Acid-Induced Rearrangement of Epoxygermacranolides: Synthesis of Furanoheliangolides and Cadinanes from Nobilin


 ,
, 
Abstract
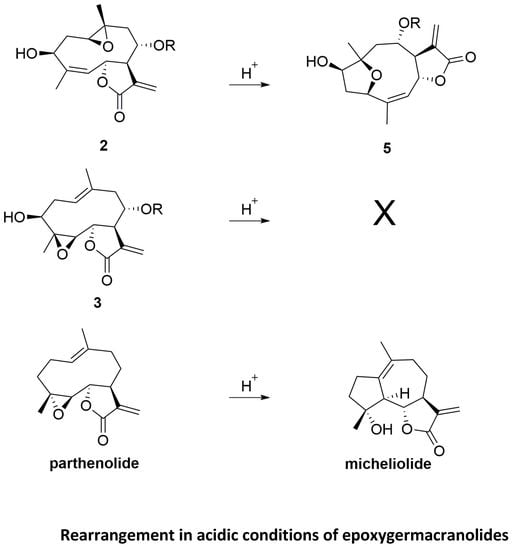
1. Introduction
2. Results and Discussion
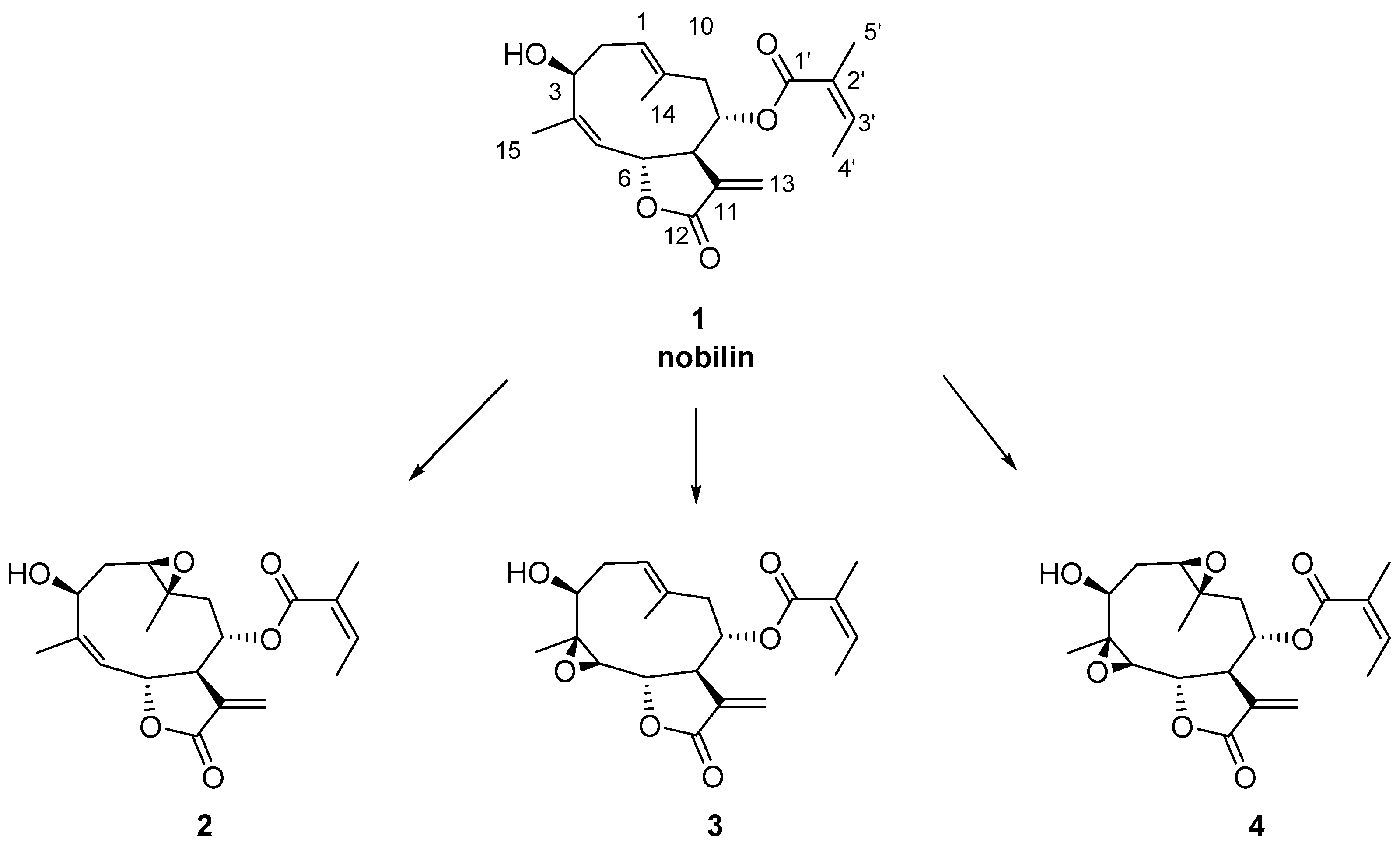
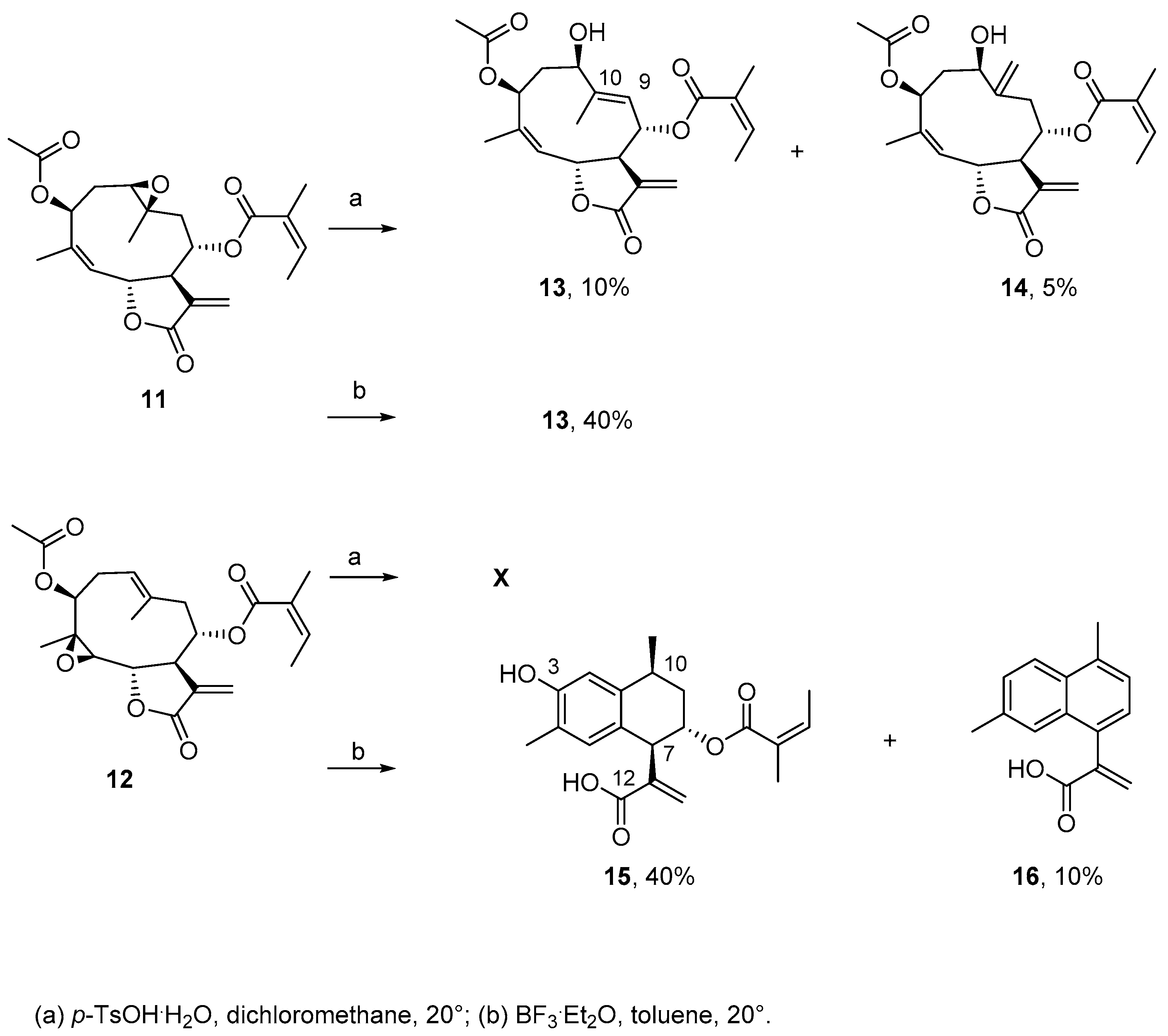
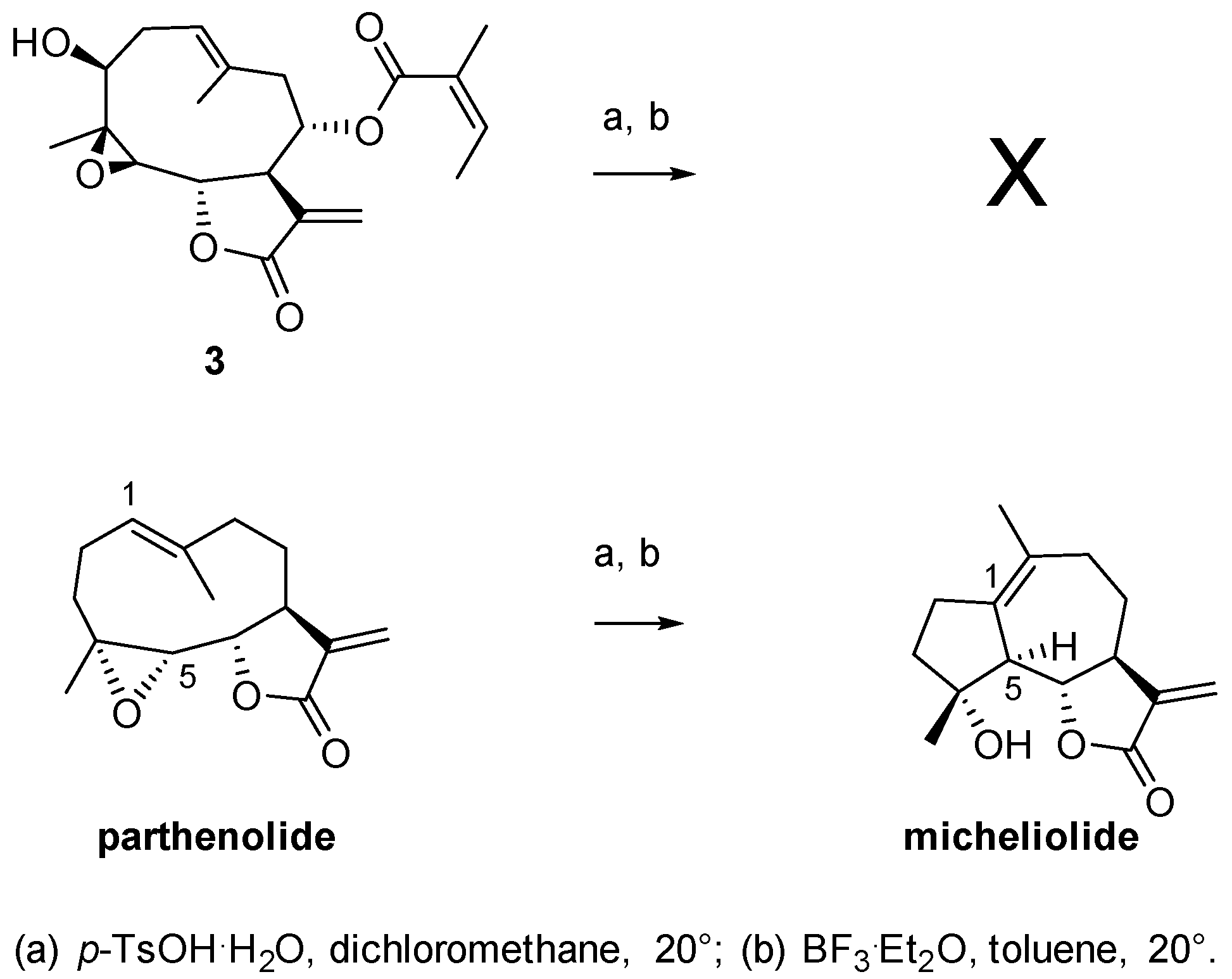
2.1. Chemical Studies
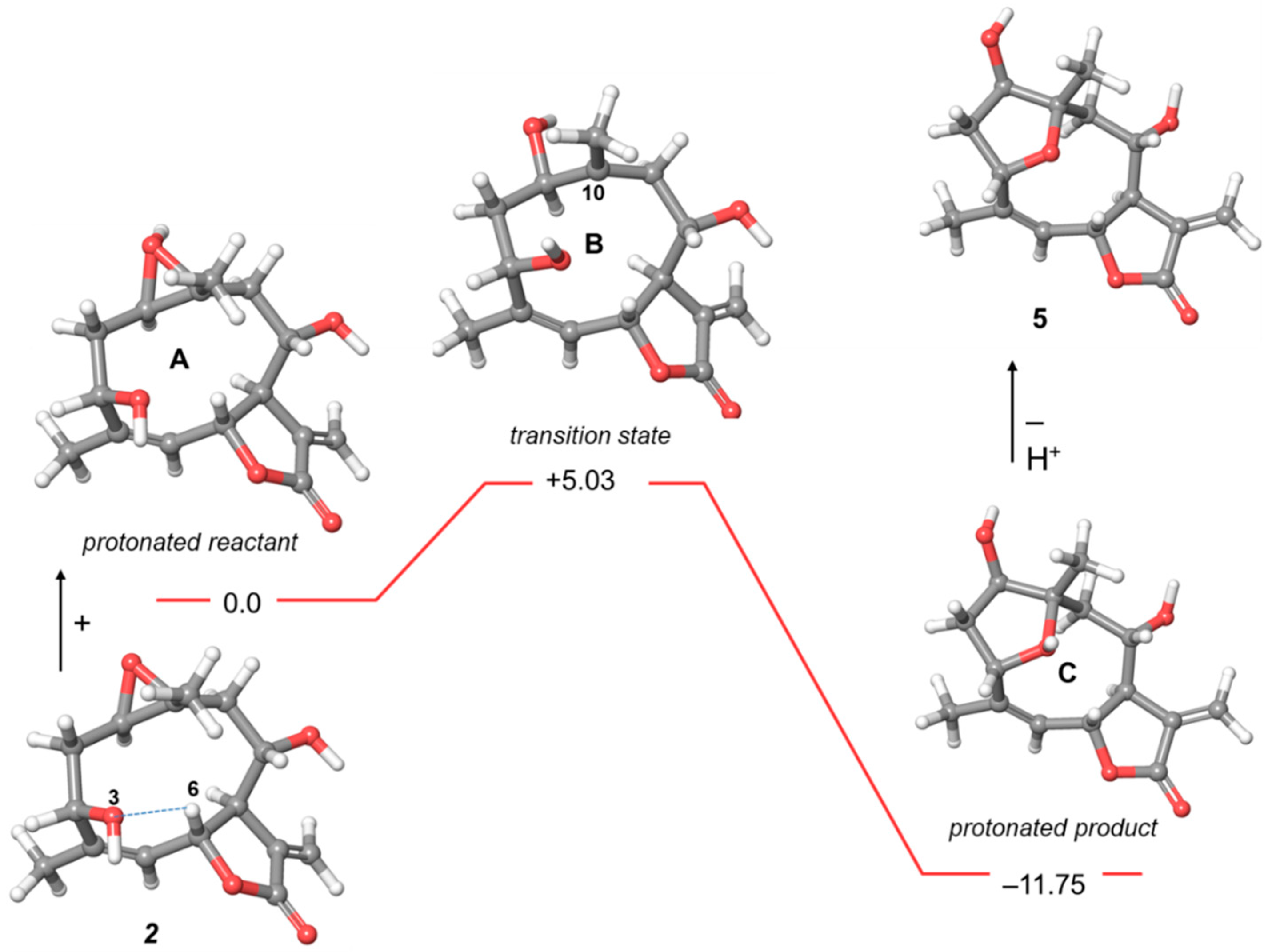
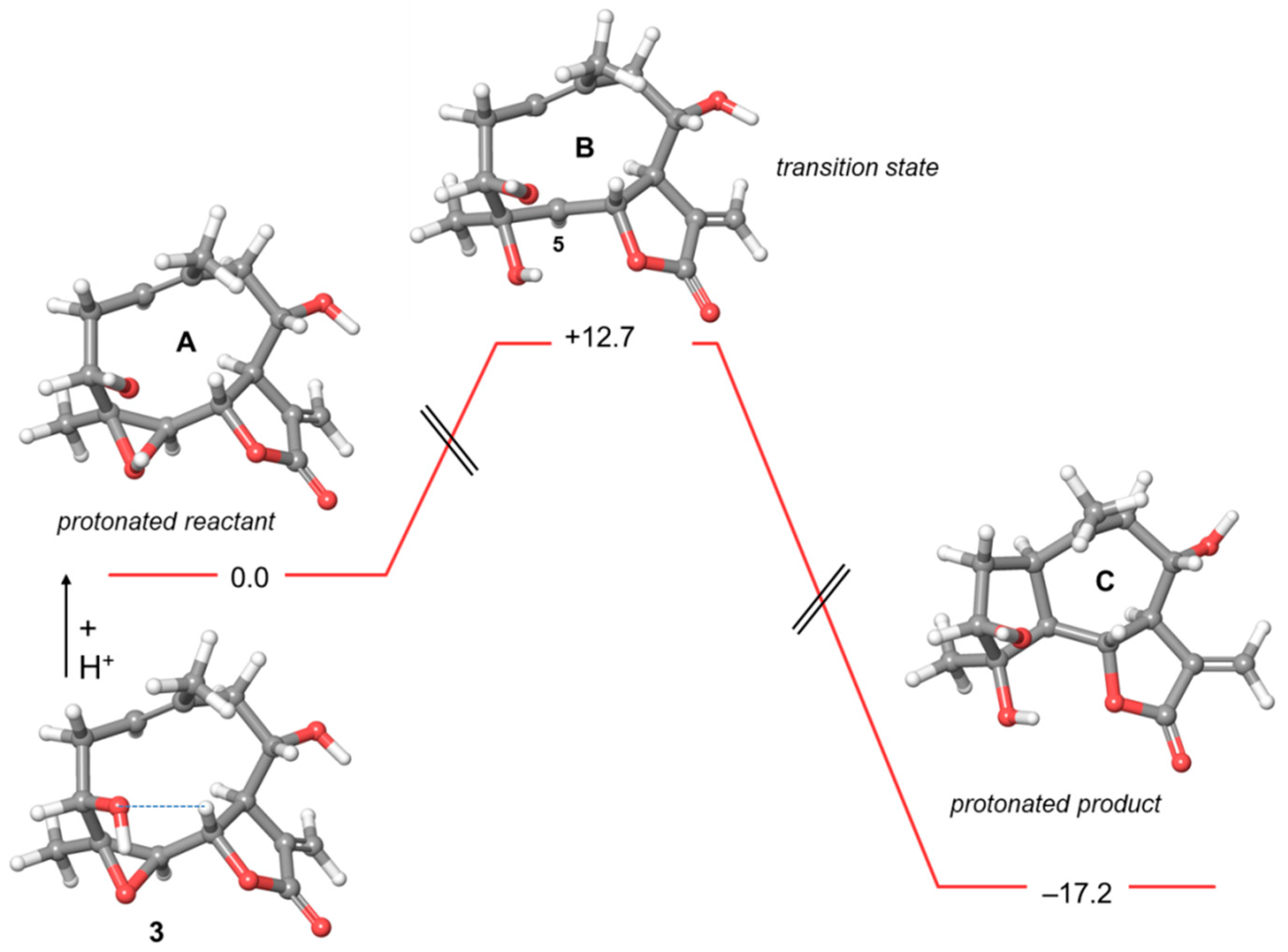
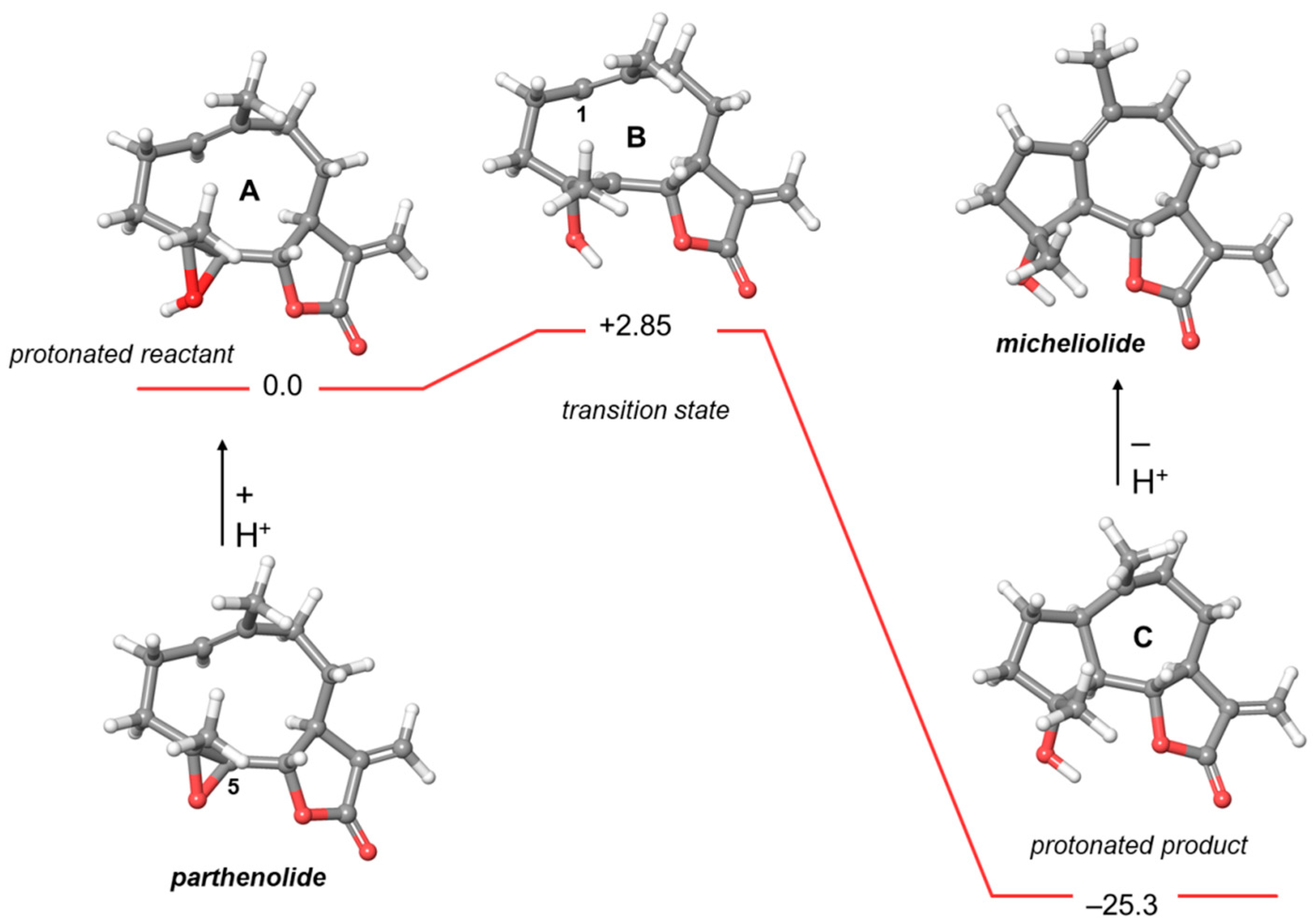
2.2. Computational Studies
2.3. In Vitro Anti-Protozoal Activity
3. Experimental Section
3.1. General Information
3.2. In Vitro Biological Testing
3.3. Isolation of Nobilin (1)
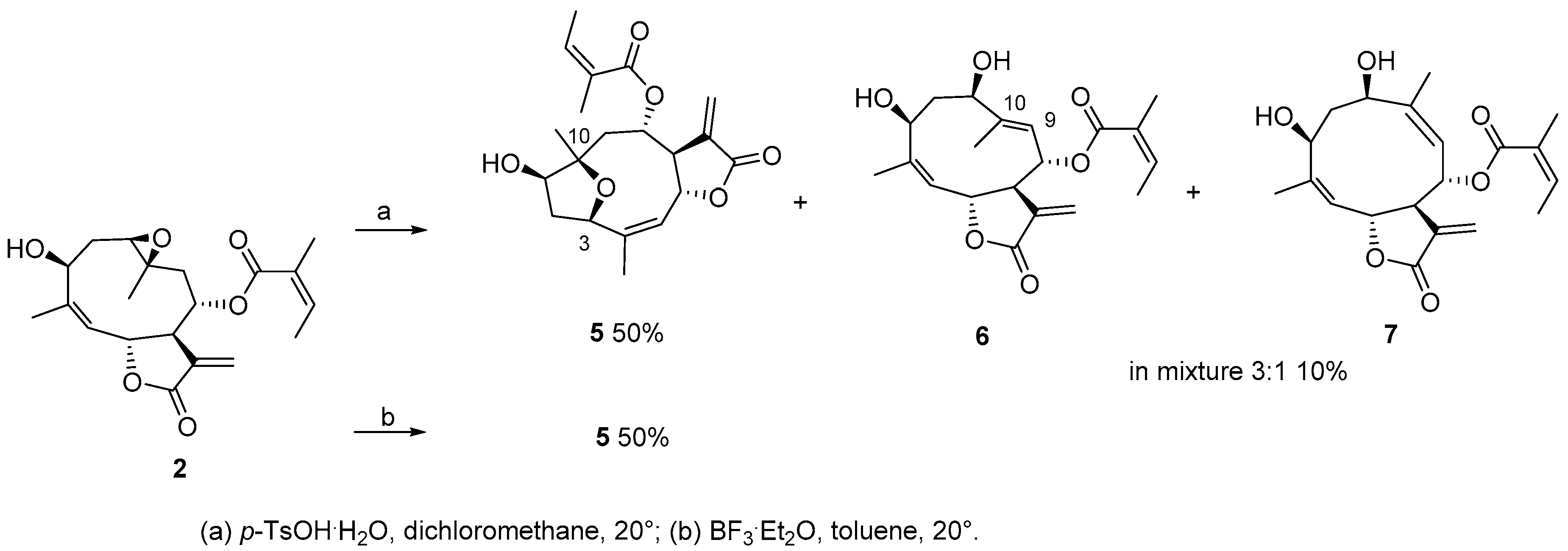
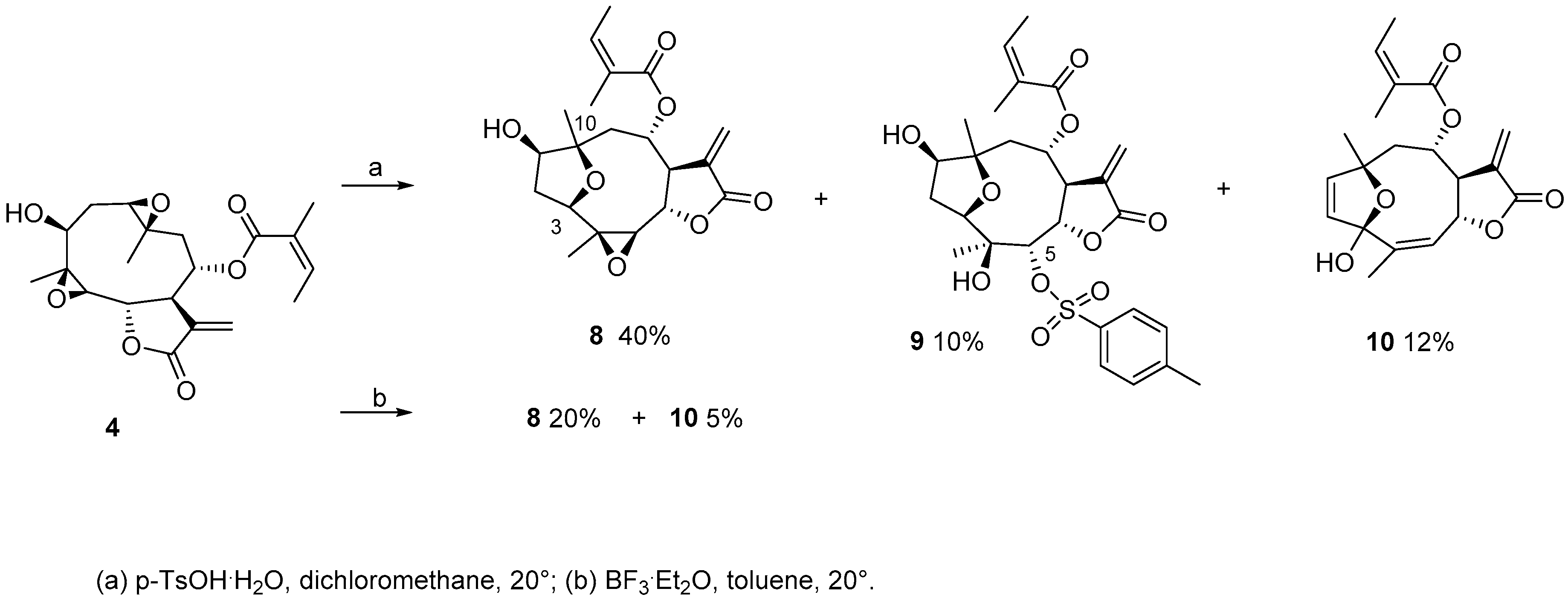
3.4. Synthesis of Compounds 2 and 4
3.5. Synthesis of Compound 3
3.6. General Procedure for Degradation with p-TsOH·H2O
3.7. General Procedure for Degradation with BF3·Et2O
3.8. General Procedure for Synthesis of Compounds 11 and 12
3.9. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids lactones: Benefits to plants and people. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef] [PubMed]
- Adio, A.M. Germacrenes a–e and related compounds: Thermal, photochemical and acid induced transannular cyclizations. Tetrahedron 2009, 65, 1533–1552. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; López-Pérez, J.-L.; Arteaga, J.F.; Catalán, J. New pathways in transannular cyclization of germacrone [germacra-1(10),4,7(11)-trien-8-one]: Evidence regarding a concerted mechanism. Org. Lett. 2009, 11, 4782–4785. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.; Molinillo, J.M.G.; Macías, F.A. Trends in the synthesis and functionalization of guaianolides. Eur. J. Org. Chem. 2015, 2093–2110. [Google Scholar] [CrossRef]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Pérez Morales, M.C.; Catalán, J.V.; Domingo, V.; Jaraíz, M.; Herrador, M.M.; Quílez del Moral, J.F.; López-Pérez, J.-L.; Barrero, A.F. Structural diversity from the transannular cyclizations of natural germacrone and epoxy derivatives: A theoretical–experimental study. Chem. Eur. J. 2013, 19, 6598–6612. [Google Scholar] [CrossRef] [PubMed]
