Identification of the Sex-Biased Gene Expression and Putative Sex-Associated Genes in Eucommia ulmoides Oliver Using Comparative Transcriptome Analyses

Abstract
1. Introduction
2. Results
2.1. NGS Sequencing and De Novo Transcriptome Assembly
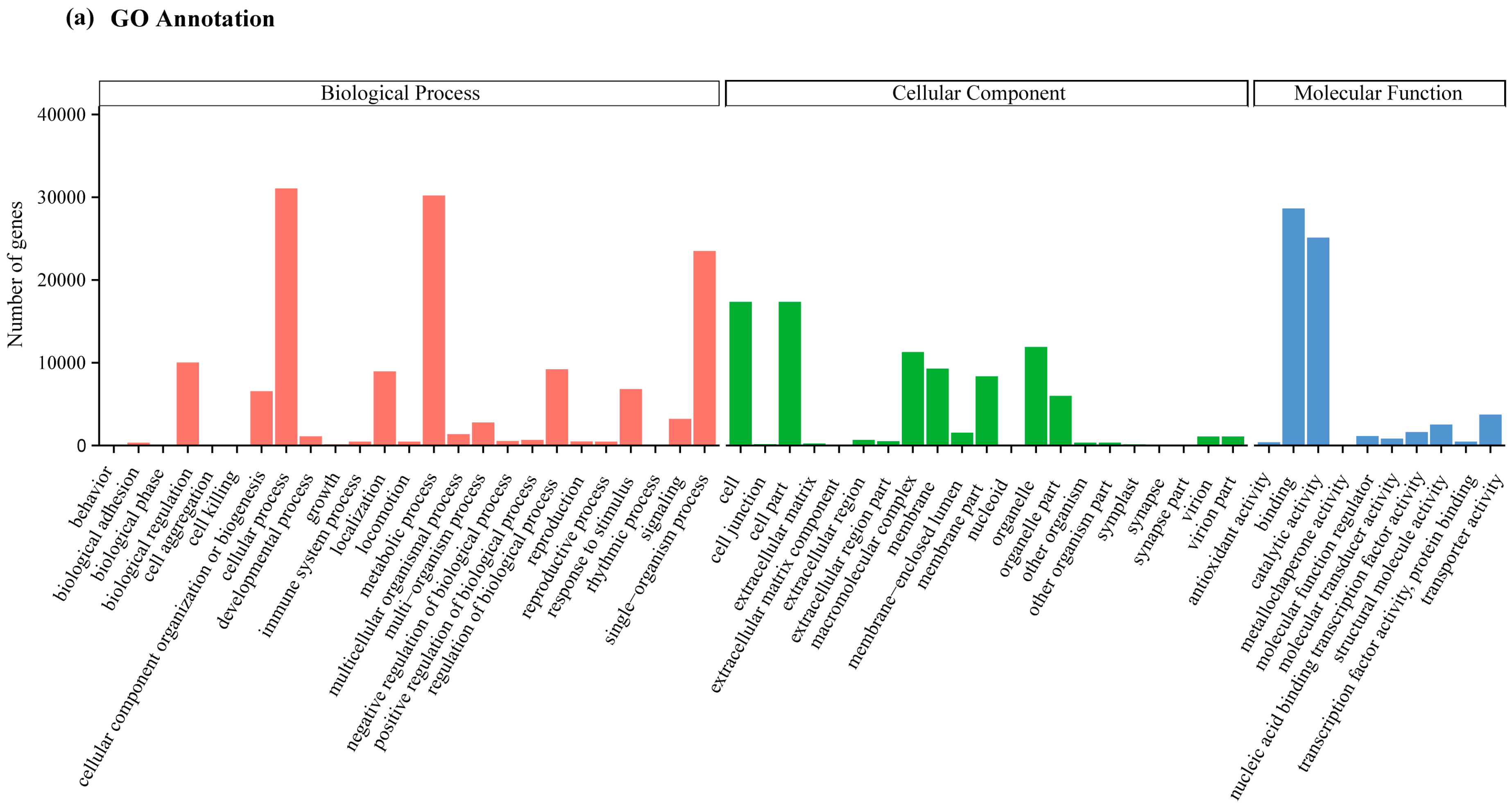
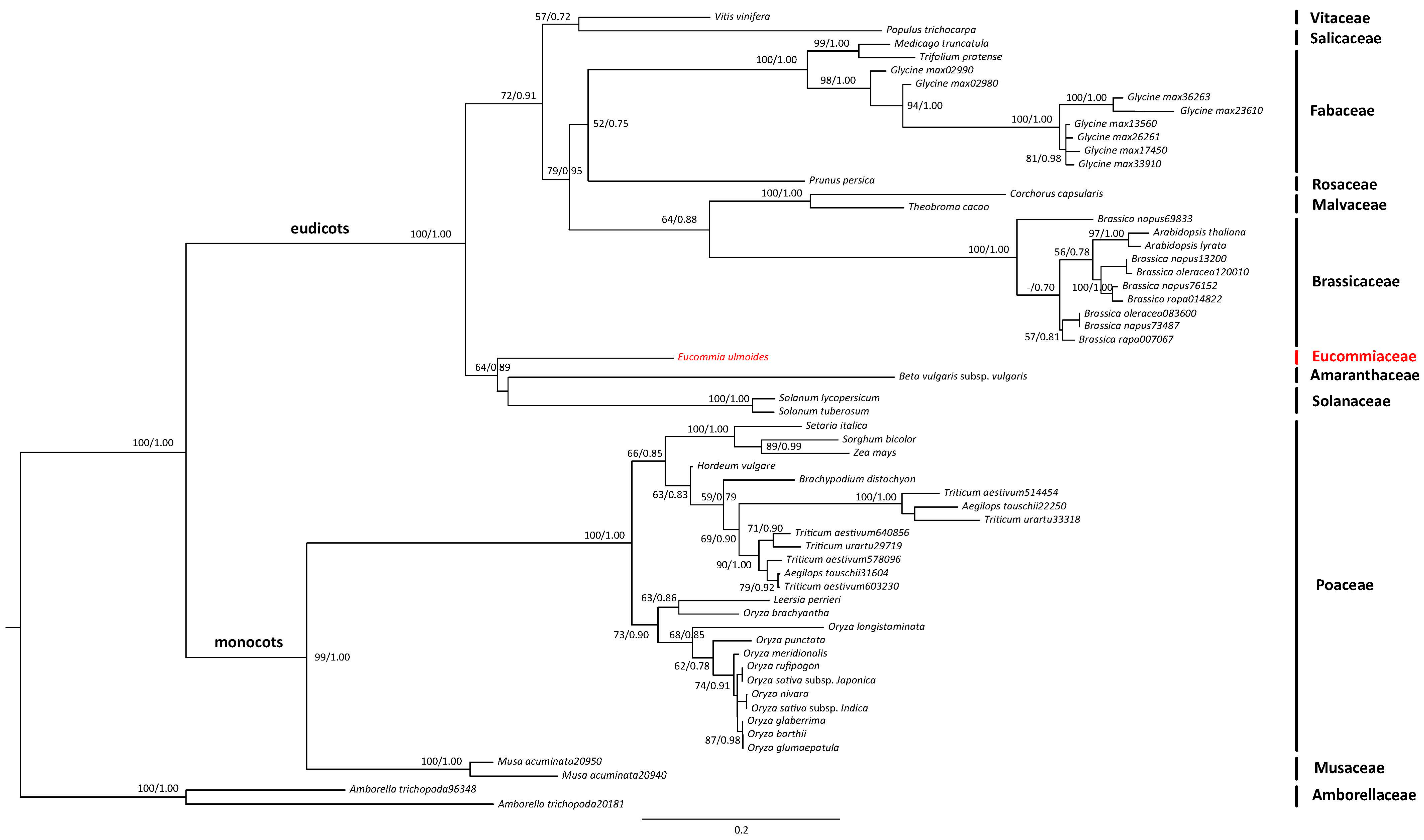
2.2. Transcriptome Annotation and Functional Classification
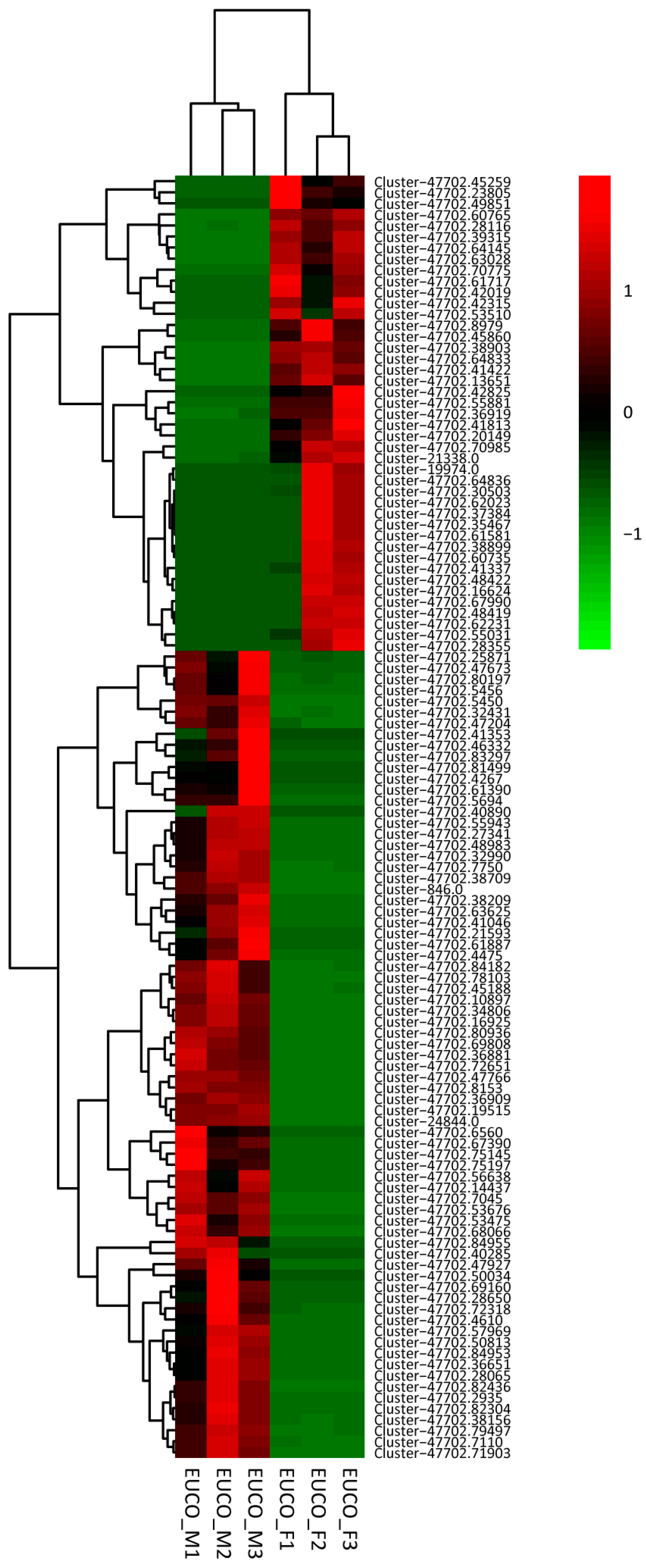
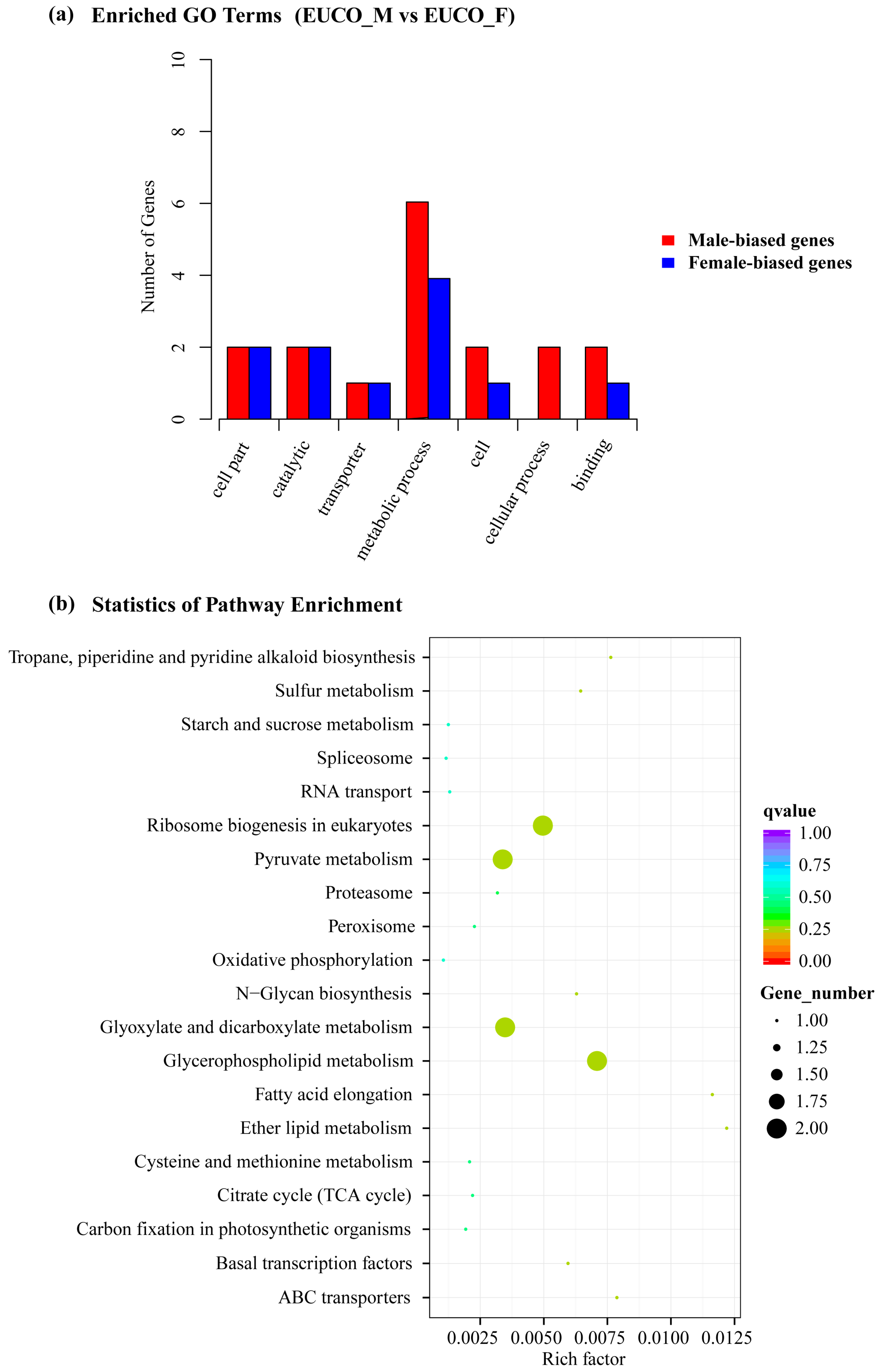
2.3. Transcriptomes Comparison between the Males and the Females
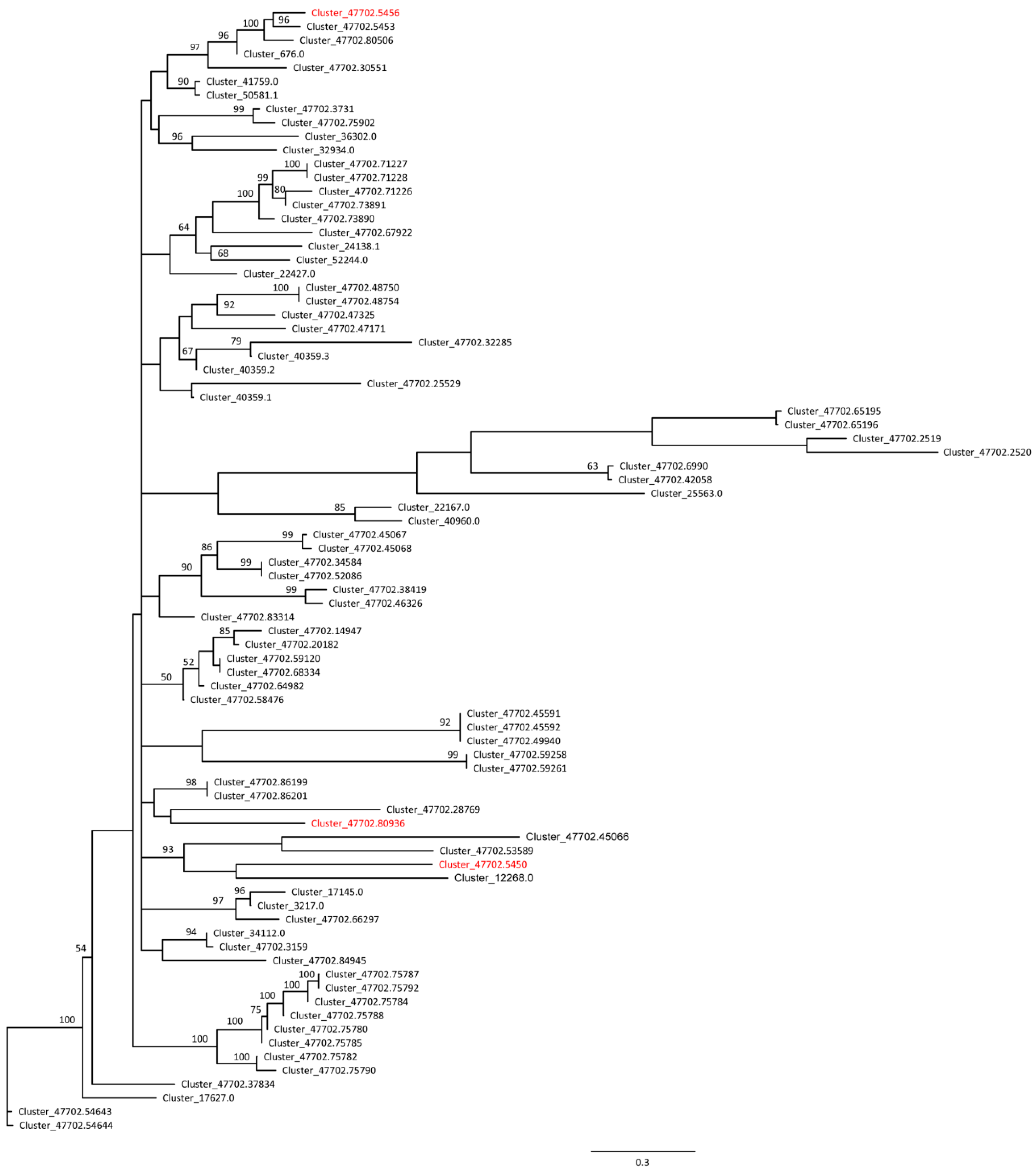
2.4. Differential Expression of MADS Box Genes in the Males and the Females
2.5. SNPs Detection in the DEGs and the Whole Transcriptomes
3. Discussion
3.1. Genes Related to Sexual Dimorphism of Gutta Content in the Leaves of E. ulmoides
3.2. A MADS Box TF Gene Involved in Sex Differentiation of E. ulmoides
3.3. Segregated SNPs in Sex-Associated Genes in E. ulmoides
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Isolation and Illumina Sequencing
4.3. Transcriptome Assembly and Gene Annotation
4.4. Gene Differential Expression Analysis
4.5. MADS Box Genes Analysis
4.6. SNPs Calling
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Byng, J.W.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Judd, W.S.; Mabberley, D.J.; Sennikov, A.N.; Soltis, D.E.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 105–121. [Google Scholar]
- Zhang, Z.Y.; Zhang, H.D.; Turland, N.J. Eucommiaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden: St. Louis, MI, USA, 2003; p. 43. [Google Scholar]
- Li, F.D.; Du, H.Y. Eucommia Ulmoides; China Press of Traditional Chinese Medicine: Beijing, China, 2001. [Google Scholar]
