HIGA: A Running History Information Guided Genetic Algorithm for Protein–Ligand Docking

Abstract
:1. Introduction
2. Results and Discussion
2.1. Data Preparation and Parameter Setting
- (1)
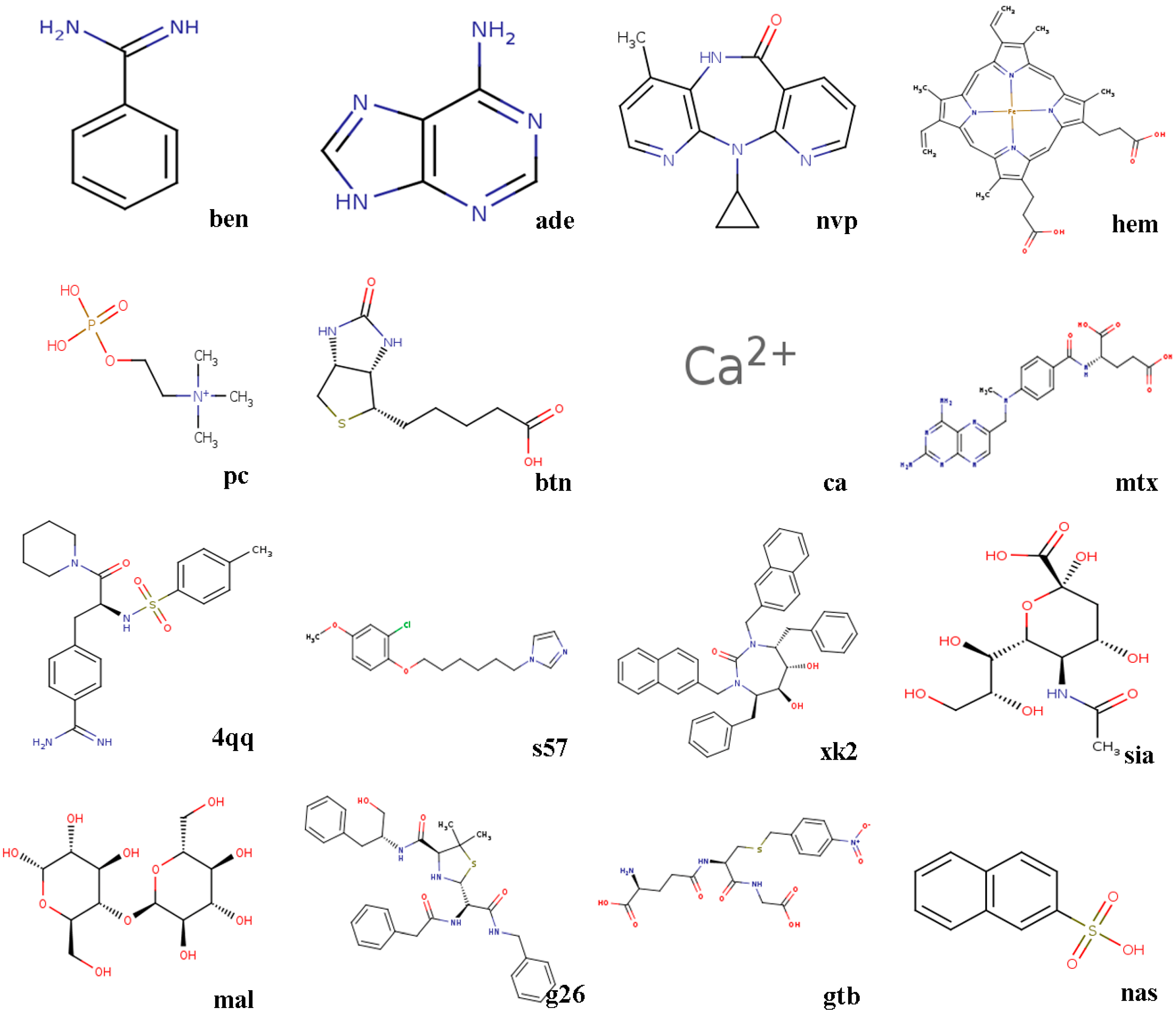
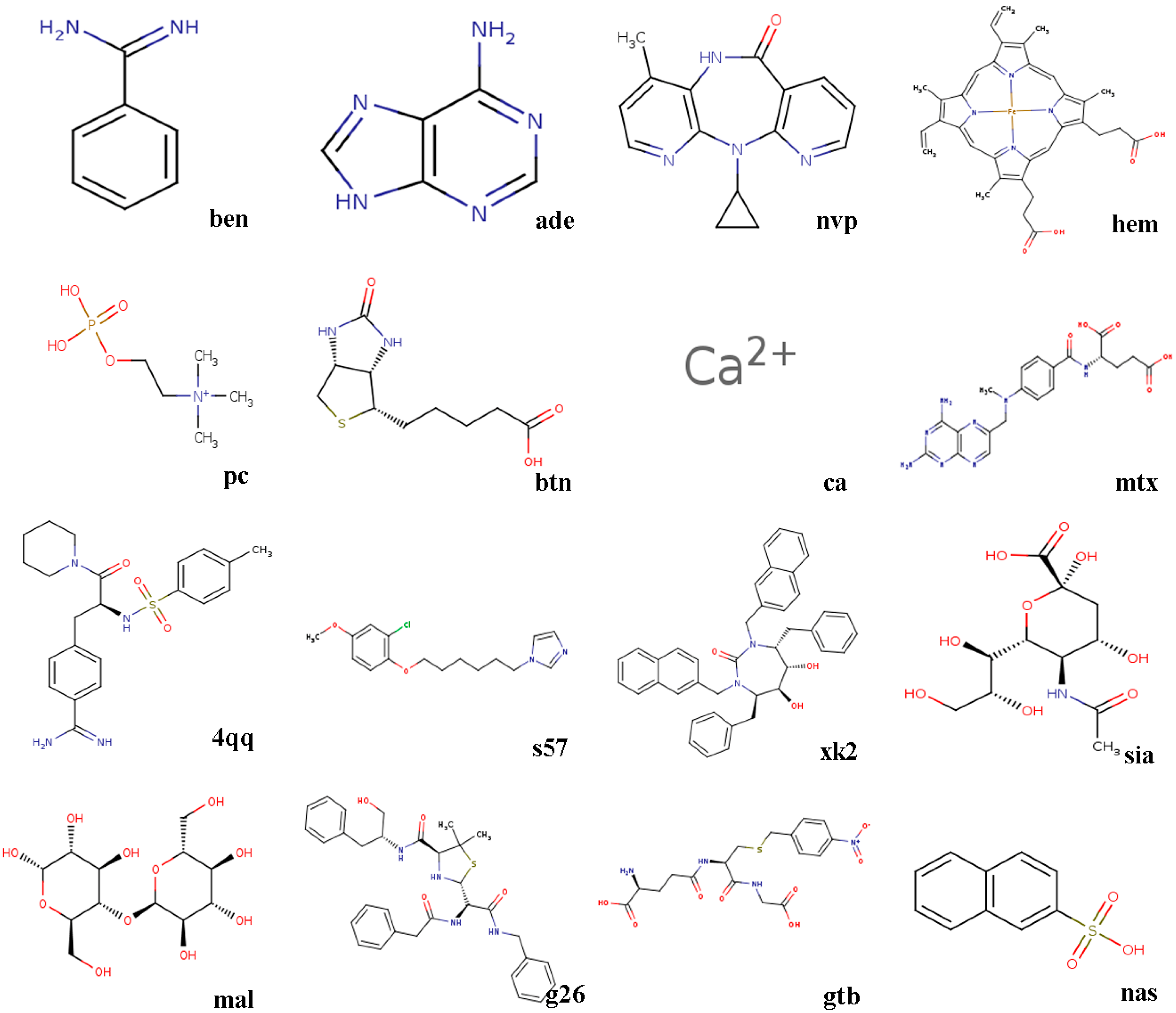
- 3ptb β-trypsin/ben (benzamidine)β-Trypsin, isolated from the pancreas of pigs, sheep, as well as cattle, is used as a protease. Benzamidine, an inhibitor, is generally utilized in suppressing proteolysis of proteins.
- (2)
- 1aha α-momorcharin/ade (adenine)α-Momorcharin originates from seeds of Momordica charantia, while adenine is a biological component of organism.
- (3)
- 3hvt HIV-1 reverse transcriptase/nvpHIV-1 reverse transcriptase (RT), a phosphate enzyme, is involved in synthesis of cDNA. Nvp is a strong, non-nucleoside RT suppressor.
- (4)
- 1phg cytochrome P450-cam/hem (protoporphyrin IX)Cytochrome P450-cam, participating in metabolism of exogenous, as well as endogenous substances, is a superfamily of heme-thiolate proteins. Protoporphyrin IX, a purple brown crystalline powder, can dissolve in methanol, while is not soluble in ether, chloroform, acetone, or water.
- (5)
- 2mcp McPC-603/pc (phosphocholine)McPC-603, a myeloma protein from mouse that binds to phosphocholine, interacts with phosphatidylcholine synthesis in tissues.
- (6)
- 1stp streptavidin/btn (biotin)Streptavidin, a protein obtained from streptomyces, harbors a similar biological features with affinity. Biotin, a member of B vitamins, plays a critical role in normal metabolism of proteins as well as fats.
- (7)
- 6rnt ribonuclease T1/ca (calcium ion)Ribonuclease T1, an endonuclease, is able to discard the non-hybridized RNA area in DNA-RNA hybrid. Calcium ion plays a vital role in human physiological functions.
- (8)
- 4dfr dihydrofolate reductase/mtx (methotrexate)Dihydrofolate reductase, has universally been utilized as a therapeutic target in anti-tumor therapy, as well as other aspects. Methotrexate, a drug with potent immunosuppressive effect, is capable of inhibiting proliferation as well as division of immune cells.
- (9)
- 1ett thrombin/4qqThrombin is a formless, white to gray, freeze-dried powder, and 4qq is regarded as a non-polymer suppressor.