- Thormann, U.; De Mieri, M.; Neuburger, M.; Verjee, S.; Altmann, P.; Hamburger, M.; Imanidis, G. Mechanism of chemical degradation and determination of solubility by kinetic modeling of the highly unstable sesquiterpene lactone nobilin in different media. J. Pharm. Sci. 2014, 103, 3139–3152. [Google Scholar] [CrossRef] [PubMed]
- De Mieri, M.; Kaiser, M.; Brun, R.; Thormann, U.; Imanidis, G.; Hamburger, M. Anti-trypanosomal cadinanes synthesized by transannular cyclization of the natural sesquiterpene lactone nobilin. Bioorg. Med. Chem. 2015, 23, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, S.; Maggio, A.; Raccuglia, R.A.; Bruno, M. Acid-induced rearrangement of epoxygermacra-8,12-olides: Synthesis and absolute configuration of guaiane and eudesmane derivatives from artemisiifolin. Eur. J. Org. Chem. 2010, 3093–3101. [Google Scholar] [CrossRef]
- Castañeda-Acosta, J.; Fischer, N.H.; Vargas, D. Biomimetic transformations of parthenolide. J. Nat. Prod. 1993, 56, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.-D.; Li, D.; Long, J.; Zhang, H.-L.; Lin, J.-P.; Qiu, C.-J.; Zhang, Q.; Chen, Y. Biomimetic semisynthesis of arglabin from parthenolide. J. Org. Chem. 2012, 77, 7103–7107. [Google Scholar] [CrossRef] [PubMed]
- Neukirch, H.; Guerriero, A.; D’Ambrosio, M. Transannular cyclization in cyclodecenes: The case study of melampolides. Eur. J. Org. Chem. 2003, 2003, 3969–3975. [Google Scholar] [CrossRef]
- Long, J.; Ding, Y.-H.; Wang, P.-P.; Zhang, Q.; Chen, Y. Protection-group-free semisyntheses of parthenolide and its cyclopropyl analogue. J. Org. Chem. 2013, 78, 10512–10518. [Google Scholar] [CrossRef] [PubMed]