- Chen, R.; Harada, Y.; Bamba, T.; Nakazawa, Y.; Gyokusen, K. Overexpression of an isopentenyl diphosphate isomerase gene to enhance trans-polyisoprene production in Eucommia ulmoides Oliver. BMC Biotechnol. 2012, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Uefuji, H.; Nishikawa, T.; Mukai, Y.; Yamashita, A.; Hattori, M.; Ogasawara, N.; Bamba, T.; Fukusaki, E.I.; Kobayashi, A. Construction and analysis of EST libraries of the trans-polyisoprene producing plant, Eucommia ulmoides Oliver. Planta 2012, 236, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, H.; Wuyun, T.N. Genome-wide identification of microRNAs and their targets in the leaves and fruits of Eucommia ulmoides using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Bamba, T.; Takeda, T.; Uefuji, H.; Harada, Y.; Li, X.; Chen, R.; Inoue, S.; Tutumi, M.; Shimizu, T. Production of Eucommia-rubber from Eucommia ulmoides Oliv. (hardy rubber tree). Plant Tissue Cult. Lett. 2009, 26, 71–79. [Google Scholar] [CrossRef]
- Du, H.Y.; Hu, W.Z.; Yu, R. The Report on Development of China’s Eucommia Rubber Resources and Industry (2014–2015); Social Sciences Academic Press: Beijing, China, 2015. [Google Scholar]
- Liu, H.; Hongyan, D.U.; Tana, W. Advances in research on biotechnology breeding of Eucommia ulmoides. Hunan For. Sci. Technol. 2016, 43, 132–136. [Google Scholar]
- Wang, B.W.; Wang, Y.Q.; Mo, H.; Luo, L.X.; Li, M.X.; Cui, K.M. Comparison of cytology, apical buds and gutta content between staminate and pistillate of Eucommia ulmoides trees. Acta Bot. Sin. 1999, 41, 11–15. [Google Scholar] [CrossRef]
- Renner, S.S. The relative and absolute frequencies of angiosperm sexual systems: Dioecy, monoecy, gynodioecy, and an updated online database. Am. J. Bot. 2014, 101, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Ming, R.; Bendahmane, A.; Renner, S.S. Sex chromosomes in land plants. Ann. Rev. Plant Biol. 2011, 62, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.; Glick, L.; Abadi, S.; Einhorn, M.; Kopelman, N.M.; Salman-Minkov, A.; Mayzel, J.; Chay, O.; Mayrose, I. The Chromosome Counts Database (CCDB): A community resource of plant chromosome numbers. New Phytol. 