- (10)
- 1hri human rhinovirus/s57Human rhinovirus causes the majority of human common cold. s57 is a member of imidazole.
- (11)
- 1hvr protease/xk2Protease, an enzyme widely found in animals as well as plants, is capable of catalyzing protein catabolism. xk2, a small molecule inhibitor, is able to decrease or even prohibit chemical reaction rate.
- (12)
- 4hmg hemagglutinin/sia (sialic acid)Hemagglutinin is the cause of coagulation of erythrocytes. Sialic acids, generated at terminal sugars, are members of acidic monosaccharides.
- (13)
- 1cdg cyclodextrin glycosyl transferase/mal (maltose)Cyclodextrin glycosyltransferase, a bacterial enzyme, is able to produce cyclodextrins. Maltose, made of starch, as well as malt, is widely utilized as nutrient as well as culture medium.
- (14)
- 1htf HIV-1 protease/g26HIV-1 protease is capable of separating newly-generated polyproteins into individual peptides. g26, a non-polymer suppressor, is an amide with easily oxidizable and highly reactive perssad.
- (15)
- 1glq glutathione S-transferase/gtb (S-(P-nitrobenzyl)Glutathione)Glutathione S-transferase, a series of enzymes, is associated with hepatic detoxification process. S-(P-nitrobenzyl) Glutathione is a critical synthesis of glutathione precursor.
- (16)
- 1tmn thermolysin/nas (2-naphthalenesulfonic acid)Thermolysin, a biological component, is featured by a more rapid hydrolysis of hydrophobic amino acids. 2-naphthalenesulfonic acid, white powder or crystal, can dissolve in water but not in alcohol, which is widely adopted in organic synthesis.
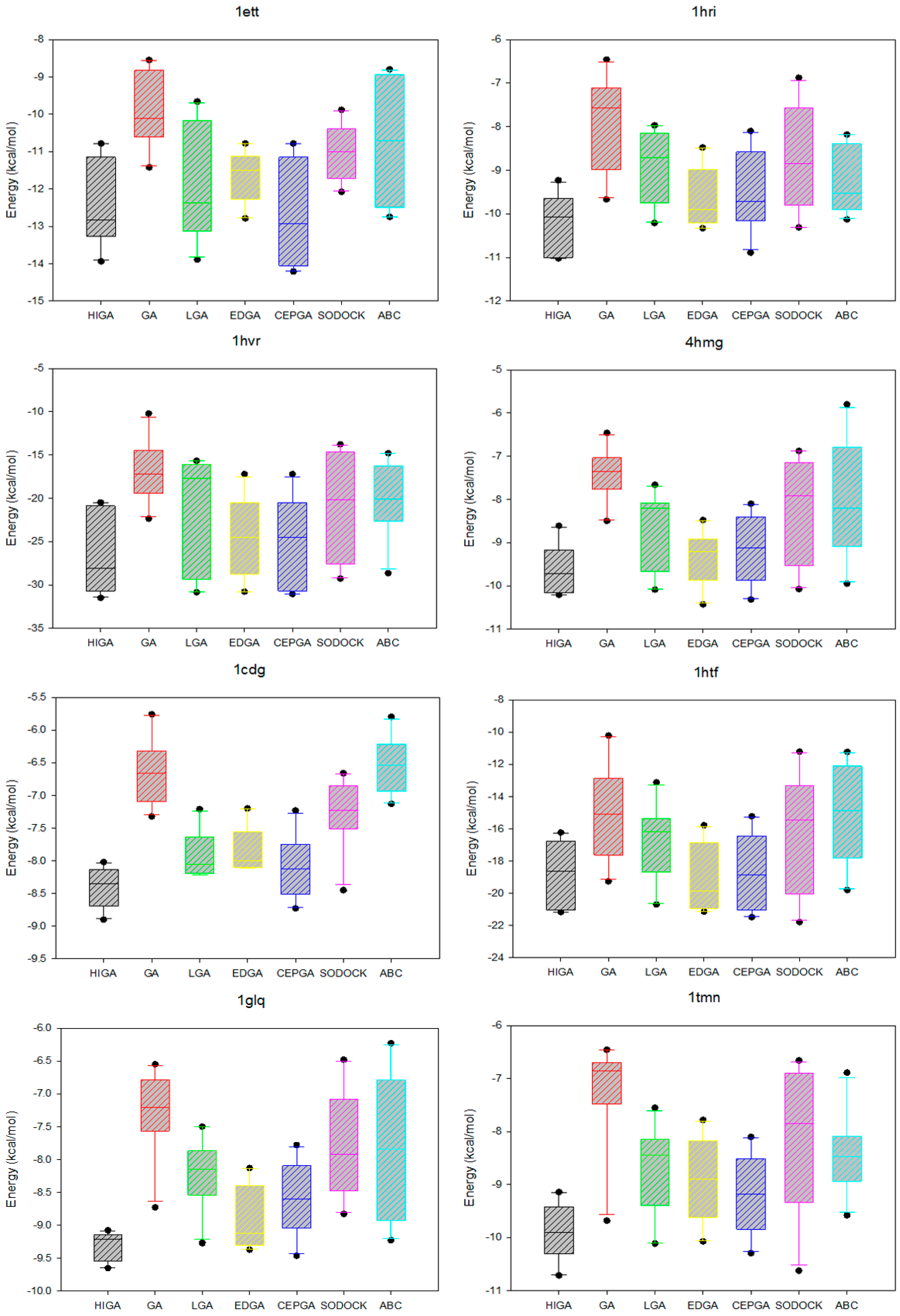
2.2. Comparison of Energy and RMSD
2.3. Cluster Analysis of Docked Conformations
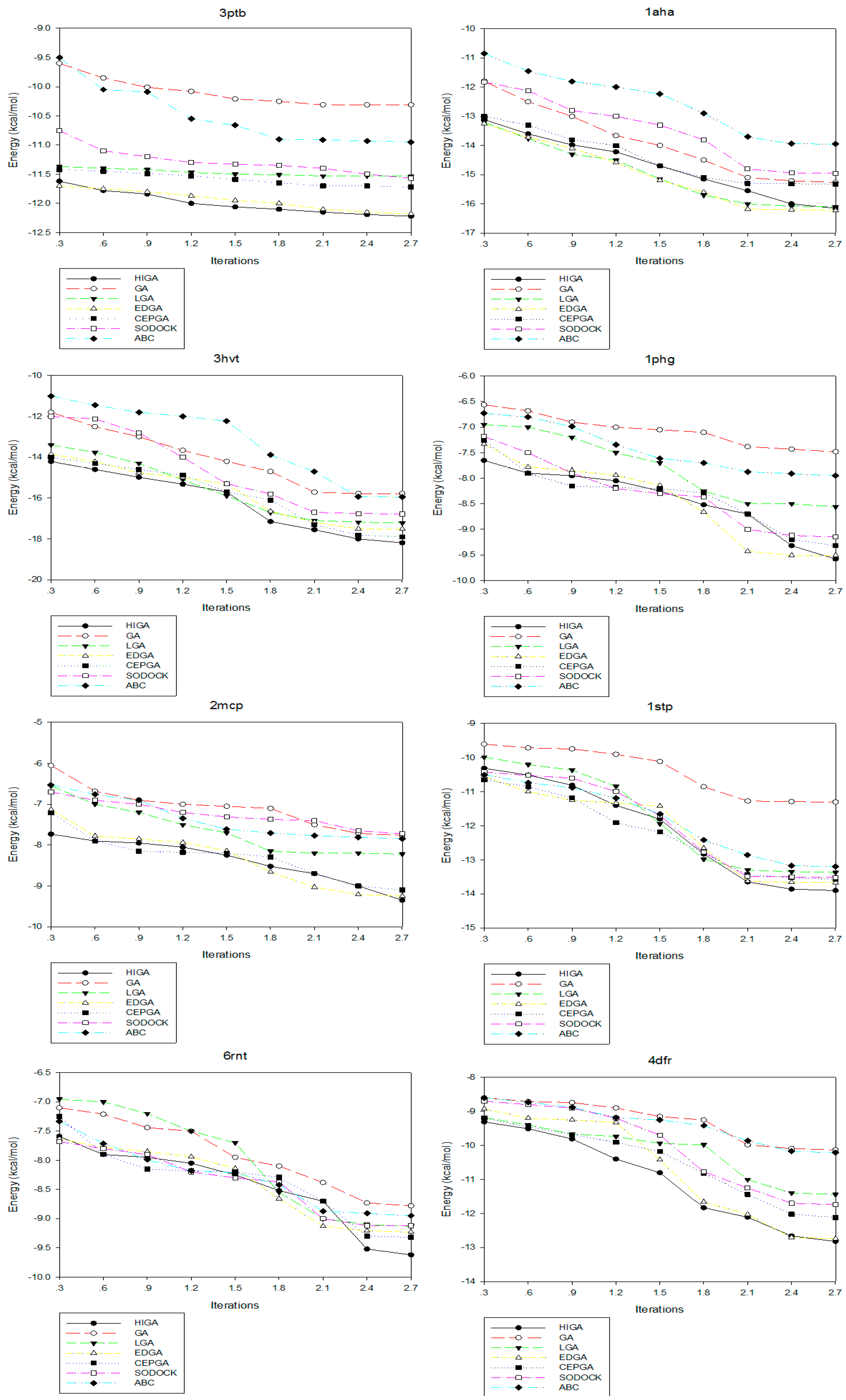
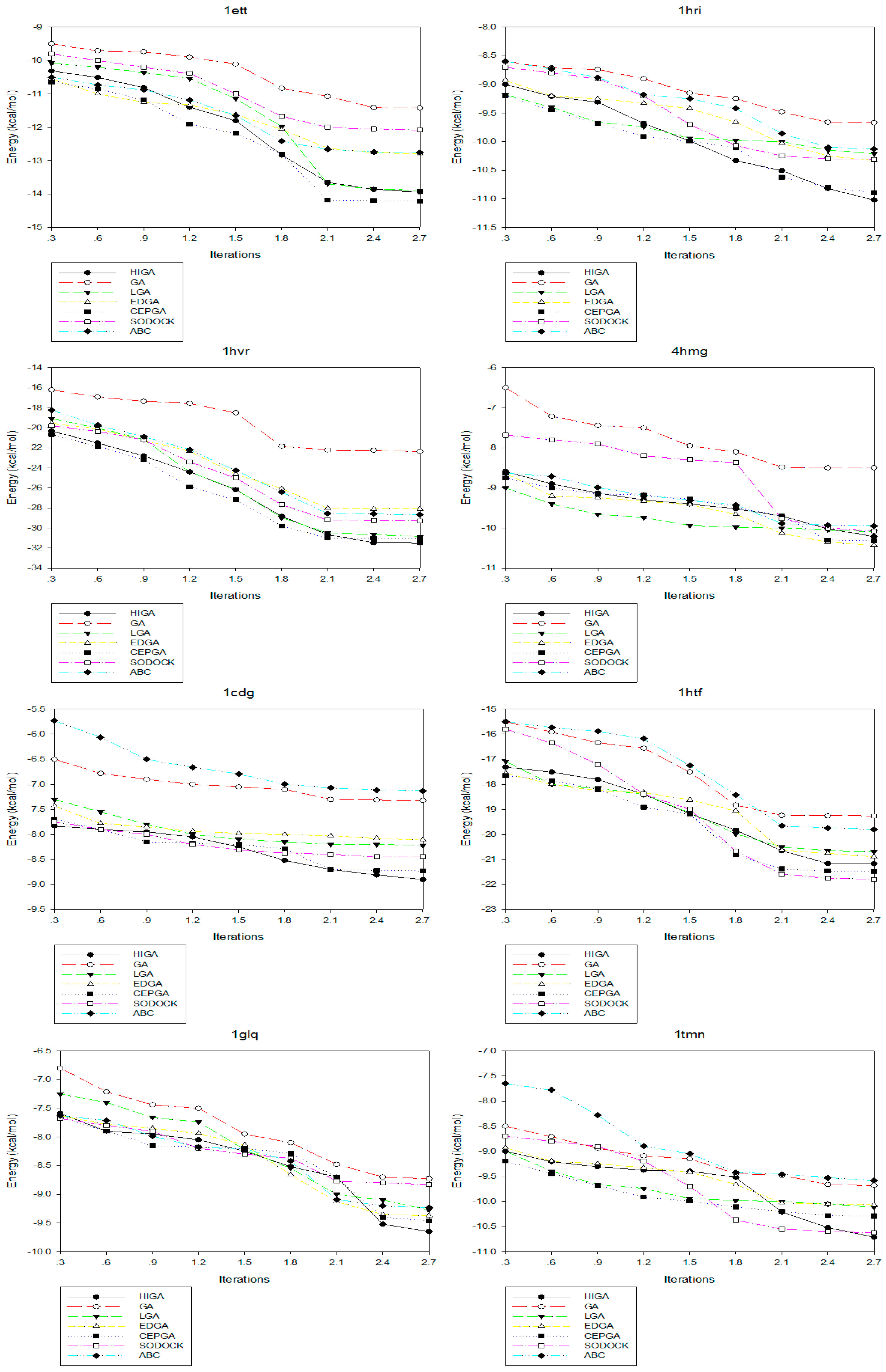
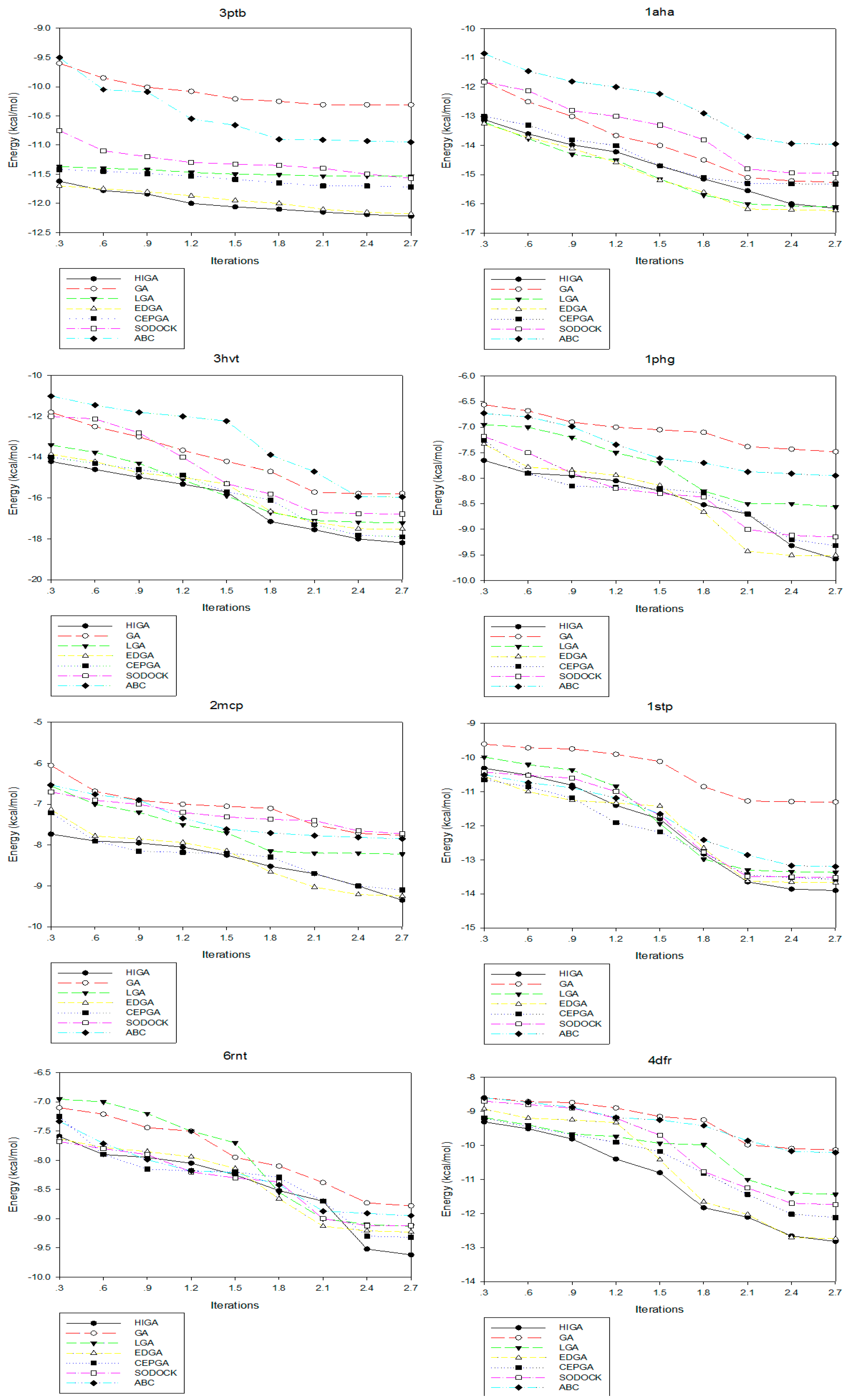
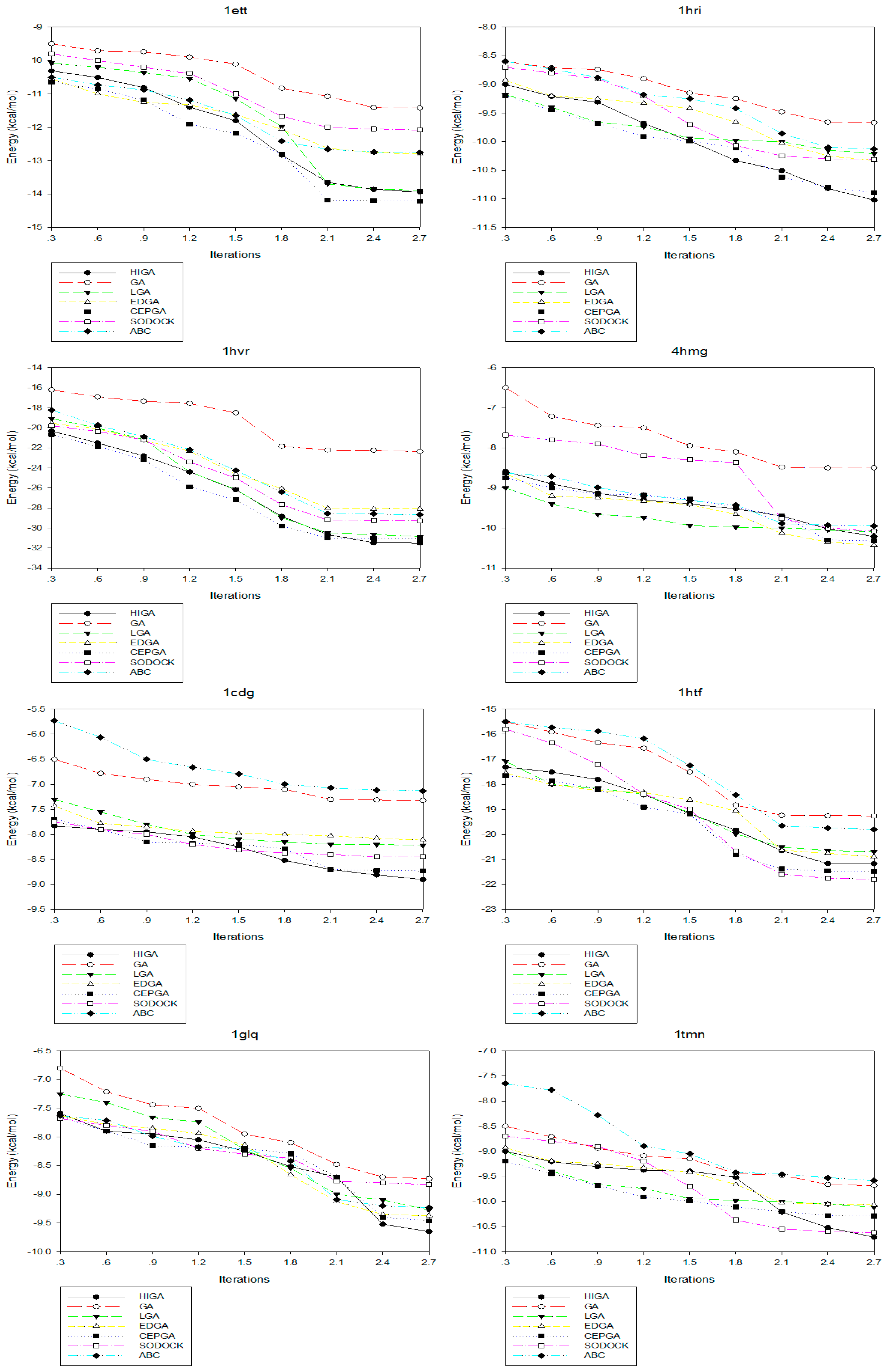
2.4. Convergence Analysis
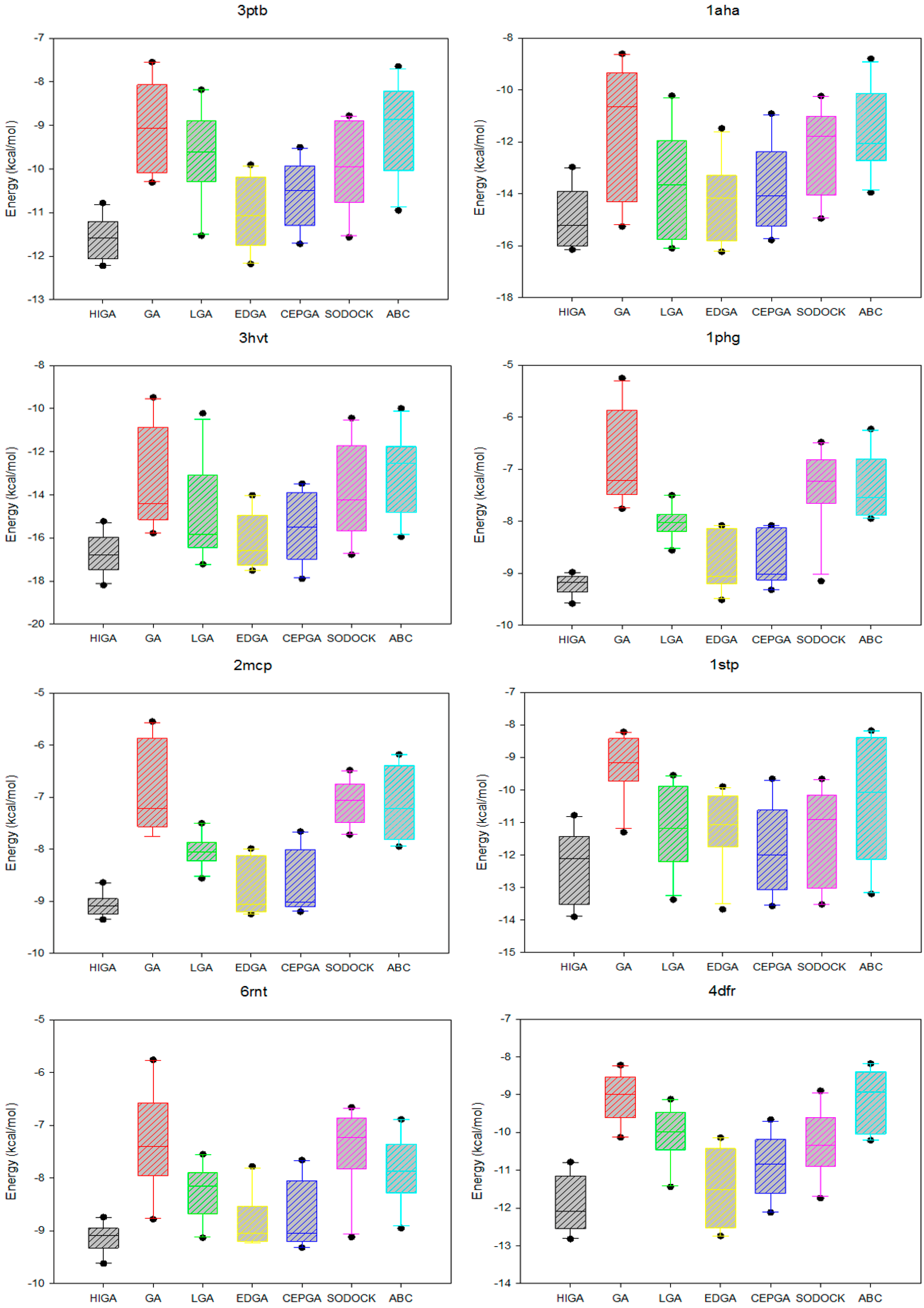
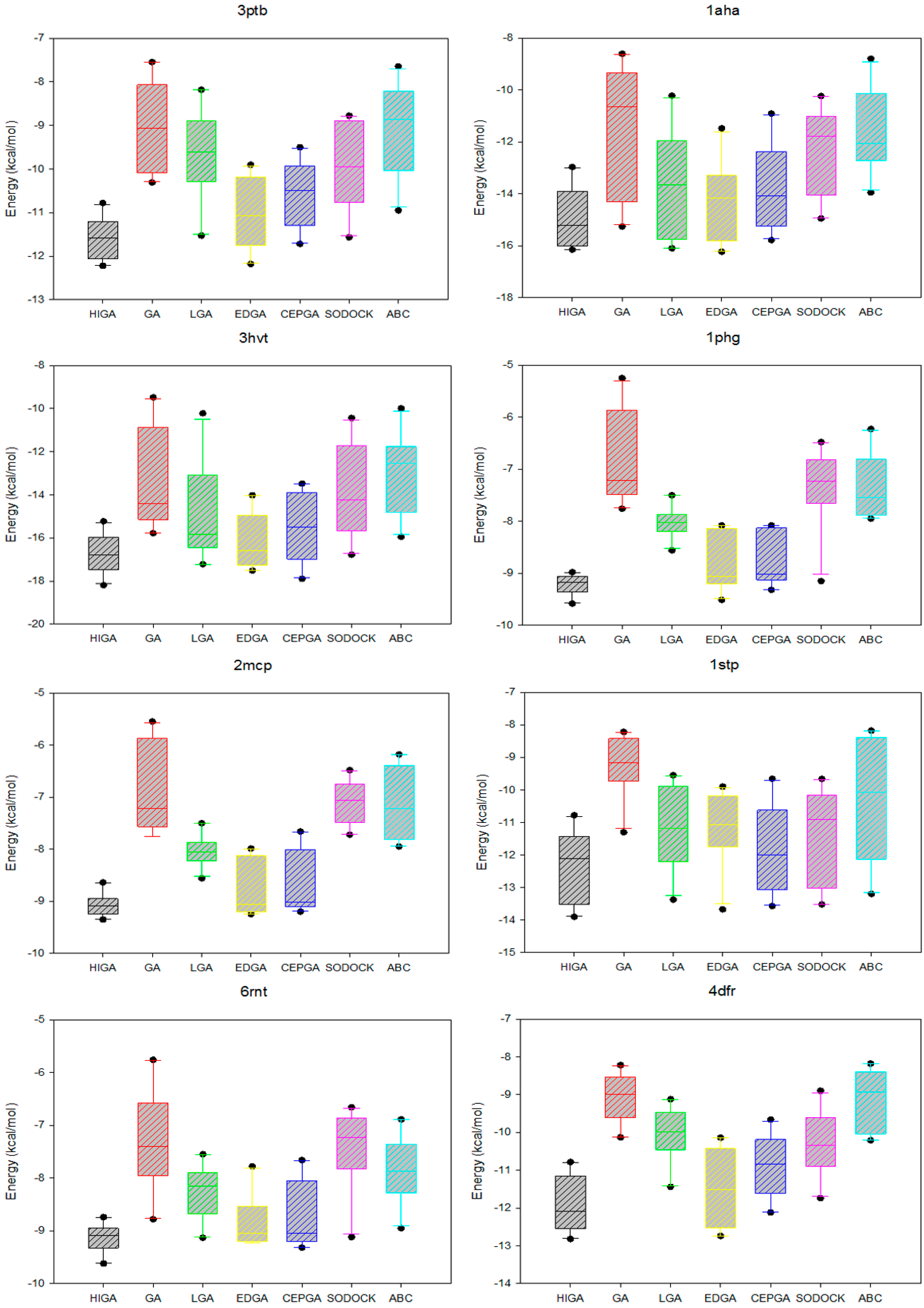
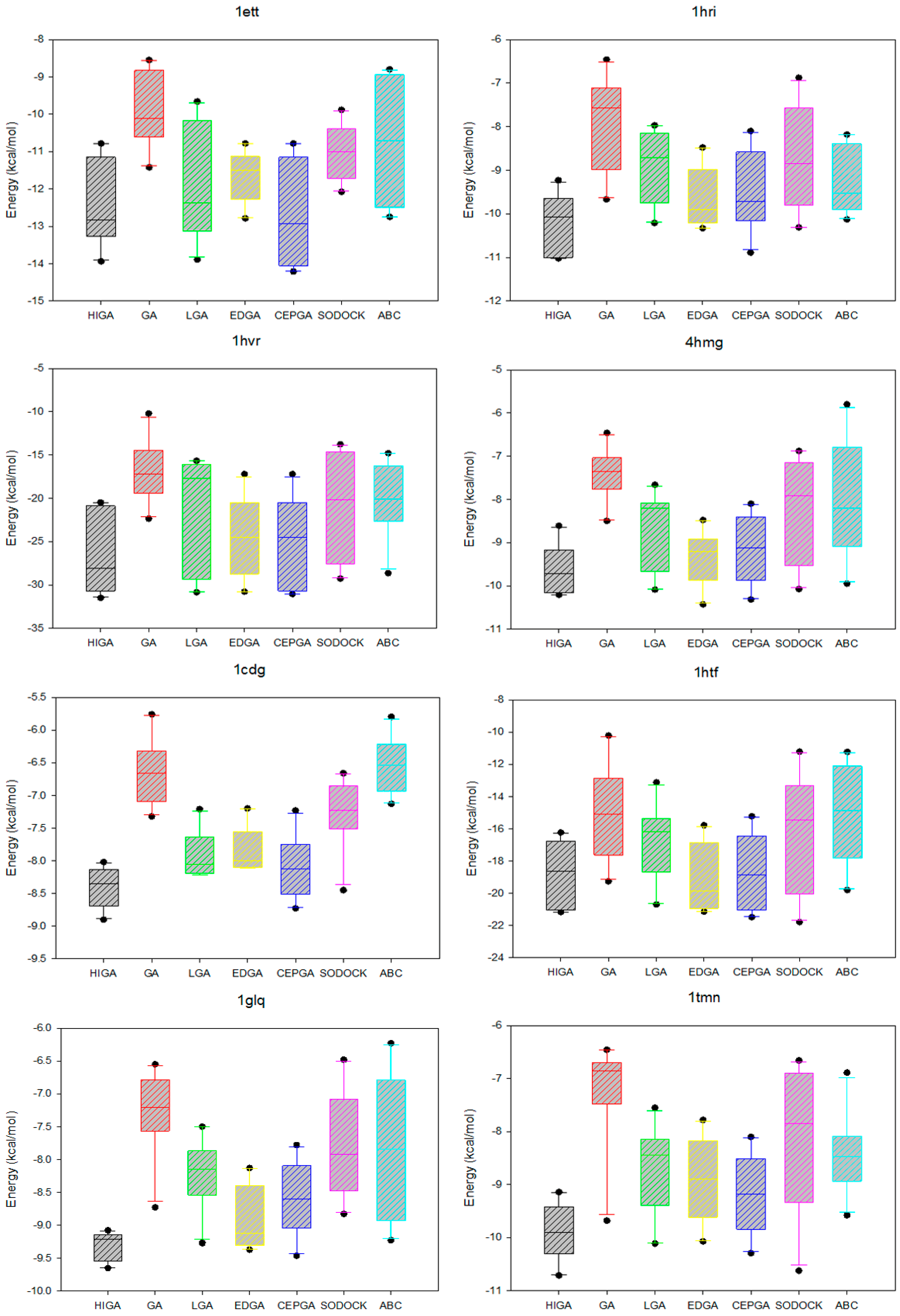
2.5. Data Distribution Analysis
2.6. Execution Time Analysis
2.7. Comparison Based on the Hypothesis Test
3. Materials and Methods
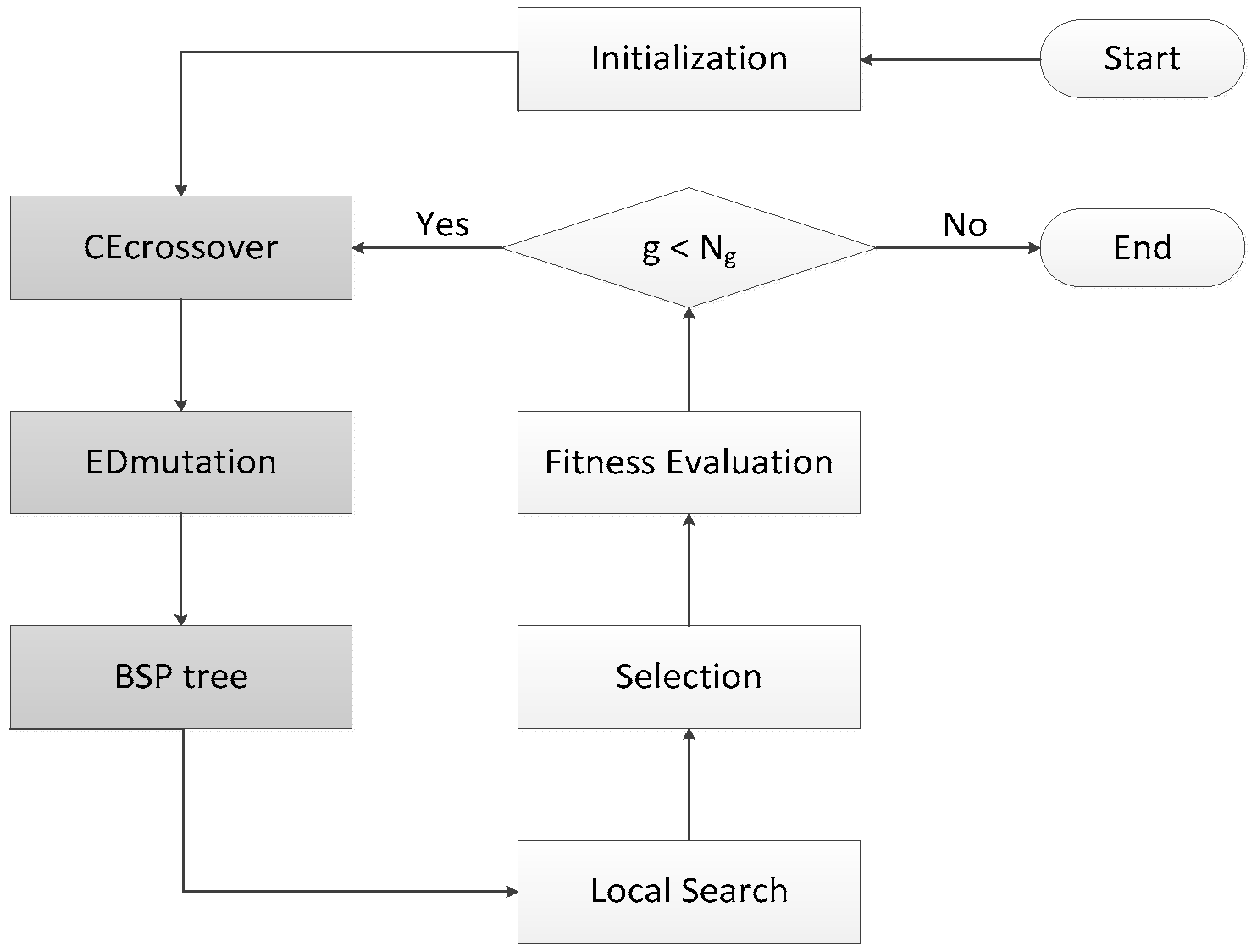
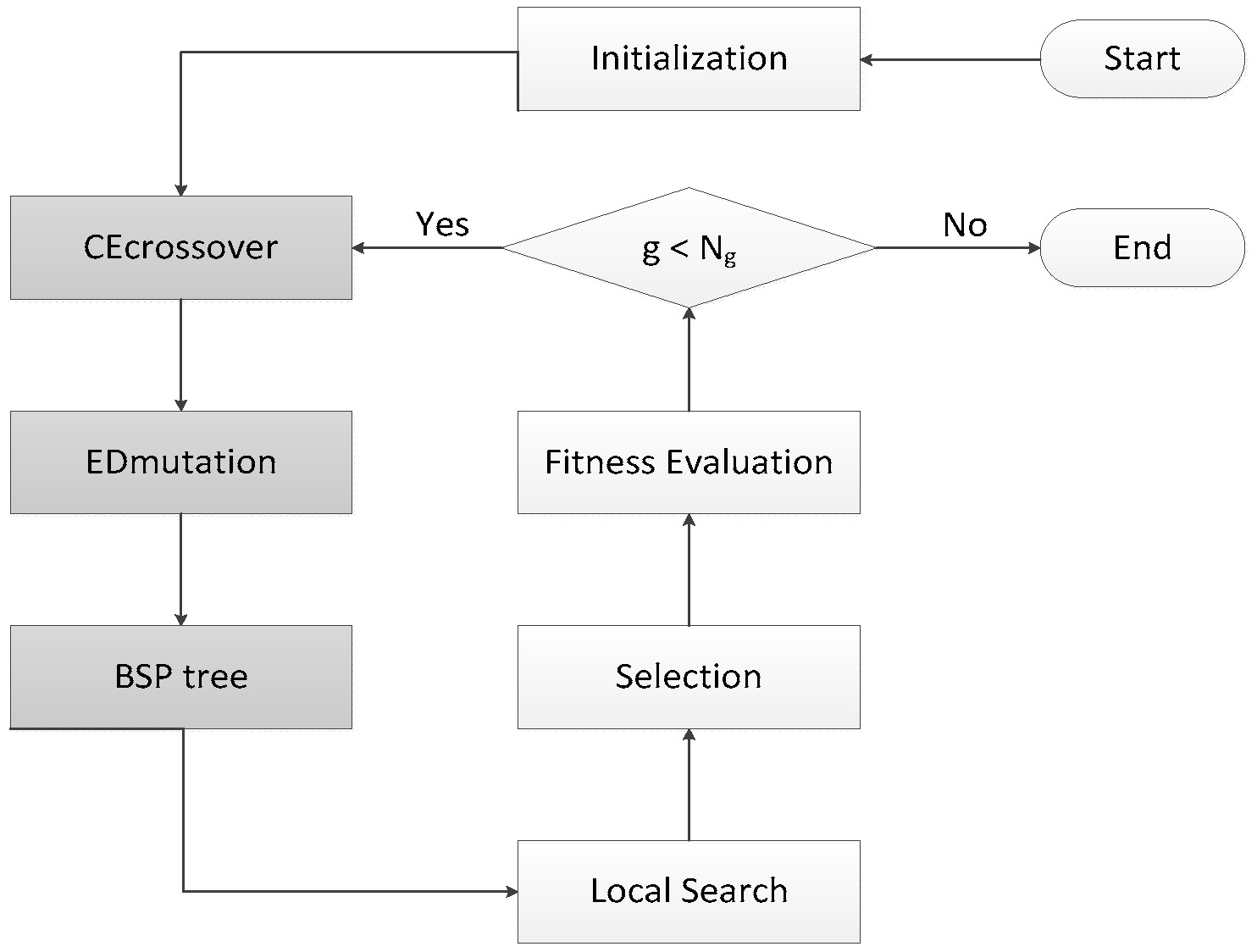
3.1. Running History Information Guided Genetic Algorithm
3.2. CE Crossover
3.3. ED Mutation
3.4. BSP Tree
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bohlooli, F.; Sepehri, S.; Razzaghi-AsI, N. Response surface methodology in drug design: A case study on docking analysis of a potent antifungal fluconazole. Comput. Biol. Chem. 2017, 67, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Wang, G.R.; Yin, Y. Improving ELM-based microarray data classification by diversified sequence features selection. Neural Comput. Appl. 2016, 27, 155–166. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Y.H.; Wang, G.R.; Wang, Z.H.; Gao, M. ELM-Based Large-Scale Genetic Association Study via Statistically Significant Pattern. IEEE Trans. Syst. Man Cybern. Syst. 2017, 1–14. [Google Scholar] [CrossRef]
