Synthesis of Spironucleosides: Past and Future Perspectives



Abstract
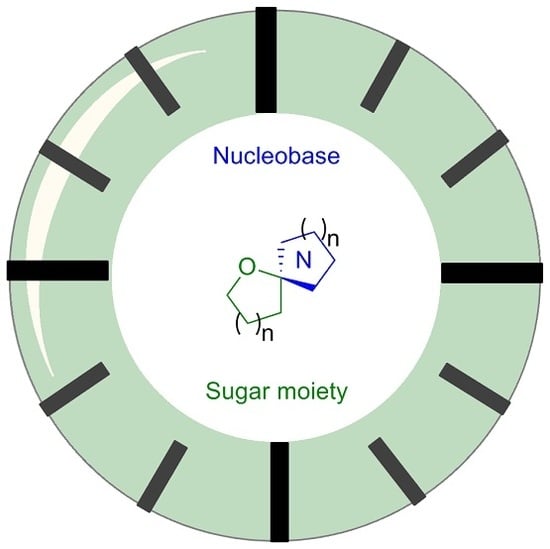
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
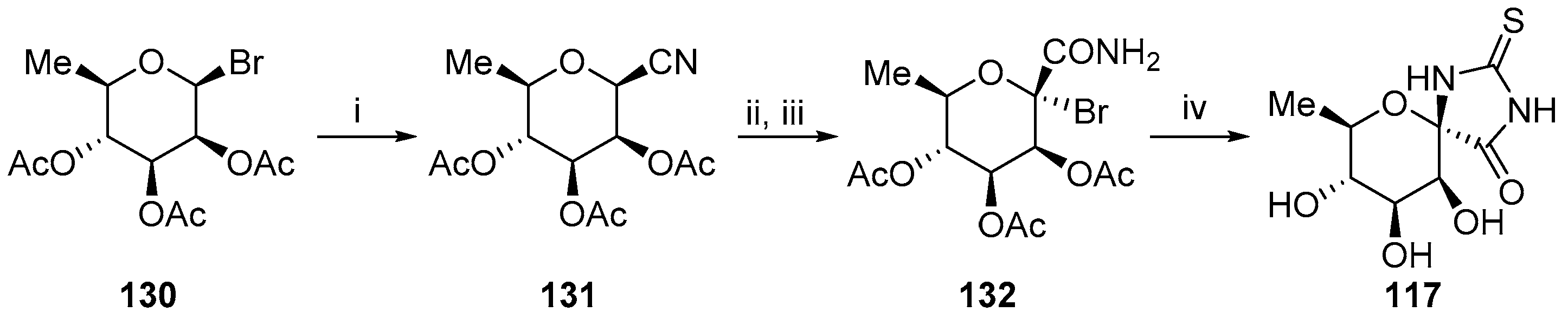{kind=link}
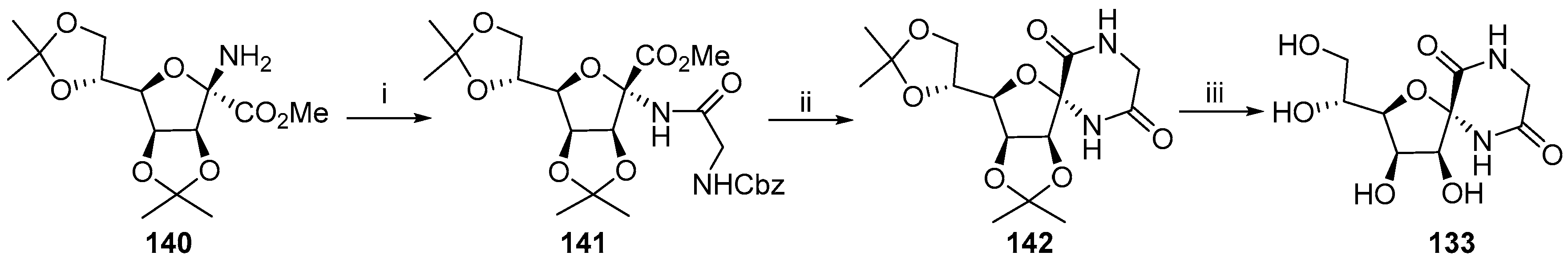