2015, 206, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.D.; Leitch, I.J. Angiosperm DNA C-Values Database (Release 8.0, December 2012). Available online: http://www.kew.org/cvalues/ (accessed on 15 April 2017).
- Zhang, Y.D.; He, L.X.; Li, H.; Cheng, J. Karyotype analysis of Eucommia ulmoides. J. Gansu For. Sci. Technol. 2008, 33, 1–4. [Google Scholar]
- Du, H.Y. China Eucommia Pictorial; China Forestry Publishing House: Beijing, China, 2014. [Google Scholar]
- Charlesworth, B.; Charlesworth, D. A model for the evolution of dioecy and gynodioecy. Am. Nat. 1978, 112, 975–997. [Google Scholar] [CrossRef]
- Bergero, R.; Charlesworth, D. The evolution of restricted recombination in sex chromosomes. Trends Ecol. Evol. 2009, 24, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Boualem, A.; Bendahmane, A.; Ming, R. Genomics of sex determination. Curr. Opin. Plant Biol. 2014, 18, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Leebensmack, J. A century of sex determination in flowering plants. J. Hered. 2017, 108, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Qin, Z.W.; Zhou, X.Y.; Zhuo, F.; Du, Y.L. Transcriptome profile analysis of floral sex determination in cucumber. J. Plant Physiol. 2010, 167, 905–913. [Google Scholar]
- Bergero, R.; Charlesworth, D. Preservation of the Y transcriptome in a 10-million-year-old plant sex chromosome system. Curr. Biol. 2011, 21, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Hough, J.; Hollister, J.D.; Wang, W.; Barrett, S.C.H.; Wright, S.I. Genetic degeneration of old and young Y chromosomes in the flowering plant Rumex hastatulus. Proc. Natl. Acad. Sci. USA 2014, 111, 7713–7718. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Mercati, F.; Shan, H.Y.; Sunseri, F.; Falavigna, A.; Leebens-Mack, J. Sex-biased gene expression in dioecious garden asparagus (Asparagus officinalis). New Phytol. 2015, 207, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Darwin, C. The Different Forms of Flowers on Plants of the Same Species; John Murray: London, UK, 1877. [Google Scholar]
- Diggle, P.K.; Di, S.V.; Gschwend, A.R.; Golenberg, E.M.; Moore, R.C.; Russell, J.R.; Sinclair, J.P. Multiple developmental processes underlie sex differentiation in angiosperms. Trends Genet. 2011, 27, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H. The Initiation and Development of Floral Organs in Eucommia ulmoides (Eucommiaceae) with Systematic Significance. Master Dissertation, Peking University, Beijing, China, 2010. [Google Scholar]
- Mitchell, C.; Diggle, P. The evolution of unisexual flowers: Morphological and functional convergence results from diverse developmental transitions. Am. J. Bot. 2005, 92, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Liston, A. A new interpretation of floral morphology in Garrya (Garryaceae). Taxon 2003, 52, 271–276. [Google Scholar] [CrossRef]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. The ABC model of flower development: Then and now. Development 2012, 139, 4095–4098. [Google Scholar] [CrossRef] [PubMed]
- Zik, M.; Irish, V.F. Global identification of target genes regulated by APETALA3 and PISTILLATA floral homeotic gene action. Plant Cell 2003, 15, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D. Plant contributions to our understanding of sex chromosome evolution. New Phytol. 2015, 208, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Zahn, L.; Guindon, S.; Wall, P.K.; Kong, H.; Ma, H.; Depamphilis, C.W.; Leebens-Mack, J. Evolution of plant MADS box transcription factors: Evidence for shifts in selection associated with early angiosperm diversification and concerted gene duplications. Mol. Biol. Evol. 2009, 26, 2229–2244. [Google Scholar] [CrossRef] [PubMed]