- Allen, S.E.; Dokholyan, N.V.; Bowers, A.A. Dynamic docking of conformationally constrained macrocycles: Methods and applications. ACS Chem. Biol. 2016, 11, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Li, J.J.; Song, L.; Zeng, X.X.; Wang, G.H. Similarity computation strategies in the microRNA-disease network: A Survey. Brief. Funct. Genom. 2016, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, E.J. Machine learning optimization of cross docking accuracy. Comput. Biol. Chem. 2016, 62, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor-ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X.Q. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Jug, G.; Anderluh, M.; Tomašič, T. Comparative evaluation of several docking tools for docking small molecule ligands to DC-SIGN. J. Mol. Model. 2015, 21, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Wang, G.R.; Zhang, X.; Yu, J.X.; Wang, Z.H. Learning Phenotype Structure Using Sequence Model. IEEE Trans. Knowl. Data Eng. 2014, 26, 667–681. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Yu, J.X.; Wang, G.R.; Chen, L.; Wang, B.; Yu, G. Maximal Subspace Coregulated Gene Clustering. IEEE Trans. Knowl. Data Eng. 2008, 20, 83–98. [Google Scholar] [CrossRef]
- Zeng, X.X.; Liao, Y.L.; Liu, Y.S.; Zou, Q. Prediction and validation of disease genes using HeteSim Scores. IEEE ACM Trans. Comput. Biol. Bioinform. 2017, 14, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Gorbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Balaz, S.; Shelver, W.H. A practical approach to docking of zinc metalloproteinase inhibitors. J. Mol. Graph. Model. 2004, 22, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. Software news and update a semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2006, 10, 1145–1152. [Google Scholar]
- Jain, A.N. Scoring functions for protein-ligand docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar] [PubMed]
- Muryshev, A.E.; Tarasov, D.N.; Butygin, A.V.; Butygina, O.V.; Aleksandrov, A.B.; Nikitin, S.M. A novel scoring function for molecular docking. J. Comput. Aided Mol. Des. 2003, 17, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Bharatham, N.; Bharatham, K.; Shelat, A.A.; Bashford, D. Ligand binding more prediction by docking: Mdm2/mdmx inhibitors as a case study. J. Chem. Inf. Model. 2014, 54, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; Gu, J.F.; Zhuan, H.Y.; Kang, L.; Zhao, X.Y.; Guo, Q. Adaptive molecular docking method baesd on information entropy genetic algorithm. Appl. Soft Comput. 2015, 26, 299–302. [Google Scholar] [CrossRef]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.Y.; Yan, Z.Q.; Zheng, X.L.; Hu, L.; Yang, Y.L.; Wang, J. A comparison of various optimization algorithms of protein-ligand docking programs by fitness accuracy. J. Mol. Model. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Blum, C.; Puchinger, J.; Raidl, G.R.; Roli, A. Hybrid mataheuristics in combinatorial optimization: A survey. Appl. Soft Comput. 2011, 11, 4135–4151. [Google Scholar] [CrossRef]