{kind=link}
{kind=link}
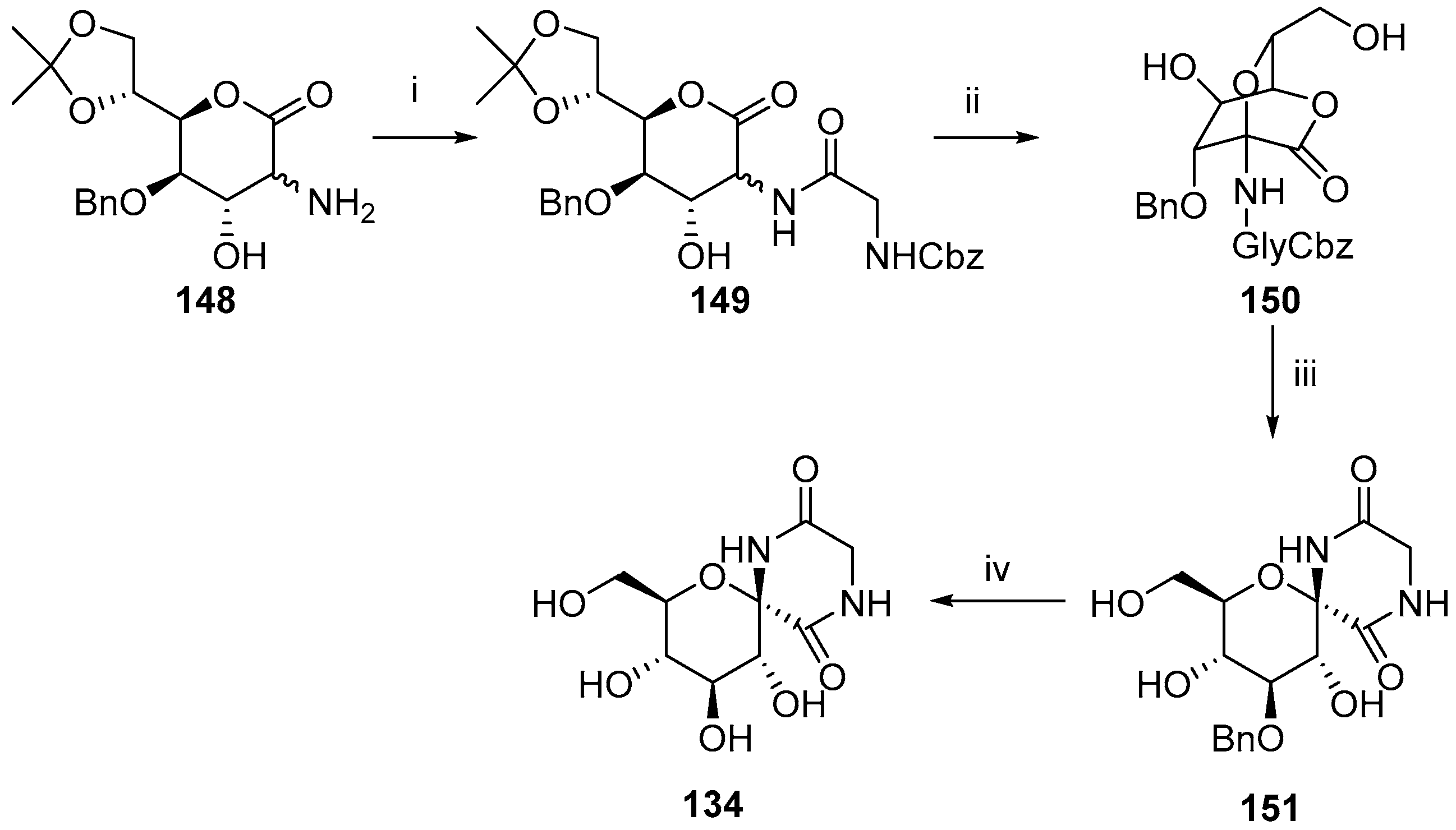{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