- Pfent, C.; Pobursky, K.J.; Sather, D.N.; Golenberg, E.M. Characterization of SpAPETALA3 and SpPISTILLATA, B class floral identity genes in Spinacia oleracea, and their relationship to sexual dimorphism. Dev. Genes Evol. 2005, 215, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Distilio, S.V.; Kramer, E.M.; Baum, D.A. Floral MADS box genes and homeotic gender dimorphism in Thalictrum dioicum (Ranunculaceae)-a new model for the study of dioecy. Plant J. Cell Mol. Biol. 2005, 41, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Zluvova, J.; Zak, J.; Janousek, B.; Vyskot, B. Dioecious Silene latifolia plants show sexual dimorphism in the vegetative stage. BMC Plant Biol. 2010, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Zemp, N.; Tavares, R.; Widmer, A. Fungal infection induces sex-specific transcriptional changes and alters sexual dimorphism in the dioecious plant Silene latifolia. PLoS Genet. 2015, 11, e1005536. [Google Scholar] [CrossRef] [PubMed]
- Mantello, C.C.; Cardososilva, C.B.; Da, S.C.; de Souza, L.M.; Scaloppi Junior, E.J.; De, S.G.P.; Vicentini, R.; de Souza, A.P. De Novo assembly and transcriptome analysis of the rubber tree (Hevea brasiliensis) and SNP markers development for rubber biosynthesis pathways. PLoS ONE 2014, 9, e102665. [Google Scholar] [CrossRef] [PubMed]
