K. Effect of Ceralifimod (ONO-4641), a Sphingosine-1-Phosphate Receptor-1 and -5 Agonist, on Magnetic Resonance Imaging Outcomes in Patients with Multiple Sclerosis: Interim Results from the Extension of the DreaMS Study. Neurology 2014, 82, P3.161. [Google Scholar]
- Kappos, L.; Arnold, D.L.; Bar-Or, A.; Camm, J.; Derfuss, T.; Kieseier, B.C.; Sprenger, T.; Greenough, K.; Ni, P.; Harada, T. Safety and efficacy of amiselimod in relapsing multiple sclerosis (MOMENTUM): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016, 15, 1148–1159. [Google Scholar] [CrossRef]
- Sanada, Y.; Mizushima, T.; Kai, Y.; Nishimura, J.; Hagiya, H.; Kurata, H.; Mizuno, H.; Uejima, E.; Ito, T. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS ONE 2011, 6, e23933. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Matsuda, C.; Kai, Y.; Nishida, T.; Nakajima, K.; Mizushima, T.; Kinoshita, M.; Yasue, T.; Sawa, Y.; Ito, T. A novel sphingosine 1-phosphate receptor agonist, 2-amino-2-propanediol hydrochloride (KRP-203), regulates chronic colitis in interleukin-10 gene-deficient mice. J. Pharmacol. Exp. Ther. 2008, 324, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Boster, A.; Fernández, Ó.; Freedman, M.S.; Pozzilli, C.; Bach, D.; Berkani, O.; Mueller, M.S.; Sidorenko, T.; Radue, E.W.; et al. Oral ponesimod in relapsing-remitting multiple sclerosis: A randomised phase II trial. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1198–1208. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules 2017, 22, 344. https://doi.org/10.3390/molecules22030344
Pyne NJ, Pyne S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules. 2017; 22(3):344. https://doi.org/10.3390/molecules22030344
Chicago/Turabian StylePyne, Nigel J., and Susan Pyne. 2017. "Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells" Molecules 22, no. 3: 344. https://doi.org/10.3390/molecules22030344
APA StylePyne, N. J., & Pyne, S. (2017). Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules, 22(3), 344. https://doi.org/10.3390/molecules22030344