- Lόpez-Camacho, E.; Godoy, M.J.; Garcỉa-Nieto, J.; Nebro, A.J.; Aldana-Montes, J.F. Solving molecular flexible docking problems with mataheuristics: A comparative study. Appl. Soft Comput. 2015, 28, 379–393. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins Struct. Funct. Genet. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.C.; Li, T.H. A combination of numeric genetic algorithm and tabu search can be applied to molecular docking. Comput. Biol. Chem. 2004, 28, 303–312. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R. Flexible ligand docking using evolutionary algorithms: Investigating the effects of variation operators and local search hybrids. Biosystems 2003, 72, 57–73. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainsk, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligang-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [PubMed]
- Chen, H.M.; Liu, B.F.; Huang, H.L.; Hwang, S.F.; Ho, S.Y. SODOCK: Swarm optimization for highly flexible protein-ligand docking. J. Comput. Chem. 2007, 28, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Jason, S.; Merkle, D.; Middendorf, M. Molecular docking with multi-objective particle swarm optimization. Appl. Soft Comput. 2008, 8, 666–675. [Google Scholar] [CrossRef]
- Ng, M.C.; Fong, S.; Siu, S.W. PSOVina: The hybrid particle swarm optimization algorithm for protein-ligand docking. J. Bioinform. Comput. Biol. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Fujimoto, K.J.; Tanaka, S. Protein-ligand docking using fitness learning-based artificial bee colony with proximity stimuli. Phys. Chem. Chem. Phys. 2015, 17, 16412–16417. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.X.; Zhang, C.S.; Ning, J.X. Genetic Algorithm with a Crossover Elitist Preservation Mechanism for Protein-Ligand Docking. AMB Express 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.X.; Zhang, C.S.; Ning, J.X. EDGA: A Population Evolution Direction Guided Genetic Algorithm for Protein-Ligand Docking. J. Comput. Biol. 2016, 23, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Yuen, S.Y.; Chow, C.K. A genetic algorithm that adaptively mutates and never revisits. IEEE. Trans. Evol. Comput. 2009, 13, 454–472. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decomez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alvarez, A.; Costa, A.M.; Vilarrasa, J. The Performance of Several Docking Programs at Reproducing Protein-Macrolide-Like Crystal Structures. Molecules 2017, 22, 136. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Fang, X.L.; Lu, Y.P.; Yang, C.Y.; Wang, S.M. The PDBbind database: Methodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.G.; Kwoh, C.K. Protein-Ligand Blind Docking Using QuickVina-W with Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithm | Success Case | Average RMSD (All Cases) | Average RMSD (RMSD < 2 Å) |
|---|---|---|---|
| HIGA | 90 | 1.81 | 1.32 |
| GA | 58 | 3.12 | 1.80 |
| LGA | 68 | 2.55 | 1.69 |
| EDGA | 77 | 2.21 | 1.62 |
| CEPGA | 81 | 2.13 | 1.59 |
| SODOCK | 73 | 2.88 | 1.79 |
| ABC | 62 | 3.25 | 1.83 |
| HIGA | GA | LGA | EDGA | CEPGA | SODOCK | ABC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB | Ligand | Torsions | Energy | RMSD | Energy | RMSD | Energy | RMSD | Energy | RMSD | Energy | RMSD | Energy | RMSD | Energy | RMSD |
| 3ptb | ben | 0 | −12.22 | 1.95 | −10.31 | 1.66 | −11.53 | 1.92 | −12.18 | 1.95 | −11.72 | 1.90 | −11.57 | 2.00 | −10.95 | 1.97 |
| 1aha | ade | 1 | −16.15 | 0.90 | −15.26 | 1.20 | −16.10 | 0.45 | −16.23 | 0.38 | −15.32 | 0.89 | −14.95 | 1.44 | −13.95 | 1.85 |