- Pulido, P.; Perello, C.; Rodriguez-Concepcion, M. New insights into plant isoprenoid metabolism. Mol. Plant 2012, 5, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H.; Parsch, J. The evolution of sex-biased genes and sex-biased gene expression. Nat. Rev. Genet. 2007, 8, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E. Sex chromosomes and the evolution of sexual dimorphism: Lessons from the genome. Am. Nat. 2009, 173, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Zemp, N.; Tavares, R.; Muyle, A.; Charlesworth, D.; Marais, G.A.; Widmer, A. Evolution of sex-biased gene expression in a dioecious plant. Nat. Plants 2016, 2, 16168. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.S.; Wan, K.L.; Isa, M.N.M.; Bahari, A.; Tan, S.H.; Harikrishna, K.; Yeang, H.Y. Insights into rubber biosynthesis from transcriptome analysis of Hevea brasiliensis latex. J. Exp. Bot. 2009, 58, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Guo, D.; Zhu, J.H.; Wang, Y.; Chen, X.T.; Peng, S.Q. Comparative transcriptome analysis of latex reveals molecular mechanisms underlying increased rubber yield in Hevea brasiliensis self-rooting juvenile clones. Front. Plant Sci. 2016, 7, 1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.K. RNA-directed DNA methylation. Curr. Opin. Plant Biol. 2011, 14, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Lorković, Z.J.; Liang, S.C.; Matzke, A.J.M.; Matzke, M. The ability to form homodimers is essential for RDM1 to function in RNA-directed DNA methylation. PLoS ONE 2014, 9, e88190. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hatlen, A.; Kelly, L.J.; Becher, H.; Wang, W.; Kovarik, A.; Leitch, I.J.; Leitch, A.R. Angiosperms are unique among land plant lineages in the occurrence of key genes in the RNA-directed DNA methylation (RdDM) pathway. Genome Biol. Evol. 2015, 7, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.F. Angiosperm Phylogeny Website, Version 12, July 2012 (and more or less continuously updated since). Available online: http://www.mobot.org/MOBOT/research/APweb/ (accessed on 15 April 2017).
