In Silico Identification and In Vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase I


 ,
,  and
and 
Abstract
:1. Introduction
2. Results
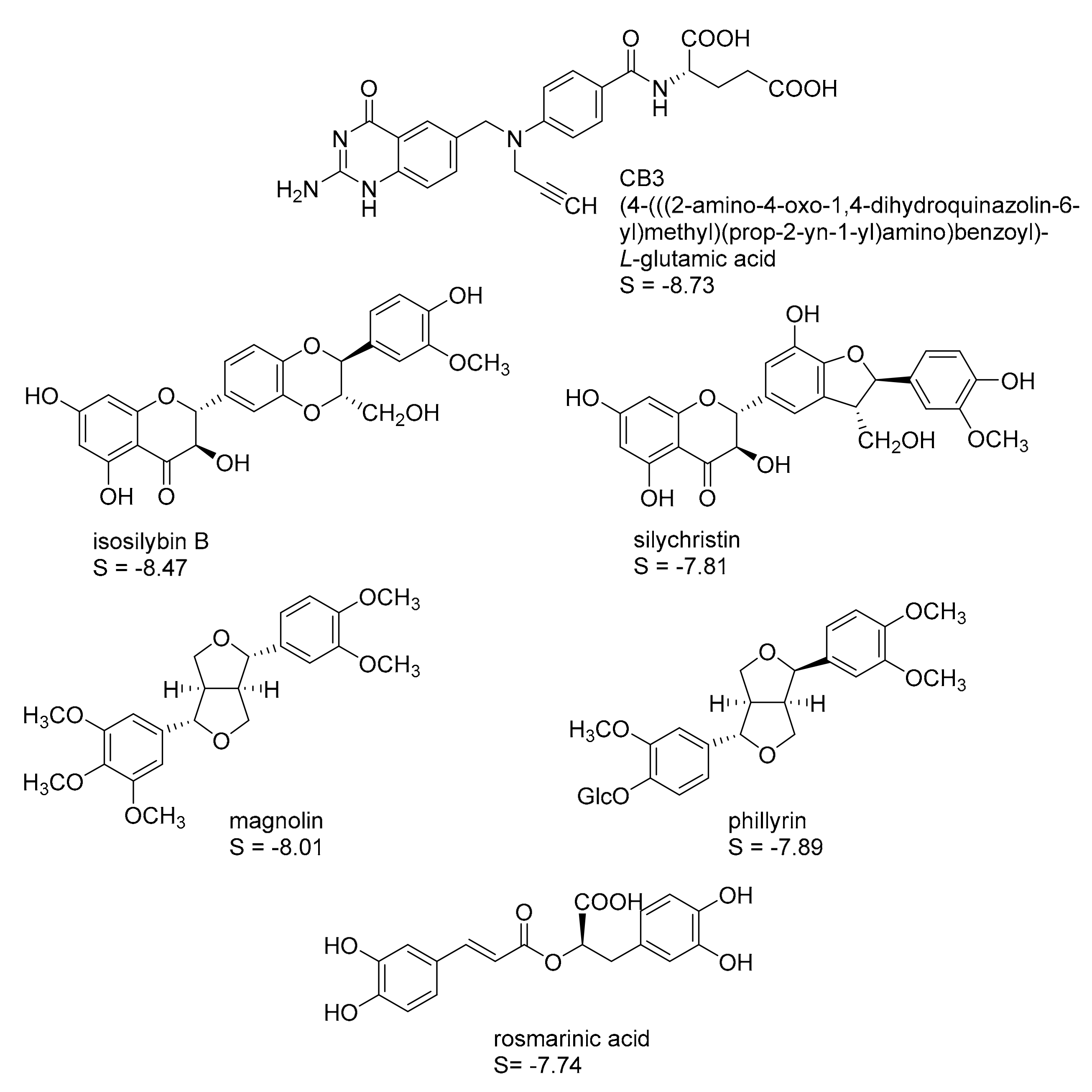
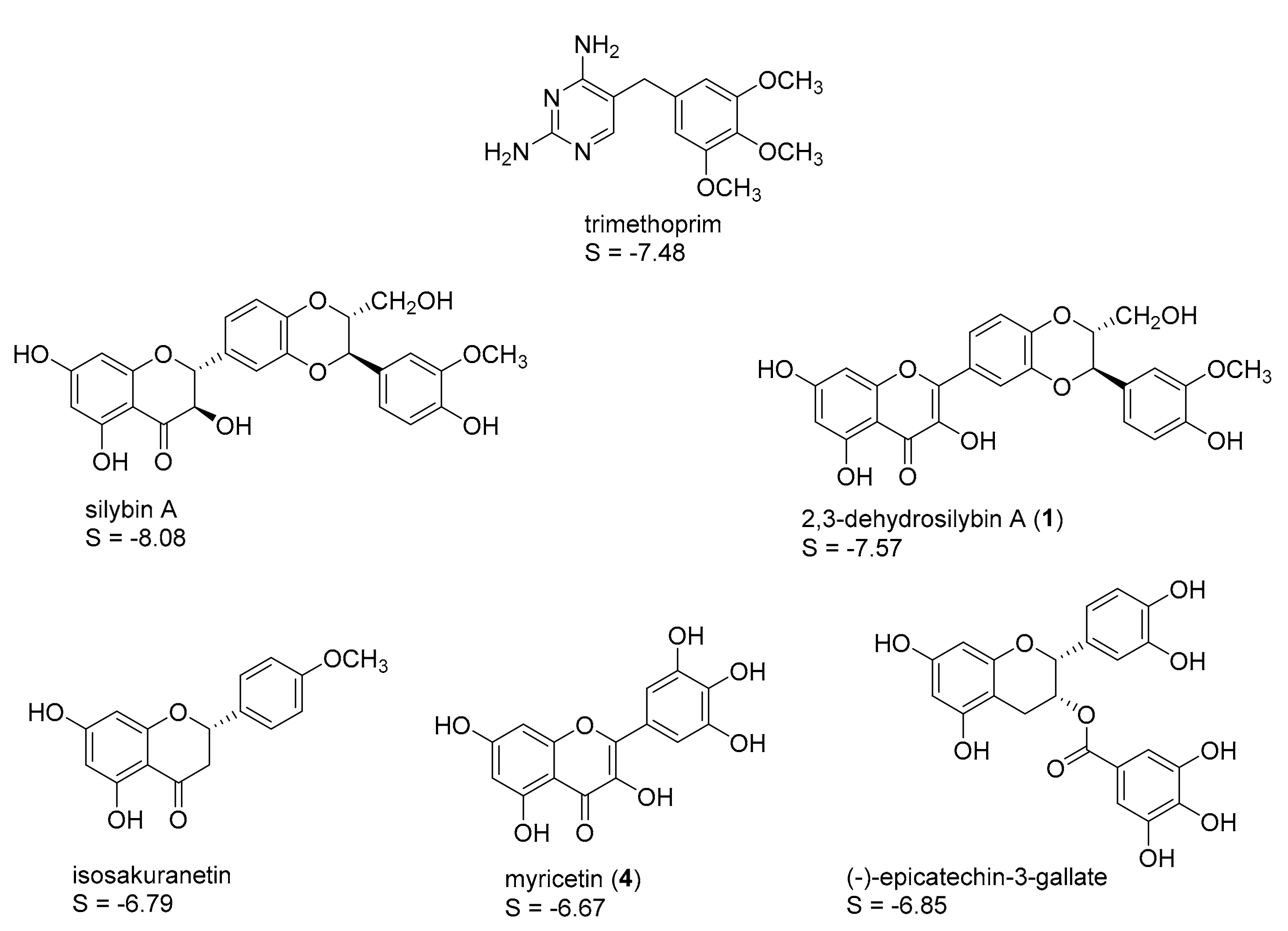
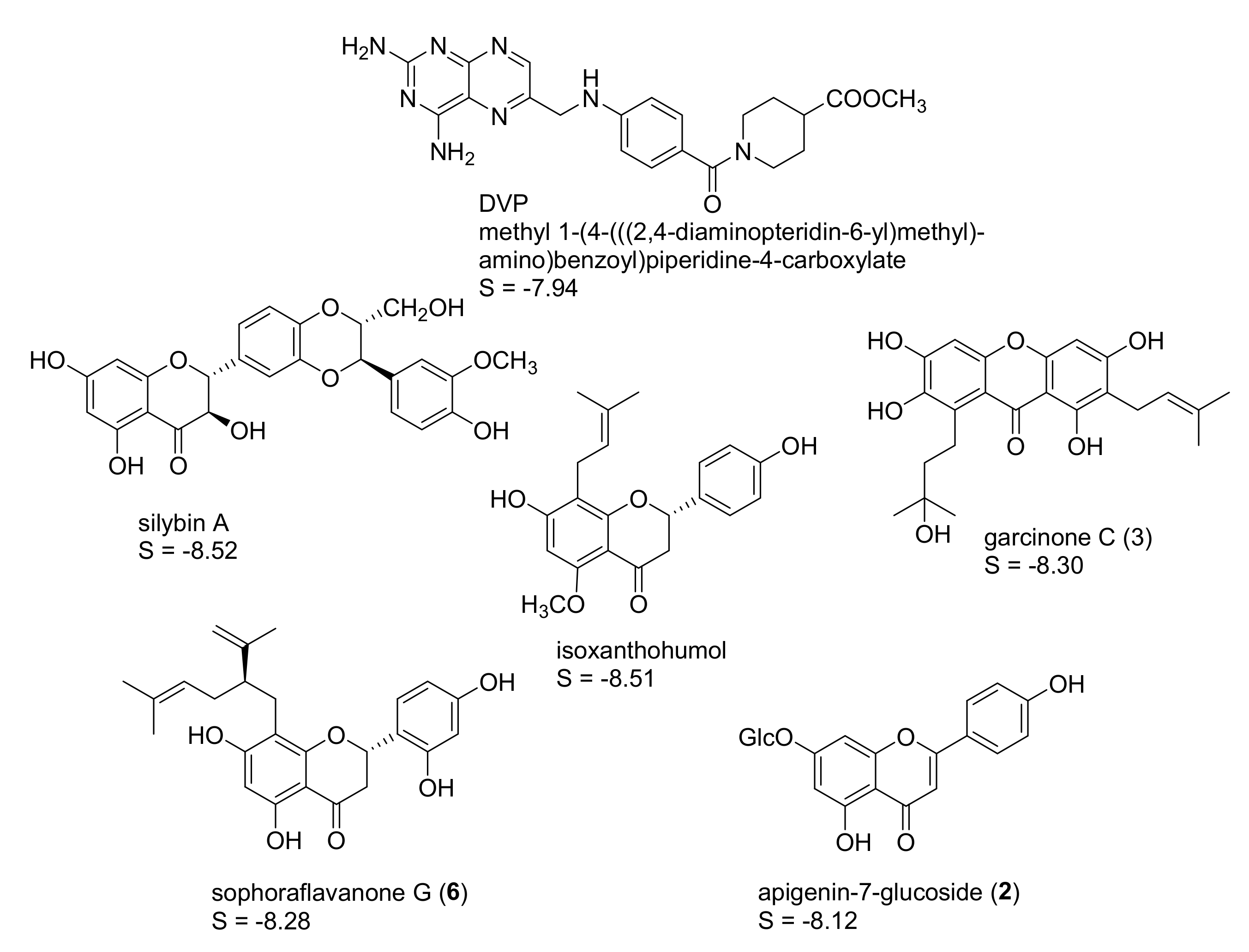
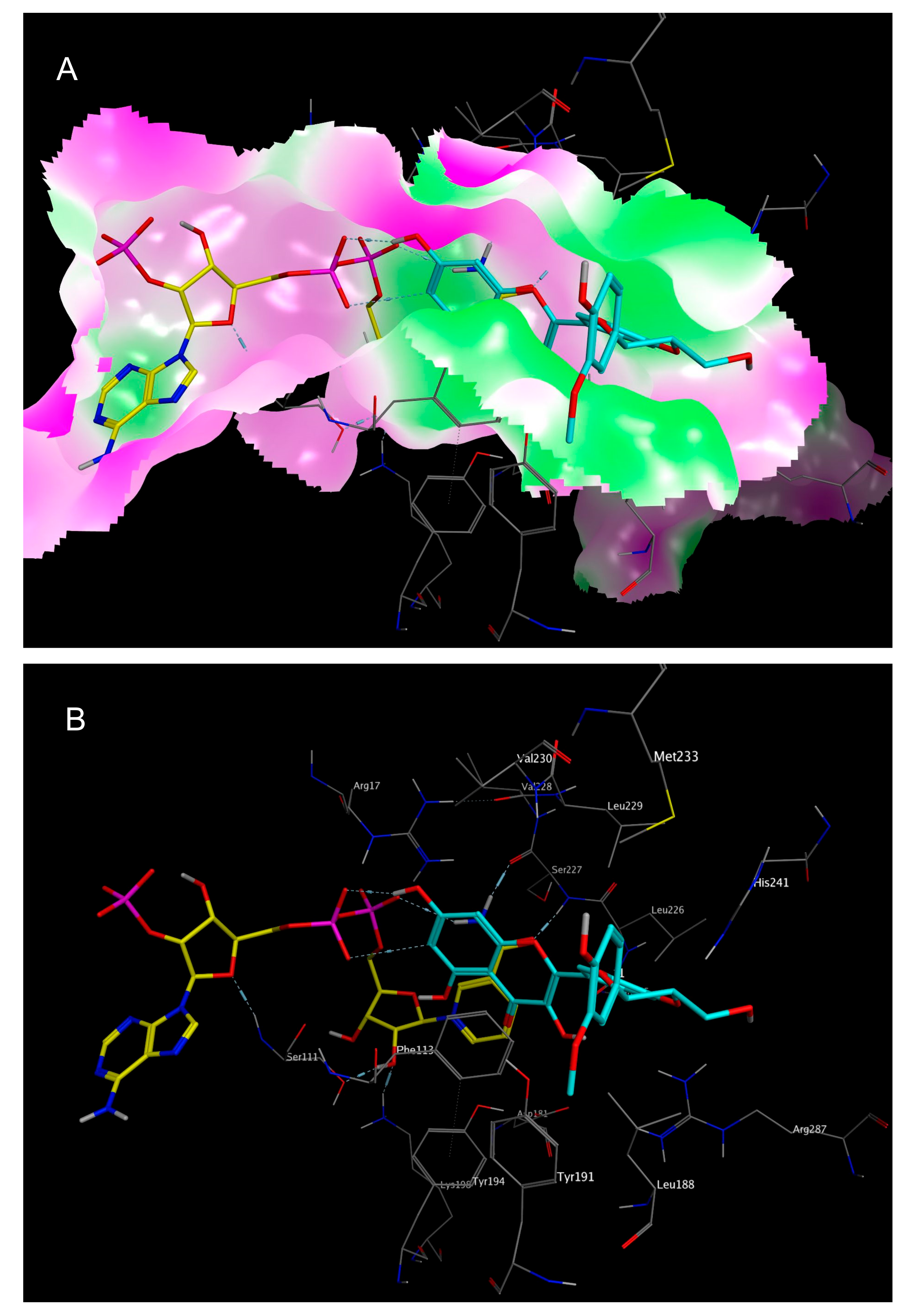