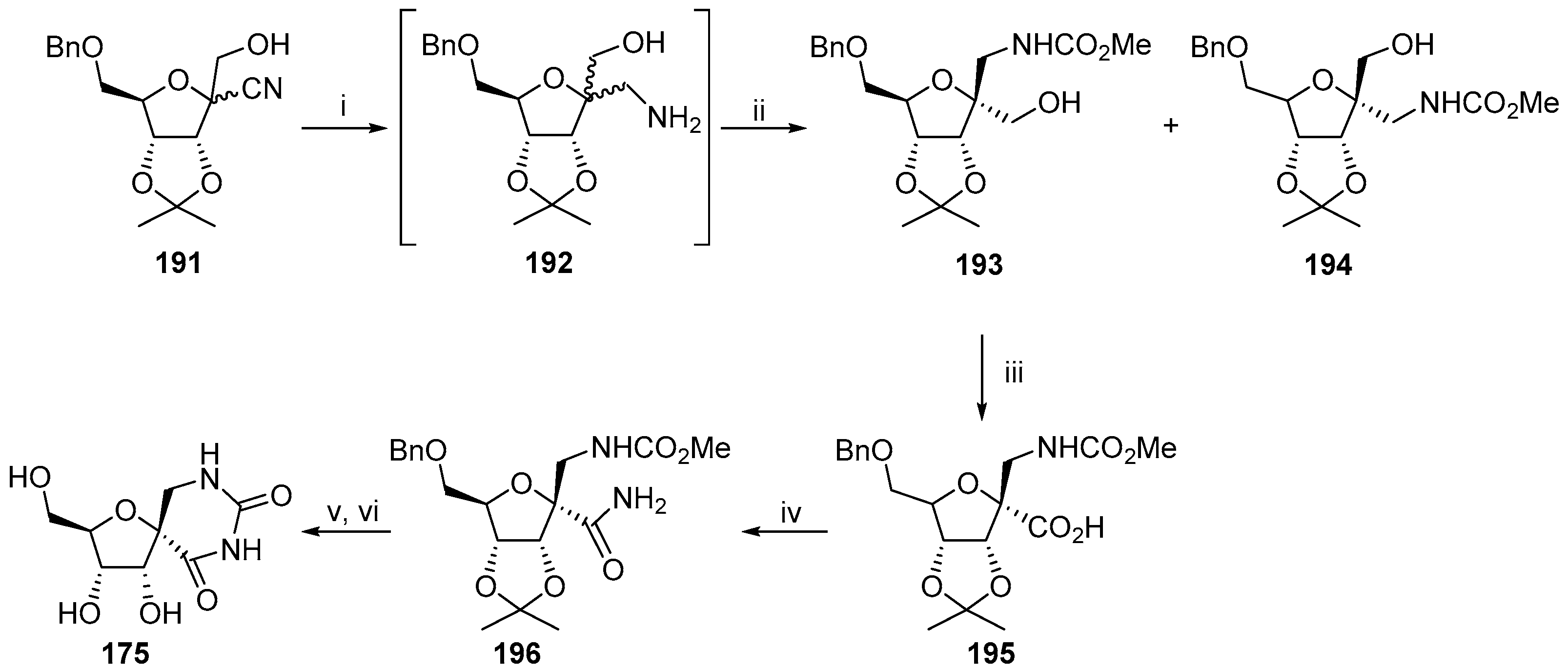{kind=link}
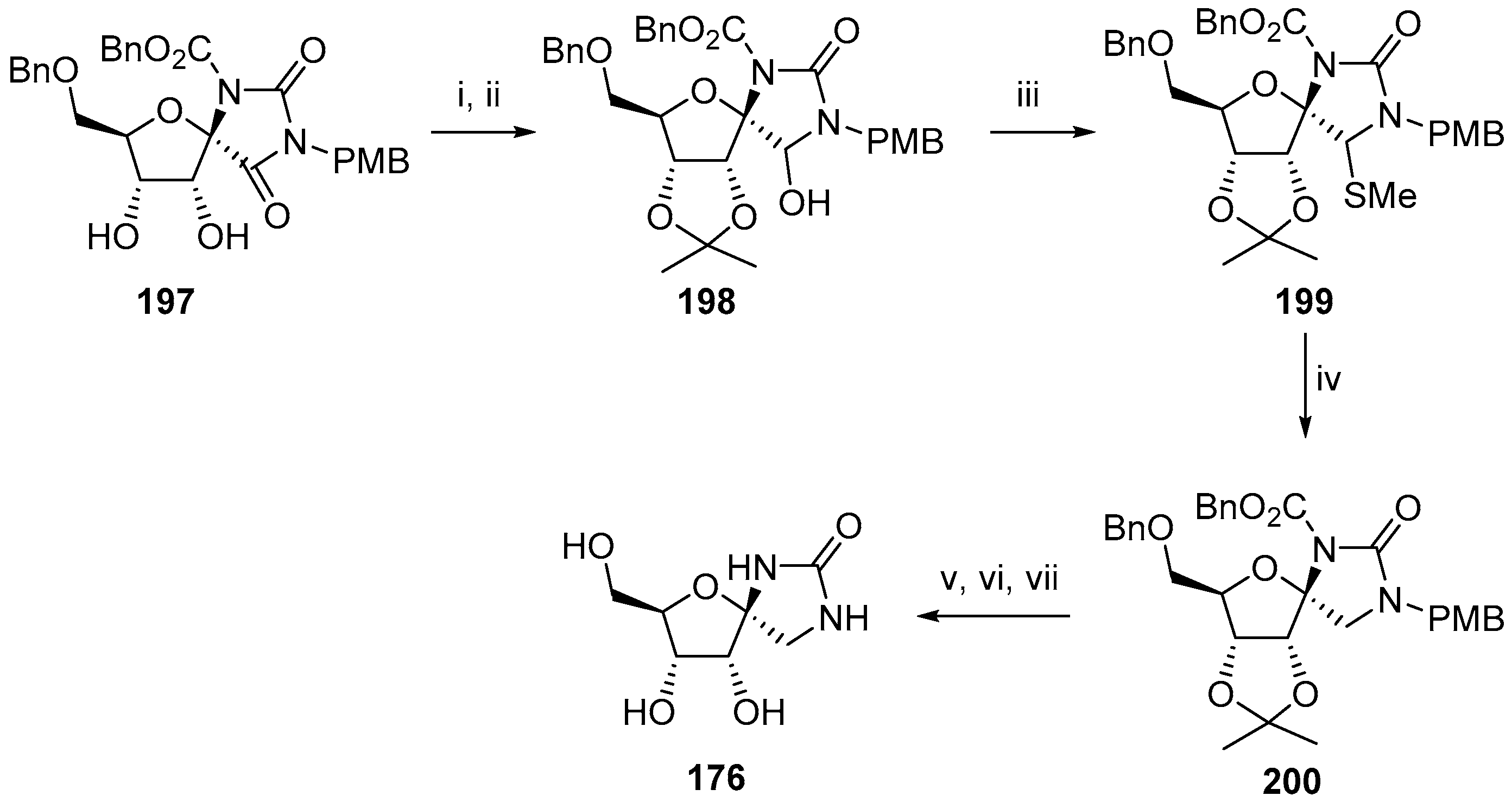{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