- Smith, S.A.; Beaulieu, J.M.; Donoghue, M.J. An uncorrelated relaxed-clock analysis suggests an earlier origin for flowering plants. Proc. Natl. Acad. Sci. USA 2010, 107, 5897–5902. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Michel, P.; Catherine, D.; Abdelhafid, B. A transposon-induced epigenetic change leads to sex determination in melon. Nature 2009, 461, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Henry, I.M.; Tao, R.; Comai, L. A Y-chromosome-encoded small RNA acts as a sex determinant in persimmons. Science 2014, 346, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.M.; Dorit, R.L.; Irish, V.F. Molecular evolution of genes controlling petal and stamen development: Duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 1998, 149, 765–783. [Google Scholar] [PubMed]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves intofloral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Par̆Enicová, L.; Folter, S.D.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.K.; Sun, F.C.; Gao, W.J.; Deng, C.L.; Lu, L.D. Relationship between MADS-box genes and sex determination and differentiation of dioecious plants. J. Anhui Agric. Sci. 2008, 36, 11653–11655. [Google Scholar]
- Tang, P.; Zhang, Q.; Yao, X. Comparative transcript profiling explores differentially expressed genes associated with sexual phenotype in kiwifruit. PLoS ONE 2017, 12, e0180542. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yin, T.; Ye, N.; Chen, Y.; Yin, T.; Liu, M.; Hassani, D. Transcriptome analysis of the differentially expressed genes in the male and female shrub willows (Salix suchowensis). PLoS ONE 2013, 8, e60181. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Zhang, G.J.; Zhang, X.J.; Yuan, J.H.; Deng, C.L.; Gao, W.J. Comparative transcriptome analysis reveals differentially expressed genes associated with sex expression in garden asparagus (Asparagus officinalis). BMC Plant Biol. 2017, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.G. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar]
- Zeng, L.; Zhang, Q.; Sun, R.; Kong, H.; Zhang, N.; Ma, H.; Zeng, L.; Zhang, Q.; Sun, R.; Kong, H. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 2014, 5, 4956. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.M.; Holappa, L.; Gould, B.; Jaramillo, M.A.; Setnikov, D.; Santiago, P.M. Elaboration of B gene function to include the identity of novel floral organs in the lower eudicot Aquilegia. Plant Cell 2007, 19, 750–766. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Mank, J.E. The birds and the bees and the flowers and the trees: Lessons from genetic mapping of sex determination in plants and animals. Genetics 2010, 186, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Heikrujam, M.; Sharma, K.; Prasad, M.; Agrawal, V. Review on different mechanisms of sex determination and sex-linked molecular markers in dioecious crops: A current update. Euphytica 2015, 201, 161–194. [Google Scholar] [CrossRef]
- Qiu, S.; Bergero, R.; Guirao-Rico, S.; Campos, J.L.; Cezard, T.; Gharbi, K.; Charlesworth, D. RAD mapping reveals an evolving, polymorphic and fuzzy boundary of a plant pseudoautosomal region. Mol. Ecol. 2016, 25, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, A.; Hefer, C.A.; Capron, A.; Kolosova, N.; Martinez-Nuñez, F.; Soolanayakanahally, R.Y.; Stanton, B.; Guy, R.D.; Mansfield, S.D.; Douglas, C.J. Recent Y chromosome divergence despite ancient origin of dioecy in poplars (Populus). Mol. Ecol. 2015, 24, 3243–3256. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Hou, S.; Feltus, F.A.; Jones, M.R.; Murray, J.E.; Veatch, O.; Lemke, C.; Saw, J.H.; Moore, R.C.; Thimmapuram, J.; et al. Low X/Y divergence in four pairs of papaya sex-linked genes. Plant J. 2008, 53, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Pucholt, P.; Wright, A.E.; Liu, C.L.; Mank, J.E.; Berlin, S. Recent sex chromosome divergence despite ancient dioecy in the willow Salix viminalis. Mol. Biol. Evol. 2017, 34, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Tennessen, J.A.; Govindarajulu, R.; Liston, A.; Ashman, T.L. Homomorphic ZW chromosomes in a wild strawberry show distinctive recombination heterogeneity but a small sex-determining region. New Phytol. 2016, 211, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.R.; Pannell, J.R. Sex determination in dioecious Mercurialis annua and its close diploid and polyploid relatives. Heredity 2015, 114, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Parsch, J. Sex-biased gene expression. Ann. Rev. Genet. 2016, 50, 29–44. [Google Scholar]
- Liu, H.; Fu, J.; Du, H.; Hu, J.; Wuyun, T. De novo sequencing of Eucommia ulmoides flower bud transcriptomes for identification of genes related to floral development. Genom. Data 2016, 9, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xing, C.; Tian, H.; Yao, X. Microsatellite genetic variation in the Chinese endemic Eucommia ulmoides (Eucommiaceae): Implications for conservation. Bot. J. Linn. Soc. 2013, 173, 775–785. [Google Scholar] [CrossRef][Green Version]