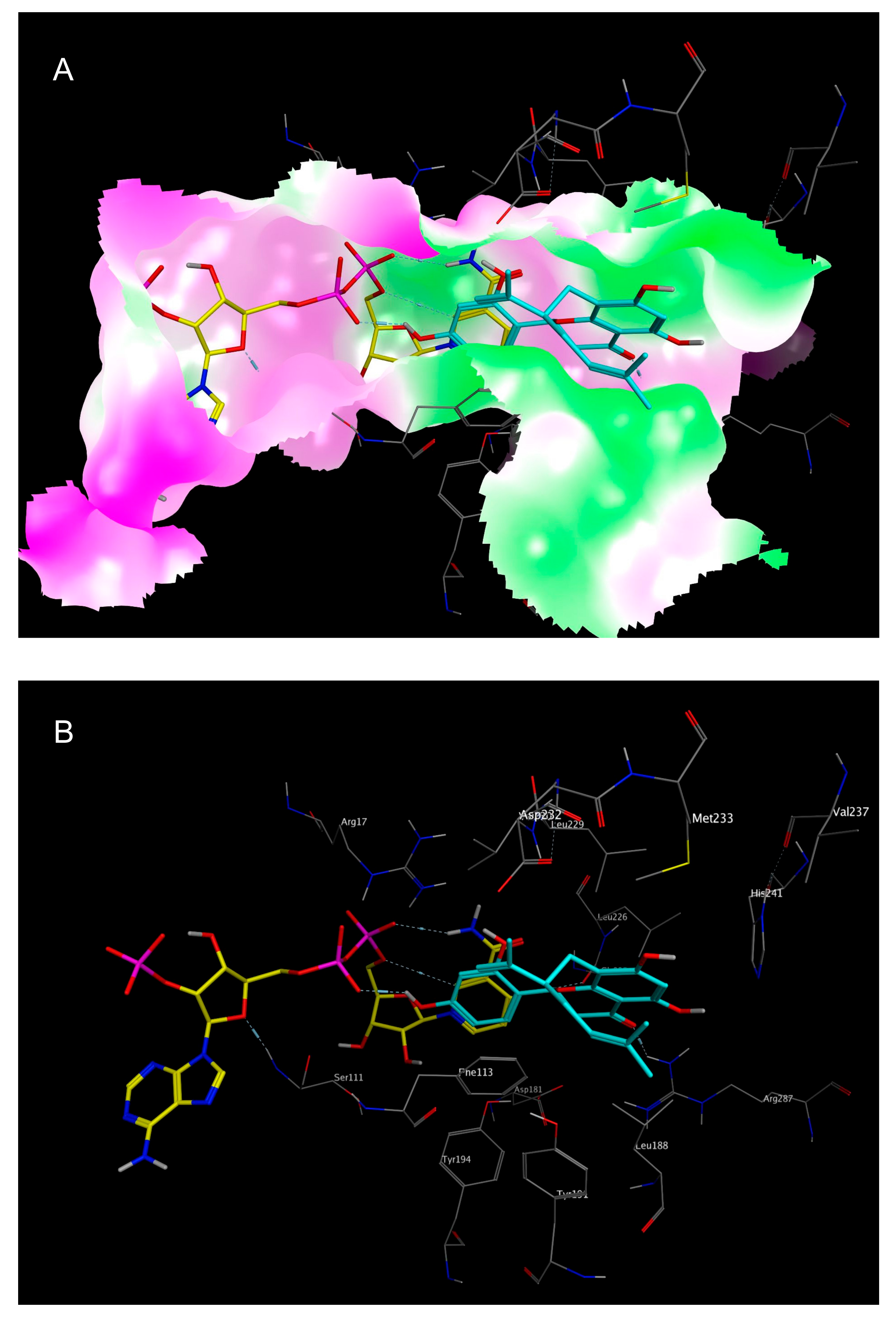
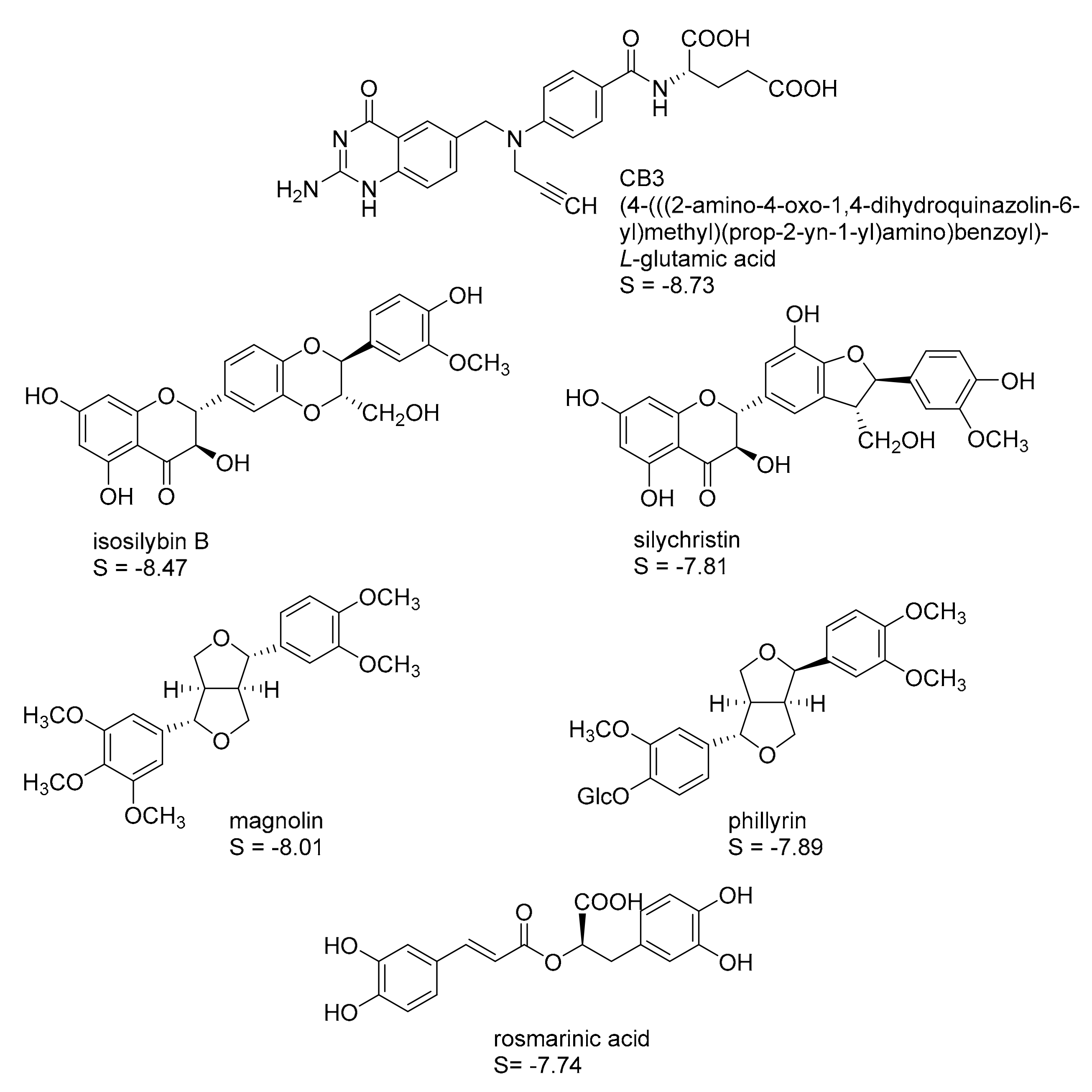
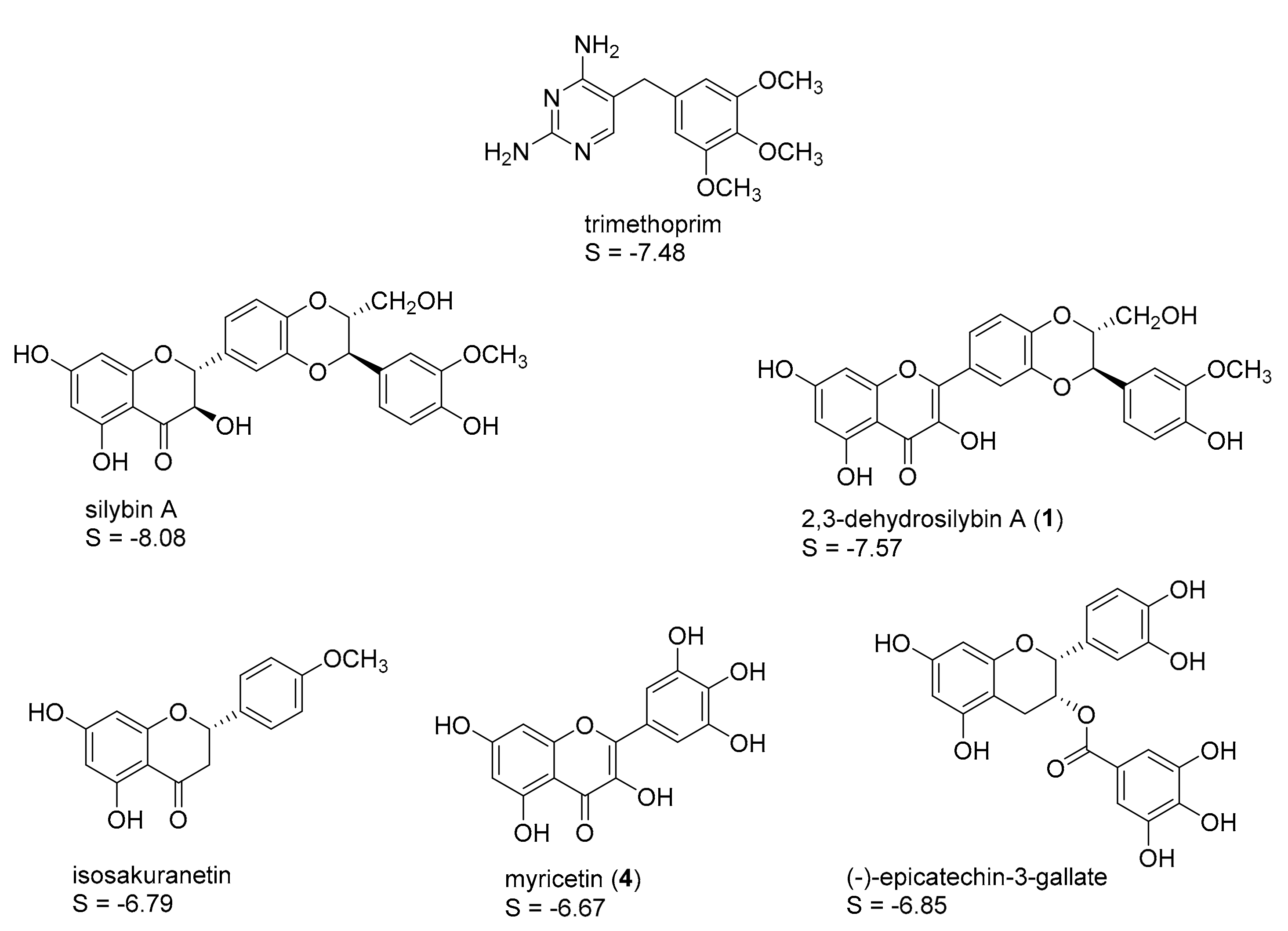
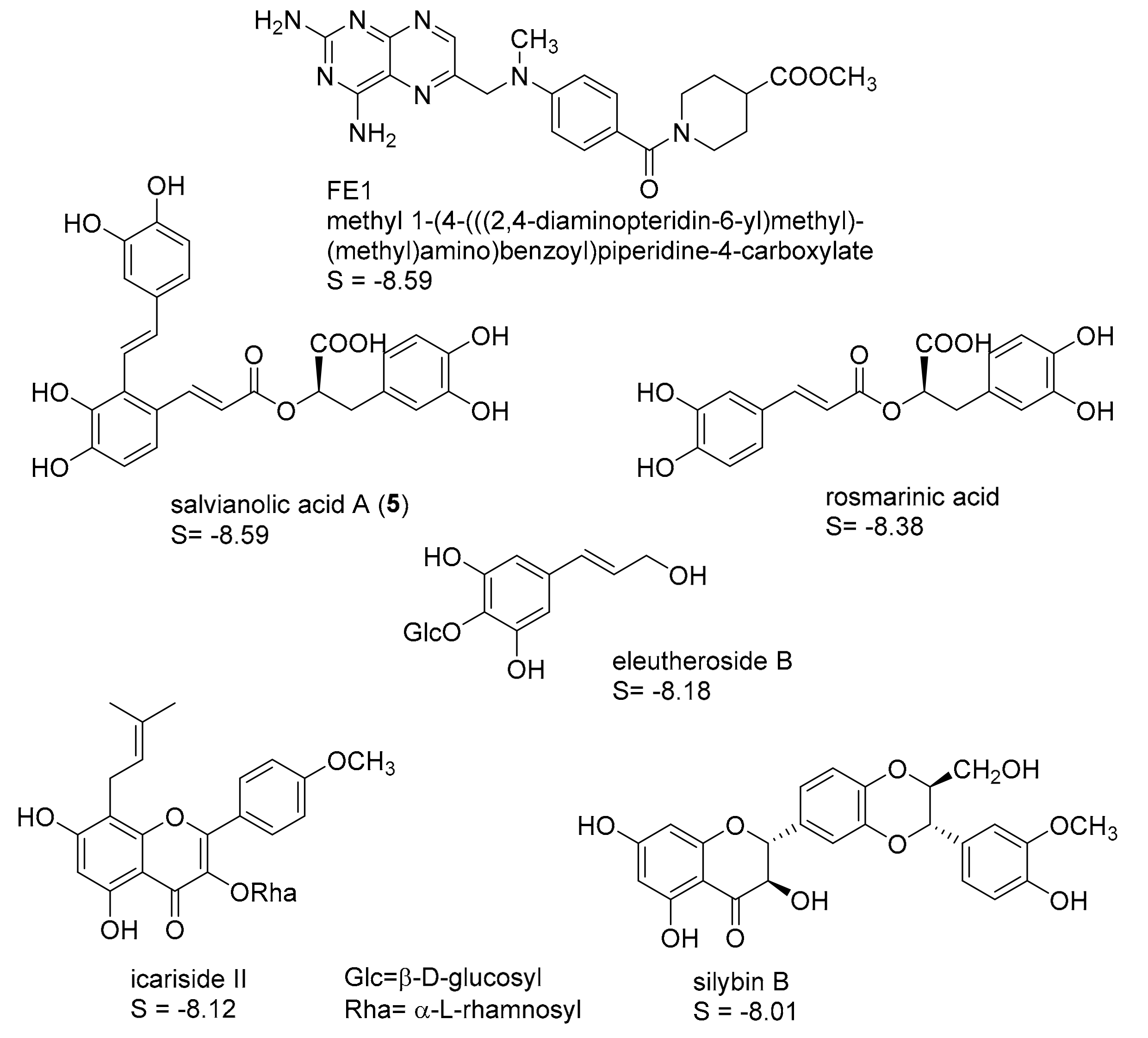
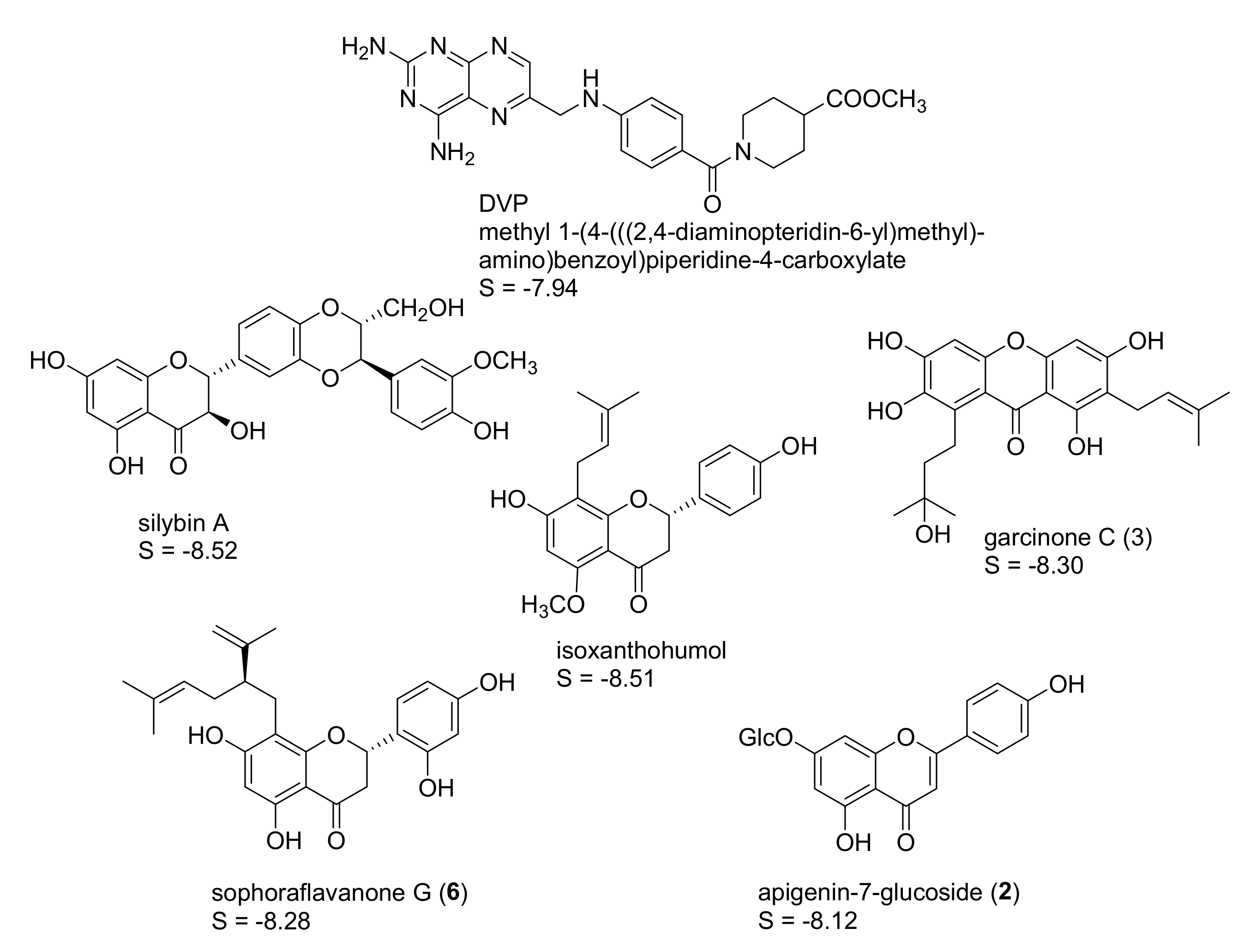
2.1. In Silico Identification of Natural Products as Potential LmPTR1 Inhibitors