| 3hvt | nvp | 2 | −18.19 | 0.45 | −15.78 | 0.40 | −17.22 | 0.33 | −17.52 | 0.53 | −17.90 | 0.30 | −16.78 | 0.58 | −15.95 | 0.68 |
| 1phg | hem | 3 | −9.58 | 0.60 | −7.48 | 1.25 | −8.56 | 0.80 | −9.51 | 0.70 | −9.32 | 0.64 | −9.15 | 1.34 | −7.95 | 1.67 |
| 2mcp | pc | 4 | −9.35 | 1.15 | −7.76 | 1.46 | −8.22 | 1.33 | −9.25 | 1.23 | −9.10 | 1.20 | −7.72 | 1.42 | −7.85 | 1.64 |
| 1stp | btn | 5 | −13.90 | 0.85 | −11.30 | 1.84 | −13.37 | 1.65 | −13.67 | 1.25 | −13.57 | 0.90 | −13.52 | 1.00 | −13.20 | 1.58 |
| 6rnt | ca | 6 | −9.62 | 0.50 | −8.78 | 0.65 | −9.13 | 0.70 | −9.23 | 0.85 | −9.32 | 0.58 | −9.12 | 1.95 | −8.95 | 1.45 |
| 4dfr | mtx | 7 | −12.82 | 1.56 | −10.13 | 0.90 | −11.44 | 1.23 | −12.74 | 1.59 | −12.12 | 1.90 | −11.74 | 1.67 | −10.21 | 1.97 |
| 1ett | 4qq | 8 | −13.94 | 1.40 | −11.42 | 1.62 | −13.89 | 1.38 | −12.79 | 1.20 | −14.21 | 1.29 | −12.08 | 1.54 | −12.75 | 1.65 |
| 1hri | s57 | 9 | −11.02 | 1.18 | −9.67 | 1.80 | −10.21 | 1.87 | −10.33 | 1.37 | −10.89 | 1.38 | −10.31 | 1.68 | −10.13 | 1.67 |
| 1hvr | xk2 | 10 | −31.50 | 0.80 | −22.35 | 1.08 | −30.85 | 0.62 | −28.10 | 0.75 | −31.06 | 0.64 | −29.29 | 0.68 | −28.65 | 0.85 |
| 4hmg | sia | 11 | −10.21 | 1.65 | −8.50 | 1.68 | −10.09 | 1.70 | −10.43 | 1.58 | −10.32 | 1.89 | −10.08 | 1.36 | −9.95 | 1.60 |
| 1cdg | mal | 12 | −8.90 | 1.65 | −7.32 | 1.69 | −8.22 | 1.94 | −8.11 | 1.54 | −8.73 | 1.48 | −8.45 | 1.80 | −7.13 | 1.12 |
| 1htf | g26 | 13 | −21.17 | 1.20 | −19.26 | 1.58 | −20.69 | 1.33 | −20.89 | 1.30 | −21.48 | 1.27 | −21.79 | 1.42 | −19.80 | 1.80 |
| 1glq | gtb | 14 | −9.65 | 1.25 | −8.73 | 1.27 | −9.27 | 1.87 | −9.37 | 1.60 | −9.46 | 1.38 | −8.83 | 1.90 | −9.23 | 1.58 |
| 1tmn | nas | 15 | −10.71 | 0.95 | −9.68 | 1.11 | −10.11 | 1.20 | −10.07 | 1.45 | −10.29 | 0.85 | −10.62 | 1.95 | −9.58 | 0.65 |
| Algorithm | HIGA | GA | LGA | EDGA | CEPGA | SODOCK | ABC |
|---|---|---|---|---|---|---|---|
| Number of clusters | 3.72 | 14.00 | 4.58 | 4.24 | 4.04 | 10.30 | 13.04 |
| Number in rank 1 | 17.04 | 8.00 | 15.72 | 15.92 | 16.20 | 11.82 | 8.70 |
| PDB | Torsions | HIGA | GA | LGA | EDGA | CEPGA | SODOCK | ABC |
|---|---|---|---|---|---|---|---|---|
| 3ptb | 0 | 1.79 | 1.56 | 1.74 | 1.77 | 1.75 | 1.89 | 1.90 |
| 1aha | 1 | 1.91 | 1.62 | 1.80 | 1.83 | 1.90 | 1.95 | 2.08 |
| 3hvt | 2 | 1.97 | 1.75 | 1.93 | 1.95 | 1.96 | 1.94 | 2.13 |
| 1phg | 3 | 2.75 | 2.21 | 2.55 | 2.61 | 2.70 | 2.23 | 2.65 |
| 2mcp | 4 | 2.72 | 2.35 | 2.62 | 2.70 | 2.68 | 2.51 | 2.69 |
| 1stp | 5 | 3.79 | 3.01 | 3.53 | 3.77 | 3.59 | 3.84 | 3.81 |
| 6rnt | 6 | 3.84 | 3.12 | 3.14 | 3.70 | 3.54 | 4.27 | 3.64 |
| 4dfr | 7 | 5.47 | 4.98 | 5.18 | 5.33 | 5.45 | 4.99 | 6.06 |
| 1ett | 8 | 8.44 | 7.83 | 8.17 | 8.28 | 8.37 | 8.29 | 7.97 |
| 1hri | 9 | 10.49 | 10.15 | 10.37 | 10.39 | 10.41 | 12.34 | 11.06 |
| 1hvr | 10 | 12.53 | 12.07 | 12.13 | 12.28 | 12.51 | 14.86 | 11.92 |
| 4hmg | 11 | 13.90 | 13.21 | 13.84 | 13.95 | 13.89 | 16.09 | 13.60 |
| 1cdg | 12 | 12.71 | 12.38 | 12.59 | 12.61 | 12.66 | 15.92 | 13.06 |
| 1htf | 13 | 13.01 | 12.49 | 12.78 | 12.99 | 12.84 | 16.26 | 13.18 |
| 1glq | 14 | 15.87 | 14.95 | 15.50 | 15.77 | 15.85 | 20.12 | 17.22 |
| Average | 7.41 | 6.91 | 7.19 | 7.33 | 7.34 | 8.5 | 7.53 |