- Holub, M.; Samek, Z. Isolation and structure of 3-epinobilin, 1,10-epoxynobilin and 3-dehydronobilin—Other sesquiterpenic lactones from the flowers of Anthemis nobilis l. Revision of the structure of nobilin and eucannabinolide. Collect. Czechoslov. Chem. Commun. 1977, 42, 1053–1064. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- De Kraker, J.-W.; Franssen, M.C.R.; de Groot, A.; Shibata, T.; Bouwmeester, H.J. Germacrenes from fresh costus roots. Phytochemistry 2001, 58, 481–487. [Google Scholar] [CrossRef]
- De Mieri, M.; Monteleone, G.; Ismajili, I.; Kaiser, M.; Hamburger, M. Antiprotozoal activity-based profiling of a dichloromethane extract from Anthemis nobilis flowers. J. Nat. Prod. 2017, 80, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Romero, A.; Gómez, A.B.; Martín-Matute, B. Acid- and iridium-catalyzed tandem 1,3-transposition/3,1-hydrogen shift/chlorination of allylic alcohols. ACS Catal. 2015, 5, 708–714. [Google Scholar] [CrossRef]
- An Zon, A.; Huis, R. Acid-catalysed isomerisation of epoxides to allylic alcohols. Recl. Trav. Chim. Pays-Bas 1981, 100, 425–429. [Google Scholar] [CrossRef]
- Macı́as, F.A.; Oliva, R.M.; Varela, R.M.; Torres, A.; Molinillo, J.M.G. Allelochemicals from sunflower leaves cv. Peredovick. Phytochemistry 1999, 52, 613–621. [Google Scholar] [CrossRef]
- Hong, Y.J.; Tantillo, D.J. Consequences of conformational preorganization in sesquiterpene biosynthesis: Theoretical studies on the formation of the bisabolene, curcumene, acoradiene, zizaene, cedrene, duprezianene, and sesquithuriferol sesquiterpenes. J. Am. Chem. Soc. 2009, 131, 7999–8015. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Maggio, A.M.; Barone, G.; Bruno, M.; Duca, D.; Rosselli, S. Conformational analysis and dft calculations of 8α-hydroxy-germacradiene-6,12-olide derivatives. J. Phys. Org. Chem. 2005, 18, 1116–1122. [Google Scholar] [CrossRef]
- Da Silva, C.F.; Batista, D.D.G.J.; De Araújo, J.S.; Batista, M.M.; Lionel, J.; de Souza, E.M.; Hammer, E.R.; da Silva, P.B.; De Mieri, M.; Adams, M.; et al. Activities of psilostachyin a and cynaropicrin against trypanosoma cruzi in vitro and in vivo. Antimicrob. Agents Chemother. 2013, 57, 5307–5314. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Fouché, G.; De Mieri, M.; Yoshimoto, Y.; Usuki, T.; Nthambeleni, R.; Parkinson, C.; van der Westhuyzen, C.; Kaiser, M.; Hamburger, M.; et al. Structure-activity relationship study of sesquiterpene lactones and their semi-synthetic amino derivatives as potential antitrypanosomal products. Molecules 2014, 19, 3523–3538. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.; Şener, B.; Kaiser, M.; Brun, R.; Tasdemir, D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs 2010, 8, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- De Pascual, J.T.; Gonzales, M.S.; Caballero, M.C.; Parra, T.; Bellido, I.S. Transannular cyclization of heliangolides. Tetrahedron Lett. 1987, 7, 821–824. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1 and 2 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 5 δH (mult, J in Hz) | 6 δH (mult, J in Hz) | 7 δH (mult, J in Hz) | 8 δH (mult, J in Hz) | 9 b δH (mult, J in Hz) | 10 δH (mult, J in Hz) |
|---|---|---|---|---|---|---|
| 1 | 3.85 (br d, 4.6) | 4.05 (dd, 10.6, 6.5) | 4.96 (dd, 10.2, 3.0) | 3.90 (d, 4.3) | 3.96 (dd, 5.2, 1.4) | 6.22 (d, 5.7) |
| 2 | 2.32–2.20 a | 1.94 a 2.36 (ddd, 14.5, 6.6, 4.5) | 2.03 (ddd, 14.5, 3.8, 3.0) 2.19 (ddd, 14.5, 10.2, 3.7) | 2.12 (dd, 13.8, 6.8) 2.44 (ddd, 13.8, 11.0, 4.3) | 1.82 (ddd, 13.2, 6.2, 1.4) 2.41 (ddd, 13.2, 11.4, 5.2) | 5.89 (d, 5.7) |
| 3 | 4.79 (dd, 8.6, 8.3) | 4.49 a | 4.49 a | 4.63 (dd, 11.0, 6.8) | 4.22 (dd, 11.4, 6.2) | - |
| 4 | - | - | - | - | - | - |
| 5 | 5.26 (dq, 7.2, 1.5) | 5.27 (dq, 10.5, 1.5) | 5.39 (dq, 10.5, 1.5) | 3.06 (d, 7.3) | 4.79 (br s) | 5.48 (dq, 5.8, 1.2) |
| 6 | 6.14 a | 6.00 (dd, 10.5, 9.5) | 6.38 (dd, 10.5, 9.2) | 5.25 (dd, 7.3, 5.5) | 5.63 (br d, 5.7) | 5.95 (ddq, 5.8, 5.2, 1.2) |
| 7 | 3.08 (dddd, 9.0, 7.5, 3.0, 3.0) | 3.09 (dddd, 9.5, 9.5, 3.5, 3.0) | 3.02 (dddd, 9.5, 9.2, 3.5, 3.0) | 3.35 (dddd, 9.0, 5.5, 2.6, 2.5) | 3.31 (m) | 3.26 (dddd, 10.5, 5.2, 2.8, 2.5) |
| 8 | 5.33 (dd, 9.5, 9.0) | 5.57 (dd, 10.2, 9.5) | 6.00 (dd, 10.5, 9.5) | 5.00 (dd, 9.8, 9.0) | 5.06 (ddd, 10.0, 6.9, 1.2) | 5.17 (ddd, 10.5, 5.0, 3.2) |