- Salgado, L.R.; Koop, D.M.; Pinheiro, D.G.; Rivallan, R.; Guen, V.L.; Nicolás, M.F.; Almeida, L.G.P.D.; Rocha, V.R.; Magalhães, M.; Gerber, A.L. De novo transcriptome analysis of Hevea brasiliensis tissues by RNA-seq and screening for molecular markers. BMC Genom. 2014, 15, 236. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, Y.; Peng, L.; Ru, M.; Liang, Z.S. Genetic diversity and population structure of Eucommia ulmoides Oliver, an endangered medicinal plant in China. Genet. Mol. Res. 2015, 14, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, D.; Li, Z.; Wei, J.; Jin, C.; Liu, M. A molecular genetic linkage map of Eucommia ulmoides and quantitative trait loci (QTL) analysis for growth traits. Int. J. Mol. Sci. 2014, 15, 2053–2074. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Y.; Li, L.; Wei, Y.; Li, Z. The first genetic linkage map of Eucommia ulmoides. J. Genet. 2014, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology, Heidelberg, Germany, 1 January 1999; pp. 138–148. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Forgy, E.W. Cluster analysis of multivariate data: Efficiency versus interpretability of classifications. Biometrics 1965, 21, 41–52. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Mao, T. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Tao, C.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Chen, J.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Gill, G.P.; Harvey, C.F.; Nihal, D.S.H.; Datson, P.M.; Tsang, G.K.; Fraser, L.G.; Crowhurst, R.N.; Mcneilage, M.A. A gene-rich linkage map in the dioecious species Actinidia chinensis (kiwifruit) reveals putative X/Y sex-determining chromosomes. BMC Genom. 2009, 10, 102. [Google Scholar]
- Oyama, R.K.; Volz, S.M.; Renner, S.S. A sex-linked SCAR marker in Bryonia dioica (Cucurbitaceae), a dioecious species with XY sex-determination and homomorphic sex chromosomes. J. Evol. Biol. 2009, 22, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Zinta, G.; Rana, S.; Shirko, P. Molecular identification of sex in Hippophae rhamnoides L. using isozyme and RAPD markers. For. Ecosyst. 2010, 12, 62–66. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Transplant. Proc. 2009, 19, 1653–1654. [Google Scholar]
- Ga, V.D.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del, A.G.; Levymoonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11–33. [Google Scholar]
Sample Availability: All samples are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample/Item * | Raw Reads | Clean Reads | Size of Clean Data (G) | Error (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|
| EUCO_M1 | 52,628,596 | 50,575,362 | 7.59 | 0.02 | 95.55 | 89.51 | 45.18 |
| EUCO_M2 | 55,644,868 | 53,387,028 | 8.01 | 0.02 | 95.44 | 89.63 | 40.91 |
| EUCO_M3 | 60,088,910 | 57,307,276 | 8.60 | 0.02 | 95.04 | 89.45 | 40.81 |
| EUCO_F1 | 52,436,122 | 49,456,850 | 7.42 | 0.02 | 95.49 | 89.50 | 43.20 |
| EUCO_F2 | 53,027,850 | 50,069,058 | 7.51 | 0.02 | 95.47 | 89.59 | 41.10 |
| EUCO_F3 | 53,303,500 | 50,531,298 | 7.58 | 0.02 | 95.46 | 89.75 | 40.56 |
| Total | 327,129,846 | 311,326,872 | 46.71 | / | / | / | / |
| Item | Values |
|---|---|
| Number of contigs | 289,704 |
| Minimum length of contigs (bp) | 201 |
| Mean length of contigs (bp) | 540 |
| Max length of contigs (bp) | 12,536 |
| N50 of contigs (bp) | 705 |
| Total length of contigs (bp) | 156,318,751 |
| Total number of unigenes | 148,595 |
| Average sequence size of unigenes (bp) | 801 |
| Length of all unigenes (bp) | 117,298,413 |
| N50 of unigenes (bp) | 1064 |
| Gene_ID | Position * | EUCO_F1 * | EUCO_F2 | EUCO_F3 | EUCO_M1 * | EUCO_M2 | EUCO_M3 | Regulation |
|---|---|---|---|---|---|---|---|---|
| Cluster-47702.80936 | 1101 | C | C | C | G | G | G | Male-biased expression |
| Cluster-47702.80197 | 360 | T | T | T | C | C | C | Male-biased expression |
| 460 | G | G | G | A | A | A | ||
| Cluster-47702.38156 | 538 | T | T | T | C | C | C | Male-biased expression |
| 596 | G | G | G | T | T | T | ||
| Cluster-47702.79497 | 297 | A | A | A | G | G | G | Male-biased expression |
| 312 | C | C | C | T | T | T | ||
| Cluster-47702.45188 | 947 | T | T | T | G | G | G | Male-biased expression |
| 968 | T | T | T | C | C | C |
| Type | Count | Occurrence per kb |
|---|---|---|
| Transition | ||
| C/T | 38,758 | 0.33 |
| A/G | 39,805 | 0.34 |
| Transversion | ||
| A/T | 11,838 | 0.10 |
| A/C | 10,256 | 0.09 |
| T/G | 10,298 | 0.09 |
| C/G | 8456 | 0.07 |
| Total | 119,411 | 1.02 |
| SNP position in codon | ||
| First | 22,235 | |
| Second | 8889 | |
| Third | 22,148 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Zhang, X. Identification of the Sex-Biased Gene Expression and Putative Sex-Associated Genes in Eucommia ulmoides Oliver Using Comparative Transcriptome Analyses. Molecules 2017, 22, 2255. https://doi.org/10.3390/molecules22122255
Wang W, Zhang X. Identification of the Sex-Biased Gene Expression and Putative Sex-Associated Genes in Eucommia ulmoides Oliver Using Comparative Transcriptome Analyses. Molecules. 2017; 22(12):2255. https://doi.org/10.3390/molecules22122255
Chicago/Turabian StyleWang, Wencai, and Xianzhi Zhang. 2017. "Identification of the Sex-Biased Gene Expression and Putative Sex-Associated Genes in Eucommia ulmoides Oliver Using Comparative Transcriptome Analyses" Molecules 22, no. 12: 2255. https://doi.org/10.3390/molecules22122255
APA StyleWang, W., & Zhang, X. (2017). Identification of the Sex-Biased Gene Expression and Putative Sex-Associated Genes in Eucommia ulmoides Oliver Using Comparative Transcriptome Analyses. Molecules, 22(12), 2255. https://doi.org/10.3390/molecules22122255