| PDB | HIGA | GA | LGA | EDGA | CEPGA | SODOCK | ABC | |
|---|---|---|---|---|---|---|---|---|
| 3ptb | HIGA | - | 0.004 | 0.010 | 0.044 | 0.035 | 0.012 | 0.008 |
| GA | 0.996 | - | 0.992 | 0.995 | 0.994 | 0.993 | 0.688 | |
| LGA | 0.990 | 0.008 | - | 0.986 | 0.982 | 0.563 | 0.425 | |
| EDGA | 0.956 | 0.005 | 0.014 | - | 0.340 | 0.017 | 0.010 | |
| CEPGA | 0.965 | 0.006 | 0.018 | 0.660 | - | 0.028 | 0.011 | |
| SODOCK | 0.988 | 0.007 | 0.437 | 0.983 | 0.972 | - | 0.306 | |
| ABC | 0.992 | 0.312 | 0.575 | 0.990 | 0.989 | 0.694 | - | |
| 1aha | HIGA | - | 0.029 | 0.062 | 0.524 | 0.044 | 0.024 | 0.017 |
| GA | 0.971 | - | 0.964 | 0.988 | 0.905 | 0.342 | 0.260 | |
| LGA | 0.938 | 0.036 | - | 0.981 | 0.460 | 0.030 | 0.024 | |
| EDGA | 0.476 | 0.012 | 0.019 | - | 0.017 | 0.010 | 0.005 | |
| CEPGA | 0.956 | 0.095 | 0.540 | 0.983 | - | 0.038 | 0.027 | |
| SODOCK | 0.976 | 0.658 | 0.970 | 0.990 | 0.962 | - | 0.470 | |
| ABC | 0.983 | 0.740 | 0.976 | 0.995 | 0.973 | 0.530 | - | |
| 3hvt | HIGA | - | 0.005 | 0.017 | 0.020 | 0.023 | 0.012 | 0.008 |
| GA | 0.995 | - | 0.795 | 0.974 | 0.993 | 0.788 | 0.537 | |
| LGA | 0.983 | 0.205 | - | 0.878 | 0.965 | 0.324 | 0.208 | |
| EDGA | 0.980 | 0.026 | 0.122 | - | 0.515 | 0.103 | 0.033 | |
| CEPGA | 0.977 | 0.007 | 0.035 | 0.485 | - | 0.029 | 0.011 | |
| SODOCK | 0.988 | 0.212 | 0.676 | 0.897 | 0.971 | - | 0.215 | |
| ABC | 0.992 | 0.463 | 0.792 | 0.967 | 0.989 | 0.785 | - | |
| 1phg | HIGA | - | 0.006 | 0.015 | 0.242 | 0.046 | 0.023 | 0.010 |
| GA | 0.994 | - | 0.682 | 0.991 | 0.983 | 0.968 | 0.545 | |
| LGA | 0.985 | 0.318 | - | 0.976 | 0.962 | 0.620 | 0.422 | |
| EDGA | 0.758 | 0.009 | 0.024 | - | 0.440 | 0.038 | 0.015 | |
| CEPGA | 0.954 | 0.017 | 0.038 | 0.560 | - | 0.045 | 0.028 | |
| SODOCK | 0.977 | 0.032 | 0.380 | 0.962 | 0.955 | - | 0.240 | |
| ABC | 0.990 | 0.455 | 0.578 | 0.985 | 0.972 | 0.760 | - | |
| 2mcp | HIGA | - | 0.002 | 0.008 | 0.036 | 0.015 | 0.001 | 0.004 |
| GA | 0.998 | - | 0.792 | 0.994 | 0.992 | 0.492 | 0.610 | |
| LGA | 0.992 | 0.208 | - | 0.988 | 0.987 | 0.092 | 0.224 | |
| EDGA | 0.964 | 0.006 | 0.012 | - | 0.442 | 0.005 | 0.011 | |
| CEPGA | 0.985 | 0.008 | 0.013 | 0.558 | - | 0.007 | 0.012 | |
| SODOCK | 0.999 | 0.508 | 0.908 | 0.995 | 0.993 | - | 0.640 | |
| ABC | 0.996 | 0.390 | 0.776 | 0.989 | 0.988 | 0.360 | - | |
| 1stp | HIGA | - | 0.001 | 0.006 | 0.048 | 0.033 | 0.014 | 0.003 |
| GA | 0.999 | - | 0.791 | 0.964 | 0.952 | 0.892 | 0.587 | |
| LGA | 0.994 | 0.209 | - | 0.942 | 0.887 | 0.624 | 0.450 | |
| EDGA | 0.952 | 0.036 | 0.058 | - | 0.414 | 0.082 | 0.043 | |
| CEPGA | 0.967 | 0.048 | 0.113 | 0.586 | - | 0.208 | 0.062 | |
| SODOCK | 0.986 | 0.108 | 0.376 | 0.918 | 0.792 | - | 0.215 | |
| ABC | 0.997 | 0.413 | 0.550 | 0.957 | 0.938 | 0.785 | - | |
| 6rnt | HIGA | - | 0.005 | 0.020 | 0.022 | 0.038 | 0.015 | 0.007 |
| GA | 0.995 | - | 0.942 | 0.965 | 0.985 | 0.695 | 0.588 | |
| LGA | 0.980 | 0.058 | - | 0.792 | 0.888 | 0.368 | 0.127 | |
| EDGA | 0.978 | 0.035 | 0.208 | - | 0.504 | 0.059 | 0.040 | |
| CEPGA | 0.962 | 0.015 | 0.112 | 0.496 | - | 0.047 | 0.025 | |
| SODOCK | 0.985 | 0.305 | 0.632 | 0.941 | 0.953 | - | 0.404 | |
| ABC | 0.993 | 0.412 | 0.873 | 0.960 | 0.975 | 0.596 | - | |