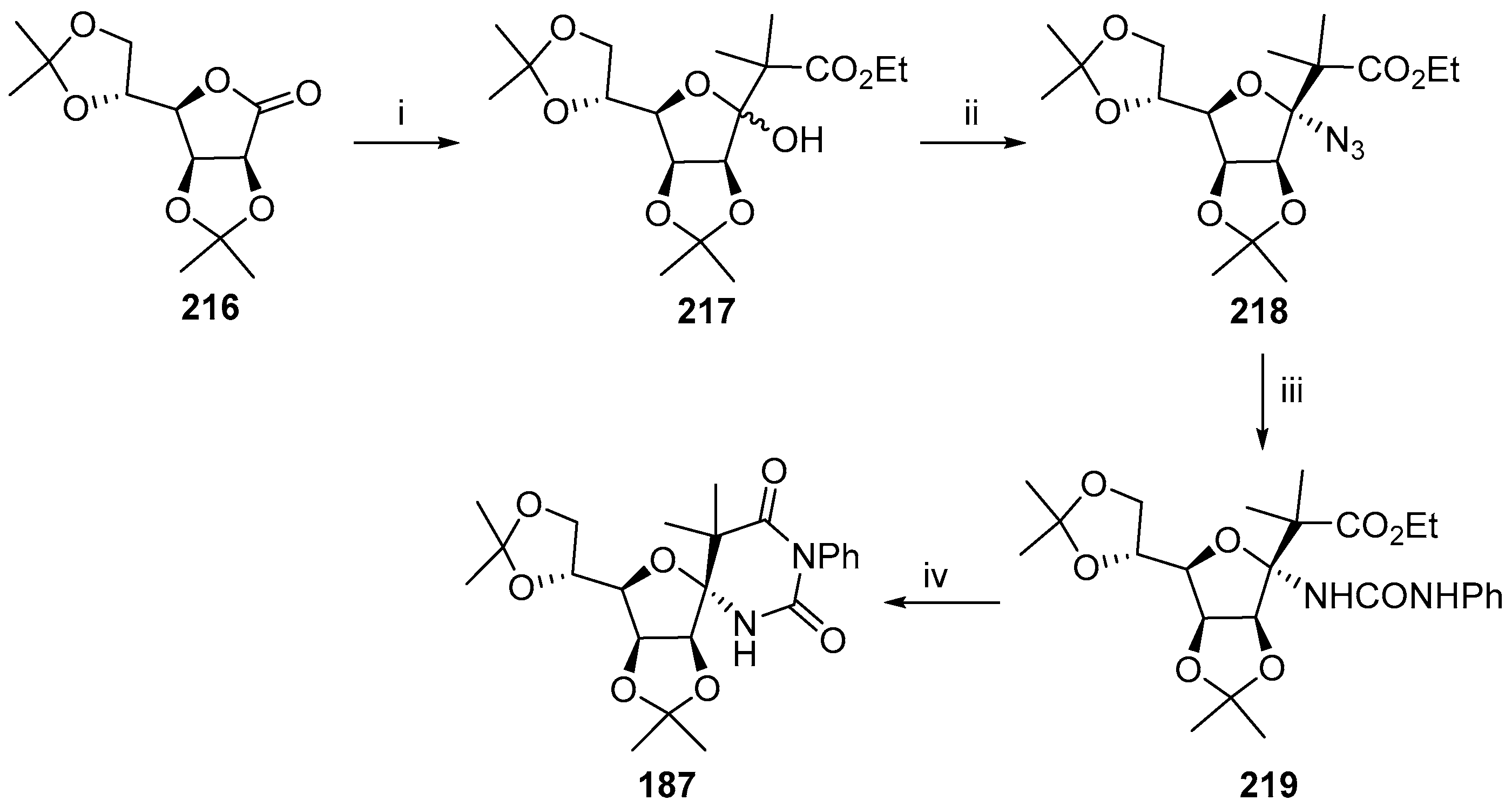{kind=link}
{kind=link}
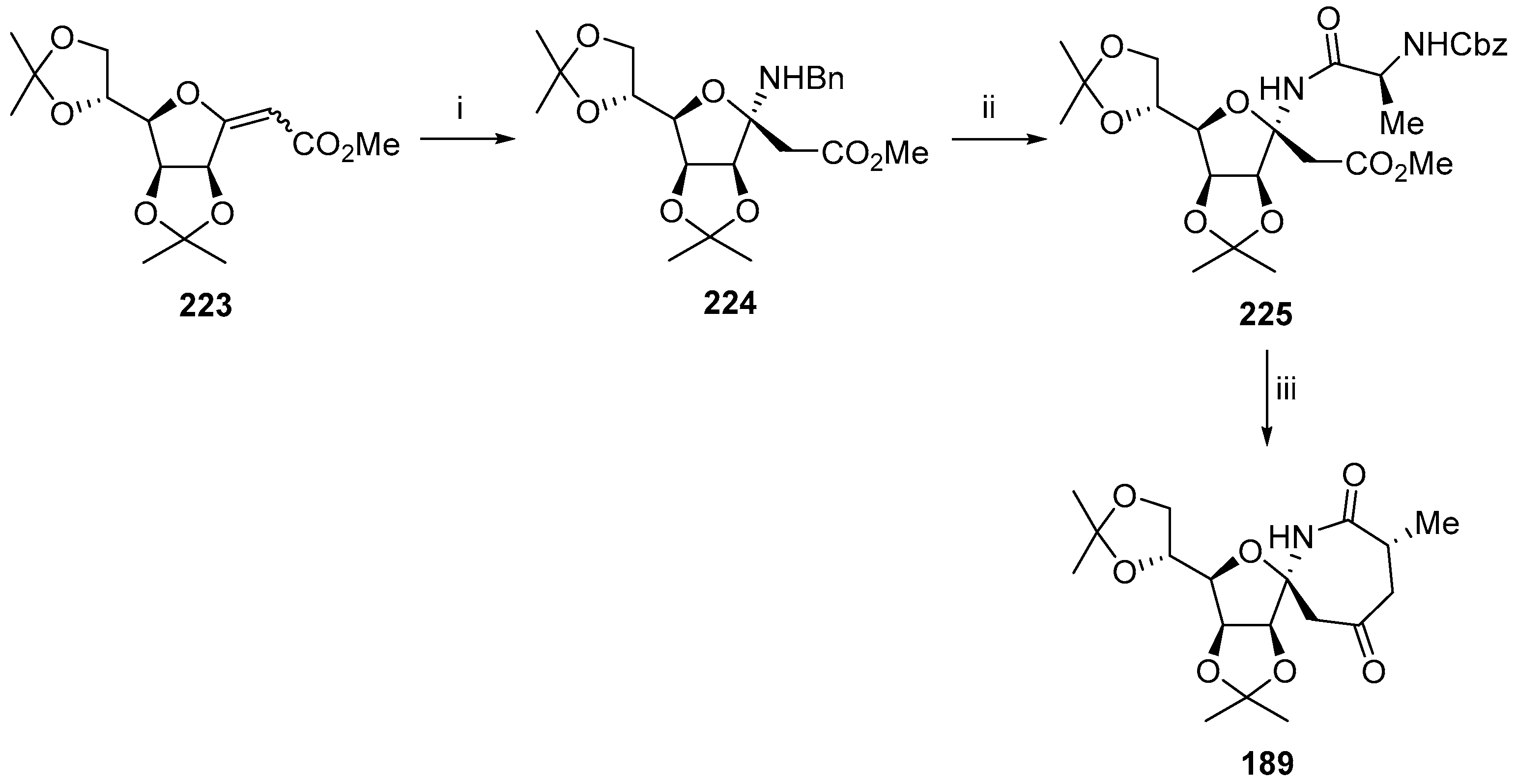{kind=link}