2.2. In Vitro Evaluation of the Identified NPs against LmPTR1
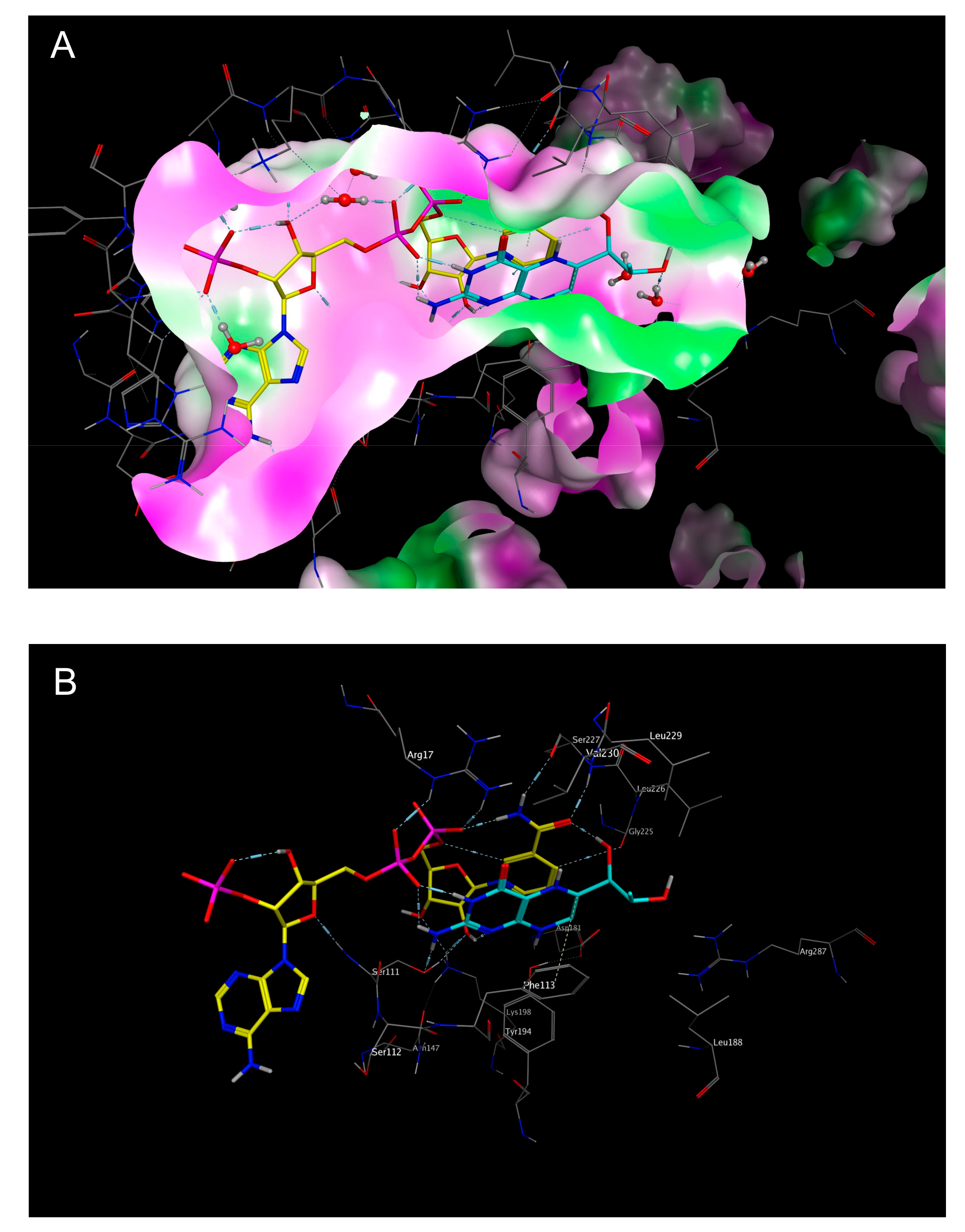
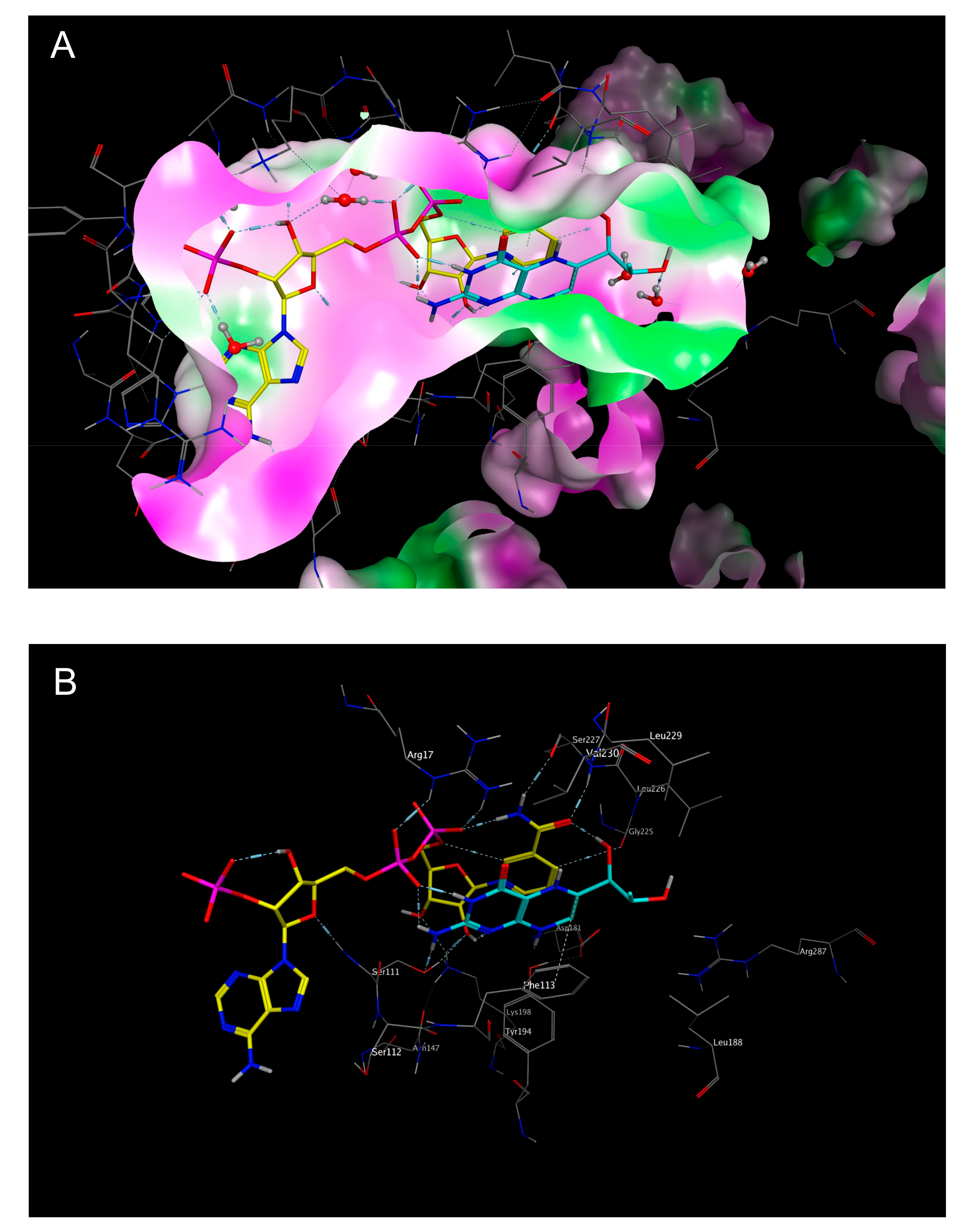
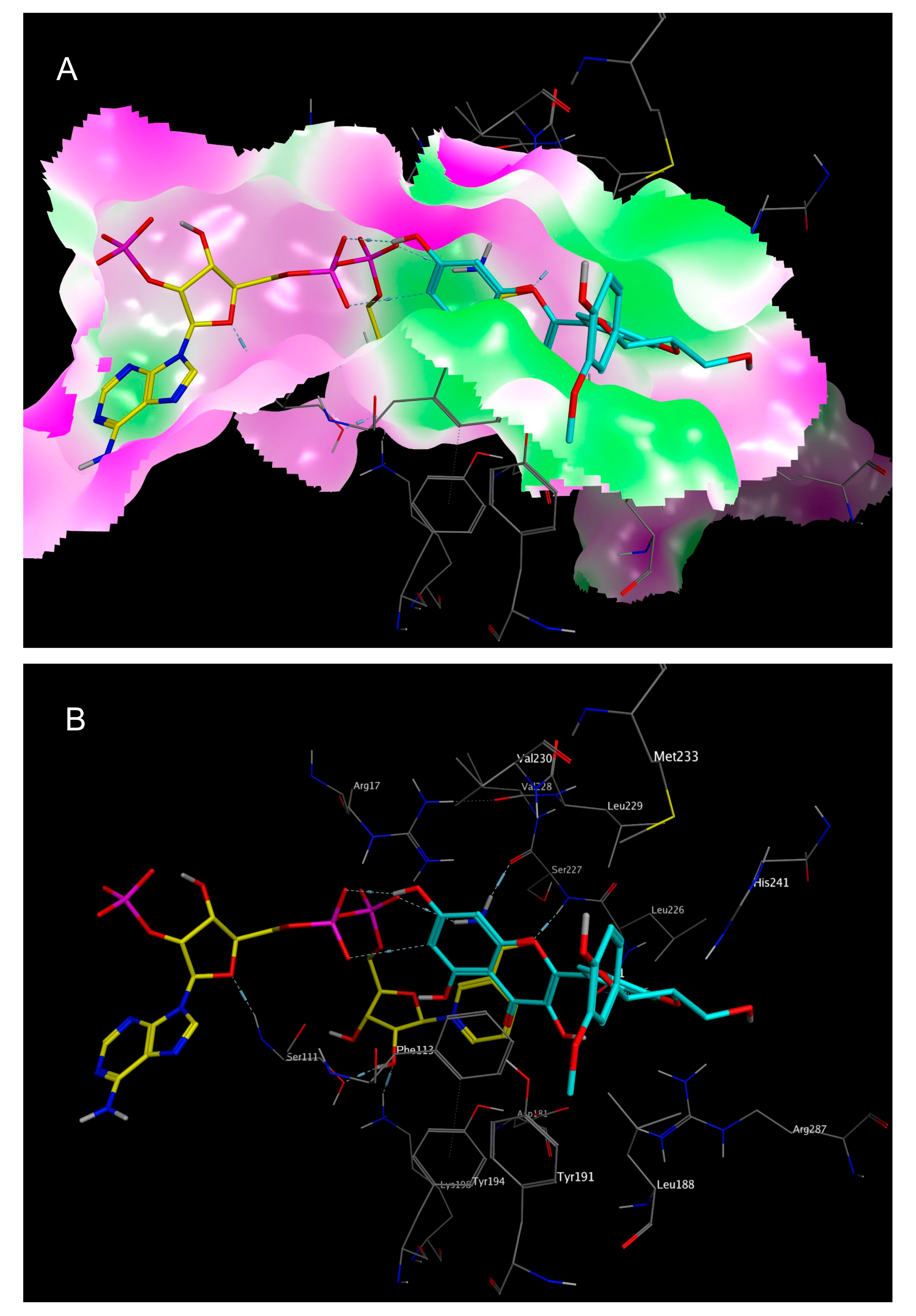
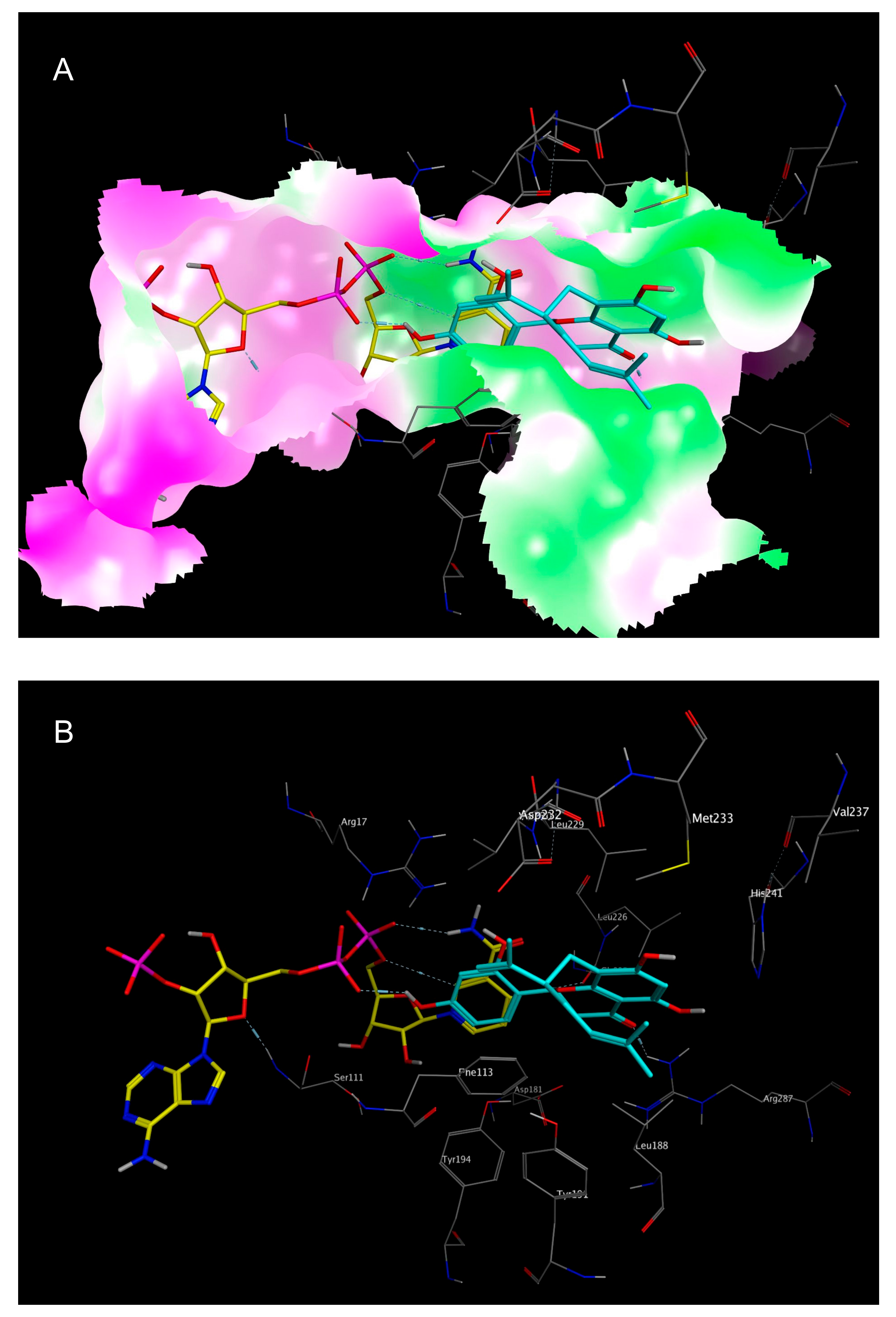
2.3. Mechanistic Considerations
3. Materials and Methods
3.1. In Silico Modelling