| 4dfr | HIGA | - | 0.002 | 0.006 | 0.035 | 0.032 | 0.021 | 0.003 |
| GA | 0.998 | - | 0.961 | 0.993 | 0.985 | 0.972 | 0.587 | |
| LGA | 0.994 | 0.039 | - | 0.966 | 0.950 | 0.624 | 0.450 | |
| EDGA | 0.965 | 0.007 | 0.034 | - | 0.490 | 0.042 | 0.013 | |
| CEPGA | 0.968 | 0.015 | 0.050 | 0.510 | - | 0.060 | 0.016 | |
| SODOCK | 0.979 | 0.028 | 0.376 | 0.958 | 0.940 | - | 0.215 | |
| ABC | 0.997 | 0.413 | 0.550 | 0.987 | 0.984 | 0.785 | - | |
| 1ett | HIGA | - | 0.008 | 0.082 | 0.060 | 0.515 | 0.044 | 0.057 |
| GA | 0.992 | - | 0.986 | 0.925 | 0.998 | 0.562 | 0.637 | |
| LGA | 0.918 | 0.014 | - | 0.482 | 0.932 | 0.073 | 0.151 | |
| EDGA | 0.940 | 0.075 | 0.518 | - | 0.950 | 0.077 | 0.205 | |
| CEPGA | 0.485 | 0.002 | 0.068 | 0.050 | - | 0.040 | 0.045 | |
| SODOCK | 0.956 | 0.432 | 0.927 | 0.923 | 0.960 | - | 0.520 | |
| ABC | 0.943 | 0.363 | 0.849 | 0.795 | 0.955 | 0.480 | - | |
| 1hri | HIGA | - | 0.001 | 0.018 | 0.025 | 0.041 | 0.021 | 0.004 |
| GA | 0.999 | - | 0.976 | 0.997 | 0.998 | 0.982 | 0.635 | |
| LGA | 0.982 | 0.024 | - | 0.862 | 0.890 | 0.723 | 0.117 | |
| EDGA | 0.975 | 0.003 | 0.138 | - | 0.504 | 0.140 | 0.015 | |
| CEPGA | 0.959 | 0.002 | 0.110 | 0.406 | - | 0.130 | 0.010 | |
| SODOCK | 0.979 | 0.018 | 0.277 | 0.860 | 0.870 | - | 0.020 | |
| ABC | 0.996 | 0.365 | 0.883 | 0.985 | 0.990 | 0.980 | - | |
| 1hvr | HIGA | - | 0.002 | 0.030 | 0.012 | 0.045 | 0.021 | 0.014 |
| GA | 0.998 | - | 0.995 | 0.562 | 0.996 | 0.942 | 0.665 | |
| LGA | 0.970 | 0.005 | - | 0.026 | 0.957 | 0.177 | 0.044 | |
| EDGA | 0.988 | 0.438 | 0.974 | - | 0.982 | 0.711 | 0.609 | |
| CEPGA | 0.955 | 0.004 | 0.043 | 0.018 | - | 0.035 | 0.024 | |
| SODOCK | 0.979 | 0.058 | 0.823 | 0.289 | 0.965 | - | 0.368 | |
| ABC | 0.986 | 0.335 | 0.956 | 0.391 | 0.976 | 0.632 | - | |
| 4hmg | HIGA | - | 0.022 | 0.072 | 0.522 | 0.514 | 0.054 | 0.032 |
| GA | 0.978 | - | 0.972 | 0.995 | 0.985 | 0.928 | 0.900 | |
| LGA | 0.928 | 0.028 | - | 0.980 | 0.945 | 0.417 | 0.214 | |
| EDGA | 0.478 | 0.005 | 0.020 | - | 0.487 | 0.017 | 0.010 | |
| CEPGA | 0.486 | 0.015 | 0.045 | 0.513 | - | 0.042 | 0.026 | |
| SODOCK | 0.946 | 0.072 | 0.583 | 0.983 | 0.958 | - | 0.240 | |
| ABC | 0.968 | 0.100 | 0.786 | 0.990 | 0.974 | 0.760 | - | |
| 1cdg | HIGA | - | 0.003 | 0.021 | 0.014 | 0.037 | 0.032 | 0.001 |
| GA | 0.997 | - | 0.883 | 0.784 | 0.995 | 0.985 | 0.408 | |
| LGA | 0.979 | 0.117 | - | 0.483 | 0.952 | 0.763 | 0.105 | |
| EDGA | 0.986 | 0.216 | 0.517 | - | 0.983 | 0.844 | 0.125 | |
| CEPGA | 0.963 | 0.005 | 0.048 | 0.017 | - | 0.059 | 0.003 | |
| SODOCK | 0.968 | 0.015 | 0.237 | 0.156 | 0.941 | - | 0.012 | |
| ABC | 0.999 | 0.592 | 0.895 | 0.875 | 0.997 | 0.988 | - | |
| 1htf | HIGA | - | 0.015 | 0.243 | 0.480 | 0.544 | 0.624 | 0.030 |
| GA | 0.985 | - | 0.973 | 0.974 | 0.985 | 0.987 | 0.637 | |
| LGA | 0.753 | 0.027 | - | 0.652 | 0.759 | 0.883 | 0.251 | |
| EDGA | 0.520 | 0.016 | 0.348 | - | 0.618 | 0.640 | 0.235 | |
| CEPGA | 0.456 | 0.015 | 0.241 | 0.382 | - | 0.538 | 0.028 | |
| SODOCK | 0.376 | 0.013 | 0.127 | 0.360 | 0.462 | - | 0.017 | |
| ABC | 0.970 | 0.363 | 0.749 | 0.765 | 0.972 | 0.983 | - | |
| 1glq | HIGA | - | 0.001 | 0.012 | 0.018 | 0.022 | 0.002 | 0.005 |