{kind=link}
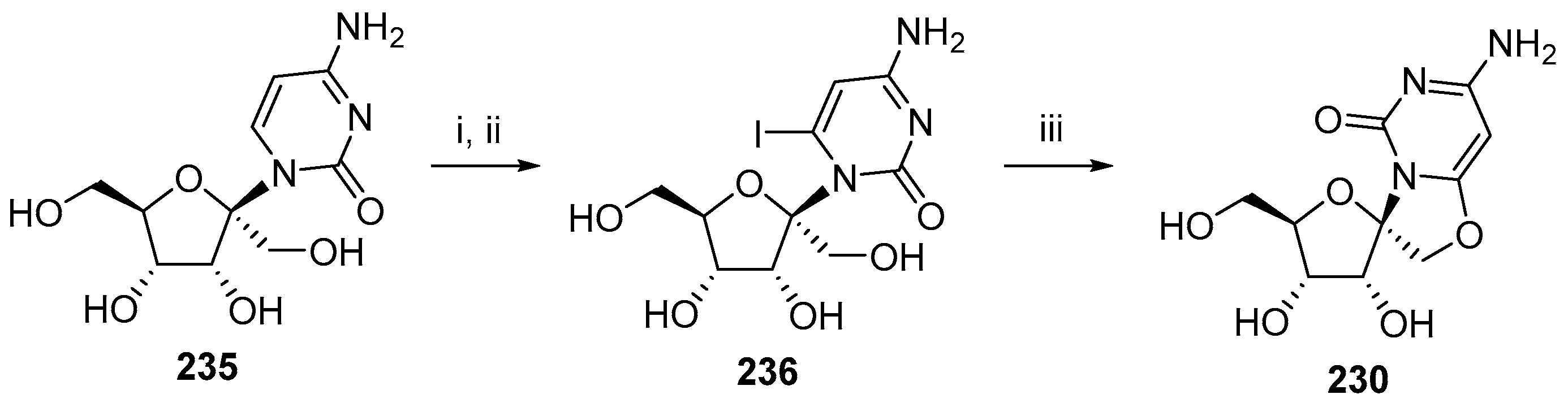{kind=link}
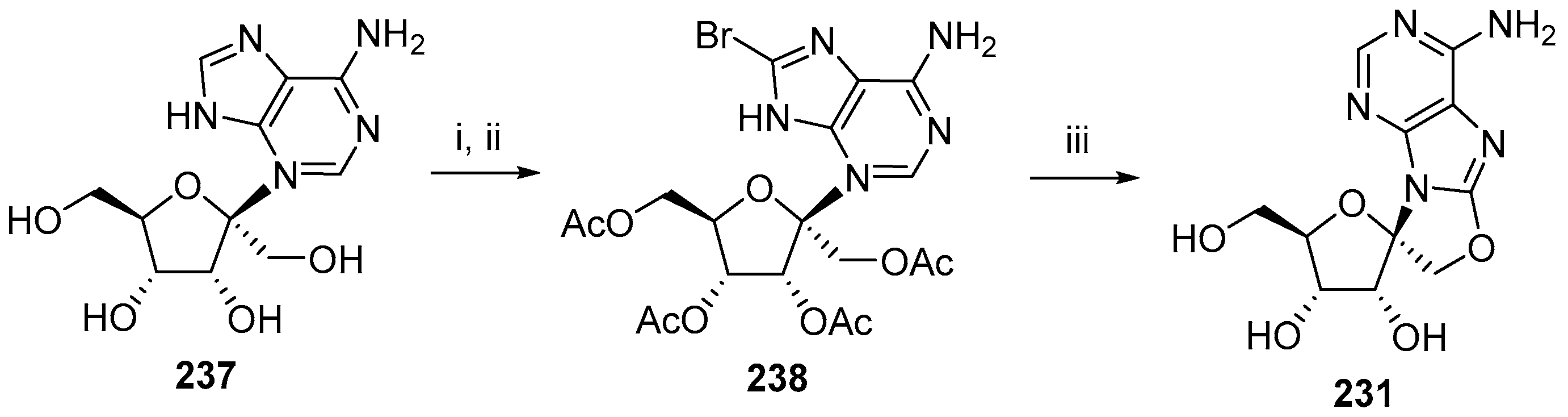{kind=link}