3.2. Database Design
3.3. Acquisition and Preparation of the Protein’s 3D-Structure
3.4. Pharmacophore Design and Virtual Screening
3.5. Docking Simulations
3.6. Tested Compounds
3.7. Transformation of E. coli Cells (BL21 (DE3)) with Vector System LmPTR1-pET-15b
3.8. Recombinant Expression and Purification of LmPTR1
3.9. LmPTR1-Inhibition Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Herrmann, F.C.; Schmidt, T.J. In silico screening of natural product databases reveals new potential leads against neglected diseases. Planta Med. 2013, 79, 1132. [Google Scholar] [CrossRef]
- Herrmann, F.C.; Lenz, M.; Jose, J.; Kaiser, M.; Brun, R.; Schmidt, T.J. In Silico identification and in vitro activity of novel natural inhibitors of Trypanosoma brucei glyceraldehyde-3-phosphate-dehydrogenase. Molecules 2015, 20, 16154–16169. [Google Scholar] [CrossRef] [PubMed]
- Nare, B.; Luba, J.; Hardy, L.W.; Beverley, S. New approaches to Leishmania chemotherapy: Pteridine reductase 1 (PTR1) as target modulator of antifolate sensitivity. Parasitology 1997, 114, 101–110. [Google Scholar]
- WHO Weekly Epidemiological Record No. 22. Available online: http://www.who.int/wer/2016/wer9122.pdf?ua=1 (accessed on 2 August 2017).