| 9 | 2.09 (dd, 14.8, 9.5) 1.70 (d, 14.8) | 5.18 (dq, 10.2, 1.5) | 5.30 (dq, 10.5, 1.5) | 1.87 a β 2.05 (br dd, 14.6, 9.8) | 1.91 a 2.13 (dd, 14.3, 10.3) | 2.14 (dd, 15.4, 5.0) 2.22 (dd, 15.4, 3.2) |
| 10 | - | - | - | - | - | - |
| 11 | - | - | - | - | - | - |
| 12 | - | - | - | - | - | - |
| 13 | 5.65 (dd, 3.0, 0.7) 6.17 a | 5.71 (d, 3.0) 6.16 (d, 3.5) | 5.85 (d, 3.0) 6.25 (d, 3.5) | 5.74 (br d, 2.5) 6.23 (br d, 2.8) | 5.41 (br d, 2.5) 5.86 (br d, 2.8) | 5.89 (br d, 2.5) 6.22 br d, 2.8) |
| 14 | 1.49 (s) | 1.95 (d, 1.5) | 1.80 (d, 1.5) | 1.37 (br s) | 1.33 (s) | 1.53 (s) |
| 15 | 1.66 (br s) | 1.76 (d, 1.5) | 1.79 (d, 1.5) | 1.27 (s) | 1.18 (br s) | 1.73 (t, 1.2) |
| 1′ | - | - | - | - | - | - |
| 2′ | - | - | - | - | - | - |
| 3′ | 6.17 (qq, 7.0, 1.5) | 6.20 (qq, 7.0, 1.5) | 6.20 (qq, 7.0, 1.5) | 6.16 (qq, 7.0, 1.5) | 6.15 (br q, 7.2) | 6.16 (br q, 7.2) |
| 4′ | 1.96 (dq, 7.0, 1.5) | 1.95 (dq 7.0, 1.5) | 1.98 (dq 7.0, 1.5) | 1.94 (dq 7.0, 1.5) | 1.91 (br d, 7.2) | 2.00 (br d, 7.2) |
| 5′ | 1.89 (quint, 1.5) | 1.89 (quint, 1.5) | 1.91 (quint, 1.5) | 1.88 (quint, 1.5) | 1.84 (quint, 1.5) | 1.91 (quint, 1.5) |
| Position | 5 δC | 6 δC | 7 δC a | 8 δC | 9 δC c | 10 δC a |
|---|---|---|---|---|---|---|
| 1 | 77.6, CH | 76.2, CH | 69.4, CH | 77.9, CH | 78.8, CH | 139.3, CH |
| 2 | 41.8, CH2 | 38.9, CH2 | 37.0, CH2 | 39.0, CH2 | 36.6, CH2 | 128.3, CH |
| 3 | 81.7, CH | 74.1, CH | 74.9, CH | 78.6, CH | 85.3, CH | 110.4, C |
| 4 | 141.9, C | 142.2, C | 145.9, C | 66.2, C | 75.2, C | 137.8, C |
| 5 | 124.0, CH | 126.7, CH | 125.5, CH | 68.7, CH | 89.3, CH | 129.6, CH |
| 6 | 78.5, CH | 76.8, CH | 78.0, CH | 82.7, CH | 82.5, CH | 76.4, CH |
| 7 | 52.2, CH | 50.9, CH | 52.1, CH | 49.09, CH | 48.0, CH | 48.2, CH |
| 8 | 74.1, CH | 74.0, CH | 72.5, CH | 73.3, CH | 74.0, CH | 71.5, CH |
| 9 | 46.8, CH2 | 126.8, CH | 121.9, CH | 49.15, CH2 | 49.3, CH2 | 42.9, CH2 |
| 10 | 87.3, C | 143.2, C | 144.1, C | 87.1, C | 85.8, C | 87.3, C |
| 11 | 139.0, C | 140.1, C | 140.0, C | 137.2, C | 137.1, C | 137.7, C |