| GA | 0.999 | - | 0.982 | 0.991 | 0.995 | 0.695 | 0.788 | |
| LGA | 0.988 | 0.018 | - | 0.955 | 0.965 | 0.163 | 0.227 | |
| EDGA | 0.982 | 0.009 | 0.045 | - | 0.510 | 0.039 | 0.042 | |
| CEPGA | 0.978 | 0.005 | 0.035 | 0.490 | - | 0.027 | 0.031 | |
| SODOCK | 0.998 | 0.305 | 0.837 | 0.961 | 0.973 | - | 0.704 | |
| ABC | 0.995 | 0.212 | 0.773 | 0.958 | 0.969 | 0.296 | - | |
| 1tmn | HIGA | - | 0.004 | 0.023 | 0.012 | 0.028 | 0.043 | 0.002 |
| GA | 0.996 | - | 0.783 | 0.692 | 0.965 | 0.983 | 0.408 | |
| LGA | 0.977 | 0.217 | - | 0.283 | 0.716 | 0.746 | 0.105 | |
| EDGA | 0.988 | 0.308 | 0.717 | - | 0.810 | 0.975 | 0.125 | |
| CEPGA | 0.972 | 0.035 | 0.284 | 0.190 | - | 0.644 | 0.028 | |
| SODOCK | 0.953 | 0.017 | 0.256 | 0.025 | 0.356 | - | 0.012 | |
| ABC | 0.998 | 0.592 | 0.895 | 0.875 | 0.972 | 0.988 | - |
| Algorithm: CE Crossover |
|---|
| Input: (1) a population with n indiviudals, (2) elitists e. |
| Output: a population after CE crossover |
| 01. For i: = 1 to n do |
| 02. Find the historial optimal individual m0 |
| 03. If the fitness of current individual mi < m0 then |
| 04. m0 = mi |
| 05. e = m0 |
| 06. preserve Mfather and Mmother |
| 07. End if |
| 08. Mfather, Mmother and e next population |
| 09. End for |
| Algorithm: ED Mutation |
|---|
| Input: (1) a population with n indiviudals, (2) balance factor β. |
| Output: a population after ED mutation |
| 01. For i: = 1 to n do |
| 02. Find the optimal solution Moptimum and the historic suboptimal solution Msub |
| 03. If θ < β then |
| 04. mi = mmin + θ (Mmax − Mmin) |
| 05. Else |
| 06. mi = mi + θ (Moptimum − Msub) +(Moptimum − mi) |
| 07. End if |
| 08. End for |
| Algorithm: BSP Tree |
|---|
| Input: (1) an individual m, (2) BSP tree T (3) revisit flag RF |
| Output: an individual m that never revisits |
| 01. Curr_node: = root node of T |
| 02. RF = 0 |
| 03. If (Curr_node has two child nodes: l and r) then |
| 04. Compare m with child node l and r |
| 05. If (m = l) or (m = r) then |
| 06. RF = 1 |
| 07. End if |
| 08. If d (l, m) < d (r, m) then |
| 09. Curr_node: = child node l |
| 10. Else |
| 11. Curr_node: = child node r |
| 12. End if |
| 13. Repeat steps 03-12 |
| 14. Else |
| 15. If (RF = 0) then |
| 16. Insert a child node to Curr_node that records m |
| 17. Else |
| 18. Creat a new child node by mutating |
| 19. End if |
| 20. End if |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, B.; Zhang, C.; Zhao, Y. HIGA: A Running History Information Guided Genetic Algorithm for Protein–Ligand Docking. Molecules 2017, 22, 2233. https://doi.org/10.3390/molecules22122233
Guan B, Zhang C, Zhao Y. HIGA: A Running History Information Guided Genetic Algorithm for Protein–Ligand Docking. Molecules. 2017; 22(12):2233. https://doi.org/10.3390/molecules22122233
Chicago/Turabian StyleGuan, Boxin, Changsheng Zhang, and Yuhai Zhao. 2017. "HIGA: A Running History Information Guided Genetic Algorithm for Protein–Ligand Docking" Molecules 22, no. 12: 2233. https://doi.org/10.3390/molecules22122233
APA StyleGuan, B., Zhang, C., & Zhao, Y. (2017). HIGA: A Running History Information Guided Genetic Algorithm for Protein–Ligand Docking. Molecules, 22(12), 2233. https://doi.org/10.3390/molecules22122233