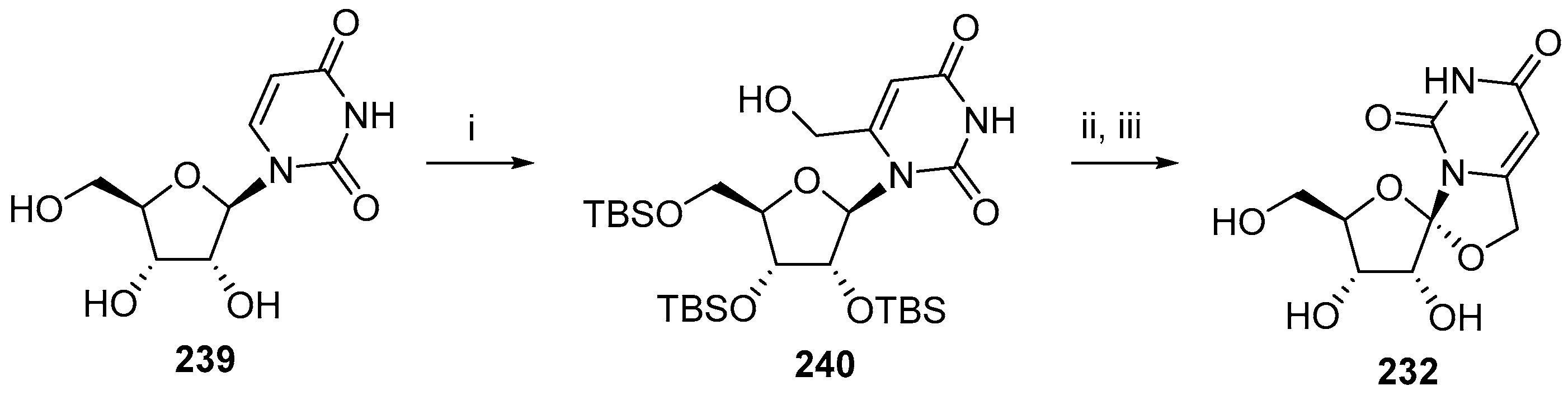{kind=link}
{kind=link}
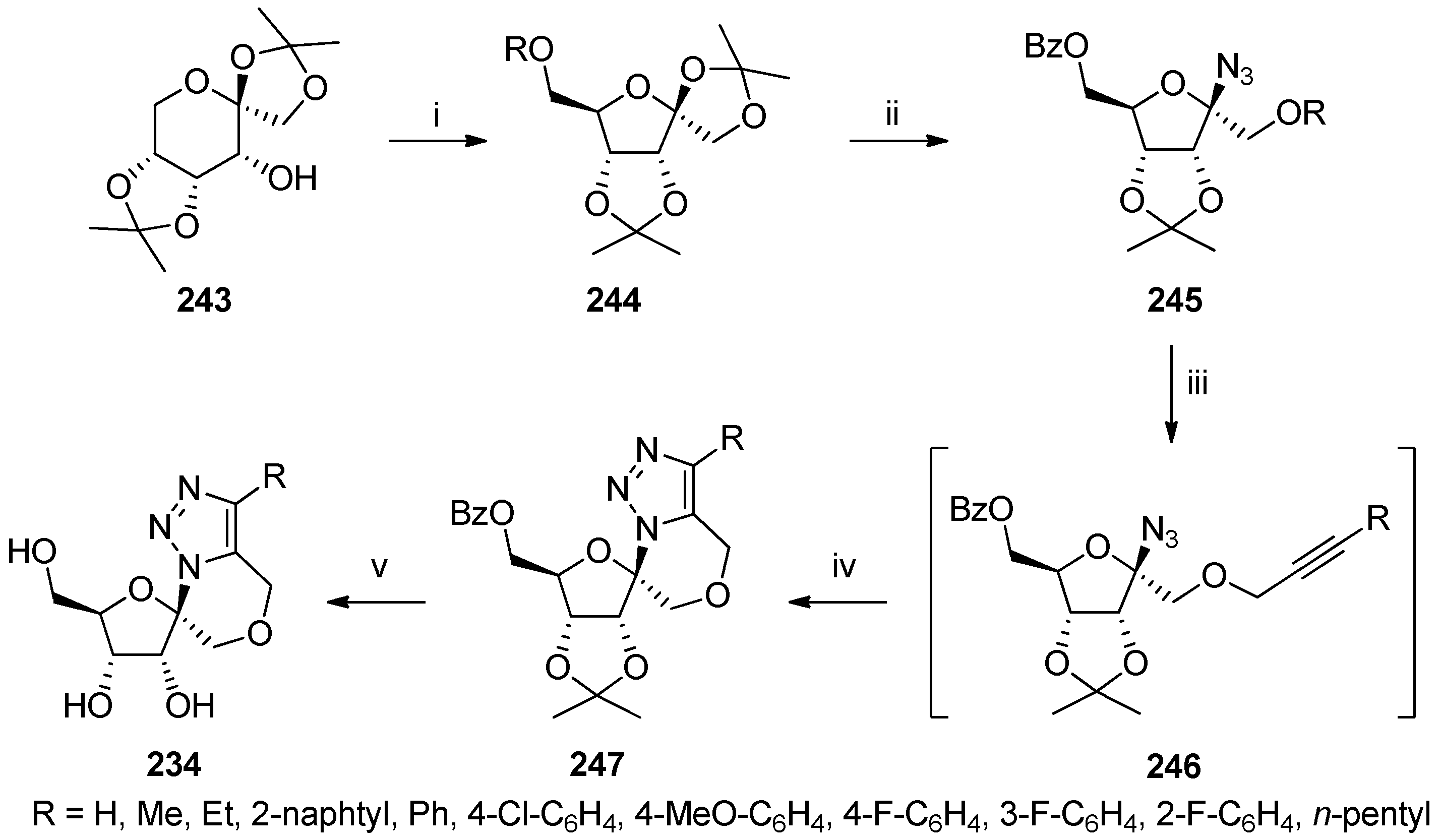{kind=link}