| 12 | 171.8, C | 172.0, C | 172.0, C | 171.7, C | 171.3, C | 169.6, C |
| 13 | 124.8, CH2 | 121.8, CH2 | 121.8, CH2 | 127.0, CH2 | 125.5, CH2 | 123.8, CH2 |
| 14 | 19.6, CH3 | 11.7, CH3 | 19.5, CH3 | 20.7, CH3 | 22.6, CH3 | 28.0, CH3 |
| 15 | 20.2, CH3 | 22.8, CH3 | 23.6, CH3 | 20.5 b, CH3 | 24.2, CH3 | 20.2, CH3 |
| 1′ | 167.9, C | 168.2, C | 168.2, C | 168.0, C | 168.3, C | 167.4, C |
| 2′ | 128.7, C | 128.7, C | 128.6, C | 128.7, C | 127.0, C | 126.2, C |
| 3′ | 140.4, CH | 139.9, CH | 140.0, CH | 14.03, CH | 140.3, CH | 139.4, CH |
| 4′ | 16.1, CH3 | 16.0, CH3 | 16.0, CH3 | 16.0, CH3 | 16.0, CH3 | 15.7, CH3 |
| 5′ | 20.7, CH3 | 20.8, CH3 | 20.9, CH3 | 20.6 b, CH3 | 20.5, CH3 | 20.6, CH3 |
| Position | 13 δH (mult, J in Hz) | 14 δH (mult, J in Hz) | 15 δH (mult, J in Hz) | 16 δH (mult, J in Hz) |
|---|---|---|---|---|
| 1 | 4.11 (dd, 10.6, 6.5) | 4.01 (dd, 11.7, 3.0) | - | - |
| 2 | 1.91 a 2.43 (ddd, 15.5, 6.5, 6.0) | 2.17 a (ddd, 15.0, 5.0, 3.0) 2.47 (ddd, 15.0, 11.7, 2.5) | 6.65 (s) | 7.91 (d, 8.5) |
| 3 | 5.49 (br d, 6.0) | 5.30 a | - | 7.34 (br d, 8.5) |
| 4 | - | - | - | - |
| 5 | 5.32 (dq, 8.5, 1.2) | 5.32 (dq, 10.3, 1.2) | 6.65 (s) | 7.58 (br s) |
| 6 | 5.28 a | 5.87 (dd, 10.3, 2.0) | - | - |
| 7 | 3.17 (dddd, 9.5, 8.5, 3.5, 3.0) | 3.20 (dddd, 10.7, 2.0, 1.7, 1.5) | 3.97 (d, 5.8) | - |
| 8 | 5.65 (dd, 10.5, 9.5) | 5.03 (td, 10.5, 4.0) | 5.50 (ddd, 7.5, 5.7, 2.7) | 7.19 (d, 7.2) |
| 9 | 5.24 (dq, 10.5, 1.2) | 2.49 a 2.90 (br dd, 13.6, 4.2) | 1.80 a 2.00 (ddd, 13.0, 7.5, 6.0) | 7.20 (d, 7.2) |
| 10 | - | - | 3.97 (sextet, 7.0) | - |
| 11 | - | - | - | - |
| 12 | - | - | - | - |
| 13 | 5.74 (d, 3.0) | 5.75 (d, 1.5) | 5.31 (br s) | 5.74 (d, 1.8) |
| 6.17 (d, 3.5) | 6.23 (d, 1.7) | 6.33 (br s) | 6.51 (d, 1.8) | |
| 14 | 1.97 (d, 1.2) | 5.48 (br s) 5.51 (br s) | 1.34 (d, 7.0) | 2.65 (s) |
| 15 | 1.85 (d, 1.2) | 1.85 (d, 1.2) | 2.10 (s) | 2.46 (br s) |
| 1′ | - | - | - | - |
| 2′ | - | - | - | - |
| 3′ | 6.19 (qq, 7.0, 1.5) | 6.15 (qq, 7.0, 1.5) | 6.03 (qq, 7.0, 1.5) | - |