- WHO Fact Sheet Leishmaniasis. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 2 August 2017).
- Kurban, A.K.; Malak, J.A.; Farah, F.S.; Chaglassian, H.T. Histopathology of cutaneous Leishmaniasis. Arch. Dermatol. 1966, 93, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Thomaz-Soccol, V.; Ferreira da Costa, E.S.; Karp, S.G.; Letti, L.A.J.; Soccol, C.R.; Thomaz, F. Recent advantages in vaccines against Leishmania based on patent applications. Recent Pat. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, P.; Kumar Gaur, D.; Jain, S. Recent development in identification of potential novel therapeutic targets against trypanosomatids. Curr. Top. Med. Chem. 2016, 16, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.J.; Beverley, S.M. Folate metabolism pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.; Drummelsmith, J.; El-Fadili, A.; Kündig, C.; Richard, D.; Roy, G. Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int. J. Parasitol. 2002, 32, 385–398. [Google Scholar] [CrossRef]
- Ravenhall, M.; Benavente, E.D.; Mipando, M.; Jensen, A.T.; Sutherland, C.J.; Roper, C.; Sepúlveda, N.; Kwiatkowski, D.P.; Montgomery, J.; Phiri, K.S.; et al. Characterizing the impact of sustained sulfadoxine/pyrimethamine use upon Plasmodium falciparum population in Malawi. Malar. J. 2016, 15, 575. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Morandi, F.; Motiejunas, D.; Nerini, E.; Henrich, S.; Luciani, R.; Venturelli, A.; Lazzari, S.; Caló, S.; Gupta, S.; et al. Virtual screening identification of nonfolate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J. Med. Chem. 2011, 54, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE) 2011.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2017.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 7 February 2016).
- Svobodová, A.; Zdarilová, A.; Walterová, D.; Vostálová, J. Flavonolignans from Silybum marianum moderate UVA-induced oxidative damage to HaCaT keratinocytes. J. Dermatol. Sci. 2007, 48, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Miquel, F.G.; Cavalheiro, A.H.; Spinola, N.F.; Ribeiro, D.L.; Barcelos, G.R.; Antunes, L.M.; Hori, J.I.; Marquele-Oliveira, F.; Rocha, B.A.; Beretta, A.A. Validation of a RP-HPLC-DAD method for Chamomile (Matricaria recutita) preparations and assessment of the marker, apigenin-7-glucoside, safety and anti-inflammatory effect. Evid. Based Complement. Altern. Med. 2015, 2015, 828437. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Qiu, S.X.; Xu, Y.; Wu, J.; Chen, Y.; Yu, Y.; Xiao, G. A new xanthone from the pericarp of Garcinia mangostana. Nat. Prod. Commun. 2013, 8, 1733–1734. [Google Scholar] [PubMed]
- Lima, M.R.; Felgueiras, M.L.; Cunha, A.; Chicau, G.; Ferreres, F.; Dias, A.C. Differential phenolic production in leaves of Vitis vinifera cv. Alvarinho affected with esca disease. Plant Physiol. Biochem. 2017, 112, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Chen, Y.; Zhang, H.; Fang, L.; Du, G. Salvianolic acid A, a component of Salvia miltiorrhiza, attenuates endothelial-mesenchymal transition of HPAECs induced by hypoxia. Am. J. Chin. Med. 2017, 45, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.D.; Jeong, M.R.; Jeong, S.I.; Lee, K.Y. Antibacterial activity of sophoraflavanone G isolated from the roots of Sophora flavescens. J. Microbiol. Biotechnol. 2007, 17, 858–864. [Google Scholar] [PubMed]
- Borsari, C.; Luciani, R.; Pozzi, C.; Poehner, I.; Henrich, S.; Trande, M.; Cordeiro-da-Silva, A.; Santarem, N.; Baptista, C.; Tait, A.; et al. Profiling of flavonol derivatives for the development of antitrypanosomatidic drugs. J. Med. Chem. 2016, 59, 7598–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüttelkopf, A.W.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005, 352, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 490–519. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Inhibition at c = 50 µM | IC50 (µM) |
|---|---|---|
| (−)-epicatechin-3-gallate | 24 | |
| 2,3-dehydrosilybin A (1) | 66 | 7.3 (5.6–9.5) (EC50) |
| apigenin-7-glucoside (2) | 60 | 43.5 (39.1–48.2) |
| eleutheroside B | 12 | |
| garcinone C (3) | 74 | 26.3 (21.0–33.0) |
| icariside II | 35 | |
| isosakuranetin | n.i. | |
| isosilybin B | 26 | |
| isoxanthohumol | 22 | |
| magnolin | n.i. | |
| myricetin (4) | 68 | 21.0 (18.3–24.2) |
| phillyrin | n.i. | |
| rosmarinic acid | 20 | |
| salvianolic acid A (5) | 67 | 42.2 (38.3–46.6) |
| silybin A | 14 | |
| silybin B | 19 | |
| silychristin | 24 | |
| sophoraflavanone G (6) | 90 | 19.2 (17.1–21.6) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrmann, F.C.; Sivakumar, N.; Jose, J.; Costi, M.P.; Pozzi, C.; Schmidt, T.J. In Silico Identification and In Vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase I. Molecules 2017, 22, 2166. https://doi.org/10.3390/molecules22122166
Herrmann FC, Sivakumar N, Jose J, Costi MP, Pozzi C, Schmidt TJ. In Silico Identification and In Vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase I. Molecules. 2017; 22(12):2166. https://doi.org/10.3390/molecules22122166
Chicago/Turabian StyleHerrmann, Fabian C., Nirina Sivakumar, Joachim Jose, Maria P. Costi, Cecilia Pozzi, and Thomas J. Schmidt. 2017. "In Silico Identification and In Vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase I" Molecules 22, no. 12: 2166. https://doi.org/10.3390/molecules22122166
APA StyleHerrmann, F. C., Sivakumar, N., Jose, J., Costi, M. P., Pozzi, C., & Schmidt, T. J. (2017). In Silico Identification and In Vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase I. Molecules, 22(12), 2166. https://doi.org/10.3390/molecules22122166