| 4′ | 1.99 (dq, 7.0, 1.5) | 1.96 (dq, 7.0, 1.5) | 1.81 (dq, 7.0, 1.5) | - |
| 5′ | 1.92 (quint, 1.5) | 1.90 (quint, 1.5) | 1.79 (quint, 1.5) | - |
| 1″ | - | - | - | - |
| 2″ | 2.02, (s) | 2.04, (s) | - | - |
| Position | 13 δC a | 14 δC a | 15 a δC | 16 a δC |
|---|---|---|---|---|
| 1 | 75.4, CH | 70.6, CH | 140.5, C | 132.5, C |
| 2 | 35.2, CH2 | 34.5, CH2 | 113.9, CH | 125.0, CH |
| 3 | 74.6, CH | 73.6, CH | 155.0, C | 128.4, CH |
| 4 | 137.9, C | 139.4, C | 124.0, C | 136.4, C |
| 5 | 127.0, CH | 124.8, CH | 132.0, CH | 126.1, CH |
| 6 | 76.0, CH | 75.0, CH | 126.6, C | 128.6, CH |
| 7 | 50.0, CH | 49.1, CH | 48.3, CH | 135.60, C |
| 8 | 72.8, CH | 73.6, CH | 71.9, CH | 127.4, CH |
| 9 | 127.6, CH | 39.8, CH2 | 33.6, CH2 | 126.0, CH |
| 10 | 141.7, C | 144.4, C | 31.5, CH | 135.4, C |
| 11 | 139.0, C | 135.2, C | 145.1, C | 145.5, C |
| 12 | 170.8, C | 170.0, C | 170.1, C | 171.8, C |
| 13 | 121.3, CH2 | 125.7, CH2 | 129.1, CH2 | 127.6, CH2 |
| 14 | 11.1, CH3 | 116.5, CH2 | 22.9, CH3 | 19.4, CH3 |
| 15 | 22.2, CH3 | 21.7, CH3 | 15.6, CH3 | 21.7, CH3 |
| 1′ | 167.6, C | 166.8, C | 169.4, C | - |
| 2′ | 128.0, C | 127.2, C | 129.5, C | - |
| 3′ | 139.5, CH | 138.5, CH | 138.8, CH | - |
| 4′ | 15.6, CH3 | 14.6, CH3 | 15.4, CH3 | - |
| 5′ | 20.3, CH3 | 19.0, CH3 | 20.5, CH3 | - |
| 1″ | 169.2, C | 170.0, C | - | - |
| 2″ | 20.9, CH3 | 19.7, CH3 | - | - |
| Compound | T. b. rhodesiense IC50 (μM) a | T. cruzi IC50 (μM) a | L. donovani IC50 (μM) a | P. falciparum IC50 (μM) a | L6 cells IC50 (μM) a |
|---|---|---|---|---|---|
| 1 | 2.08± 0.09 (2.4) b | 11.3± 3.3 (0.4) b | 2.69± 1.08 (1.8) b | 3.12± 0.52 (1.6) b | 4.91 ± 0.53 |
| 3 | 1.96± 0.16 (2.04) b | 4.24 (0.9) h | 13.19± 3.31 (0.3) b | 2.39 (1.7) h | 4.0 ± 0.03 |
| 4 | 2.18 ± 0.49 (2.6) b | 8.38 (0.7) h | 28.67 ± 8.95 (0.2) b | 3.20 (1.7) h | 5.56 ± 0.30 |
| 5 | 0.91 ± 0.16 (3.7) b | 6.35 (0.5) h | 8.28 ± 2.40 (0.4) b | 4.06 (0.8) h | 3.33 ± 0.28 |
| 6–7 | 1.21 ± 0.16 (3.9) b | 16.00 ± 0.34 (0.3) b | 12.88 ± 0.65 (0.4) b | 13.82 ± 1.59 (0.3) b | 4.74 ± 0.24 |
| 8 | 3.25 ± 0.50 (3.5) b | 47.9 ± 1.11 (0.24) b | 66.8 ± 7.79 (0.17) b | 31.3 ± 2.40 (0.4) b | 11.5 ± 0.72 |
| 10 | 13.4 ± 0.46 (1.51) b | 132.19 ± 0.59 (0.15) b | 35.96 ± 0.18 (0.56) b | 79.13 ± 1.02 (0.26) b | 20.20 ± 2.098 |
| 11 | 1.57 ± 0.005 (0.28) b | 11.49 ± 0.68 (0.2) b | 4.20 ± 0.28 (0.55) b | 3.93 ± 0.53 (0.59) b | 2.31 ± 0.196 |
| 12 | 0.47 ± 0.006 (2.19) b | 2.60 ± 0.69 (0.5) b | 1.30 ± 0.03 (0.79) b | 1.55 ± 0.31 (0.66) b | 1.03 ± 0.076 |
| 13 | 0.54 ± 0.013 (3.46) b | 12.76 ± 1.08 (0.15) b | 3.26 ± 0.22 (0.57) b | 9.17 ± 0.61 (0.20) b | 1.87 ± 0.026 |
| 14 | 1.20 ± 0.151 (4.4) b | 15.4 ± 1.48 (0.6) b | 4.97 ± 0.43 (7.2) b | 8.80 ± 0.94 (0.68) b | 3.95 ± 0.088 |
| 15 | 13.32 ± 0.333 (1.68) b | 36.81 ± 7.76 (0.61) b | 5.86 ± 0.59 (3.83) b | 35.66 ± 1.10 (0.63) b | 22.44 ± 0.157 |
| Positive control | 0.01 ± 0.01 c | 2.0 ± 0.2 d | 0.13 ± 0.01 e | 0.006 ± 0.01 f | 0.016 ± 0.01 g |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Mieri, M.; Smieško, M.; Ismajili, I.; Kaiser, M.; Hamburger, M. Acid-Induced Rearrangement of Epoxygermacranolides: Synthesis of Furanoheliangolides and Cadinanes from Nobilin. Molecules 2017, 22, 2252. https://doi.org/10.3390/molecules22122252
De Mieri M, Smieško M, Ismajili I, Kaiser M, Hamburger M. Acid-Induced Rearrangement of Epoxygermacranolides: Synthesis of Furanoheliangolides and Cadinanes from Nobilin. Molecules. 2017; 22(12):2252. https://doi.org/10.3390/molecules22122252
Chicago/Turabian StyleDe Mieri, Maria, Martin Smieško, Isidor Ismajili, Marcel Kaiser, and Matthias Hamburger. 2017. "Acid-Induced Rearrangement of Epoxygermacranolides: Synthesis of Furanoheliangolides and Cadinanes from Nobilin" Molecules 22, no. 12: 2252. https://doi.org/10.3390/molecules22122252
APA StyleDe Mieri, M., Smieško, M., Ismajili, I., Kaiser, M., & Hamburger, M. (2017). Acid-Induced Rearrangement of Epoxygermacranolides: Synthesis of Furanoheliangolides and Cadinanes from Nobilin. Molecules, 22(12), 2252. https://doi.org/10.3390/molecules22122252