1. Introduction
2. Synthesis of Hydantoins
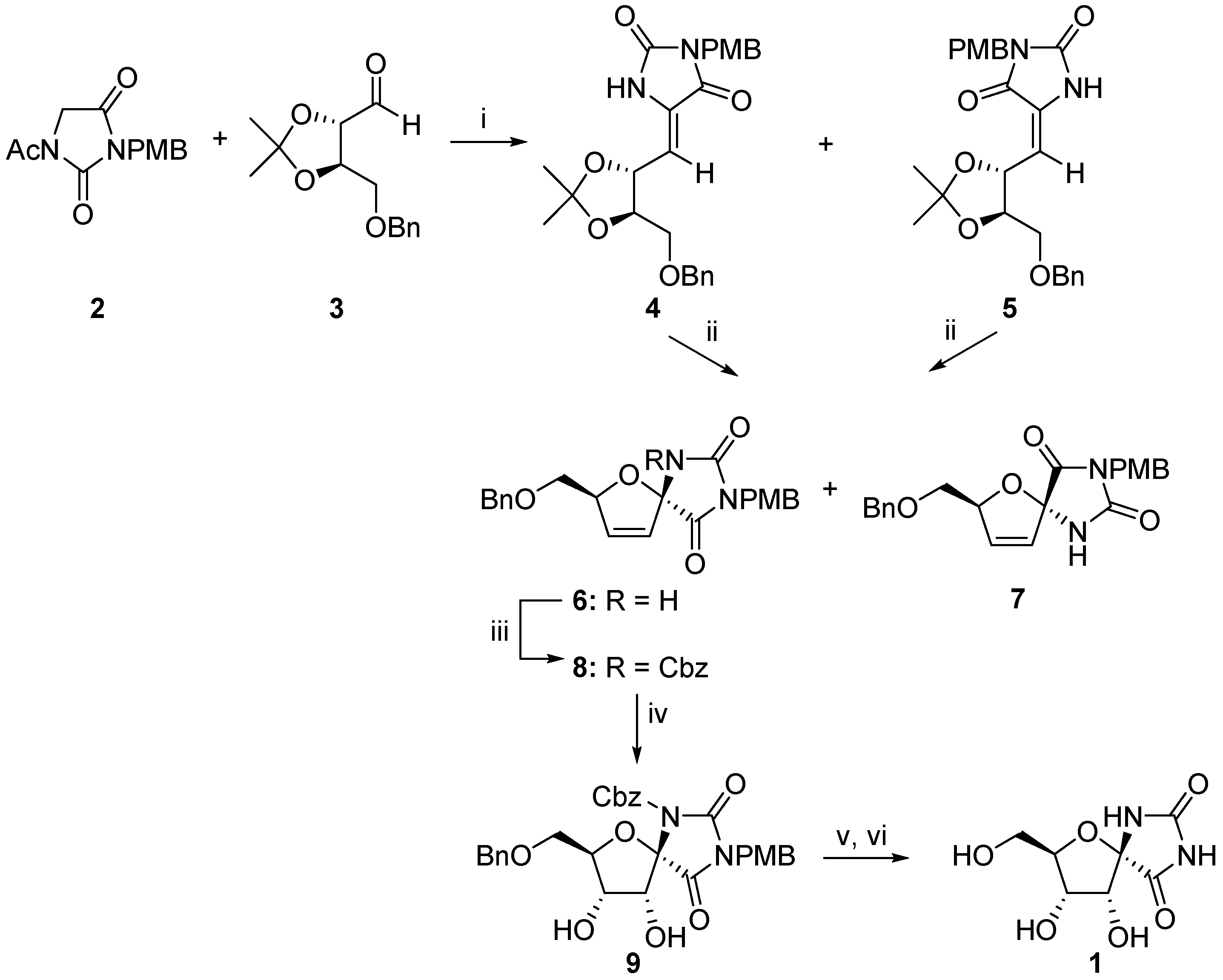
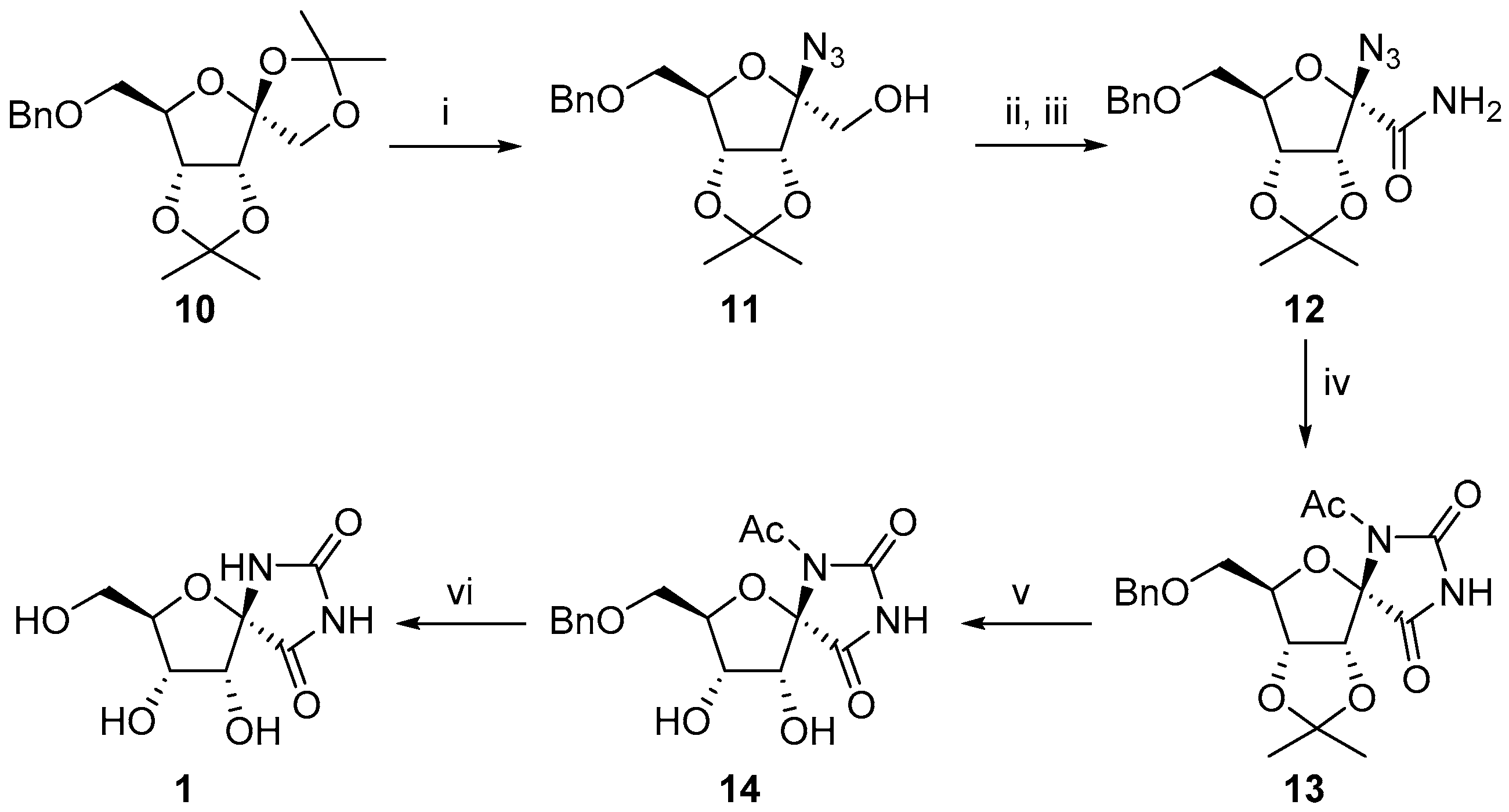
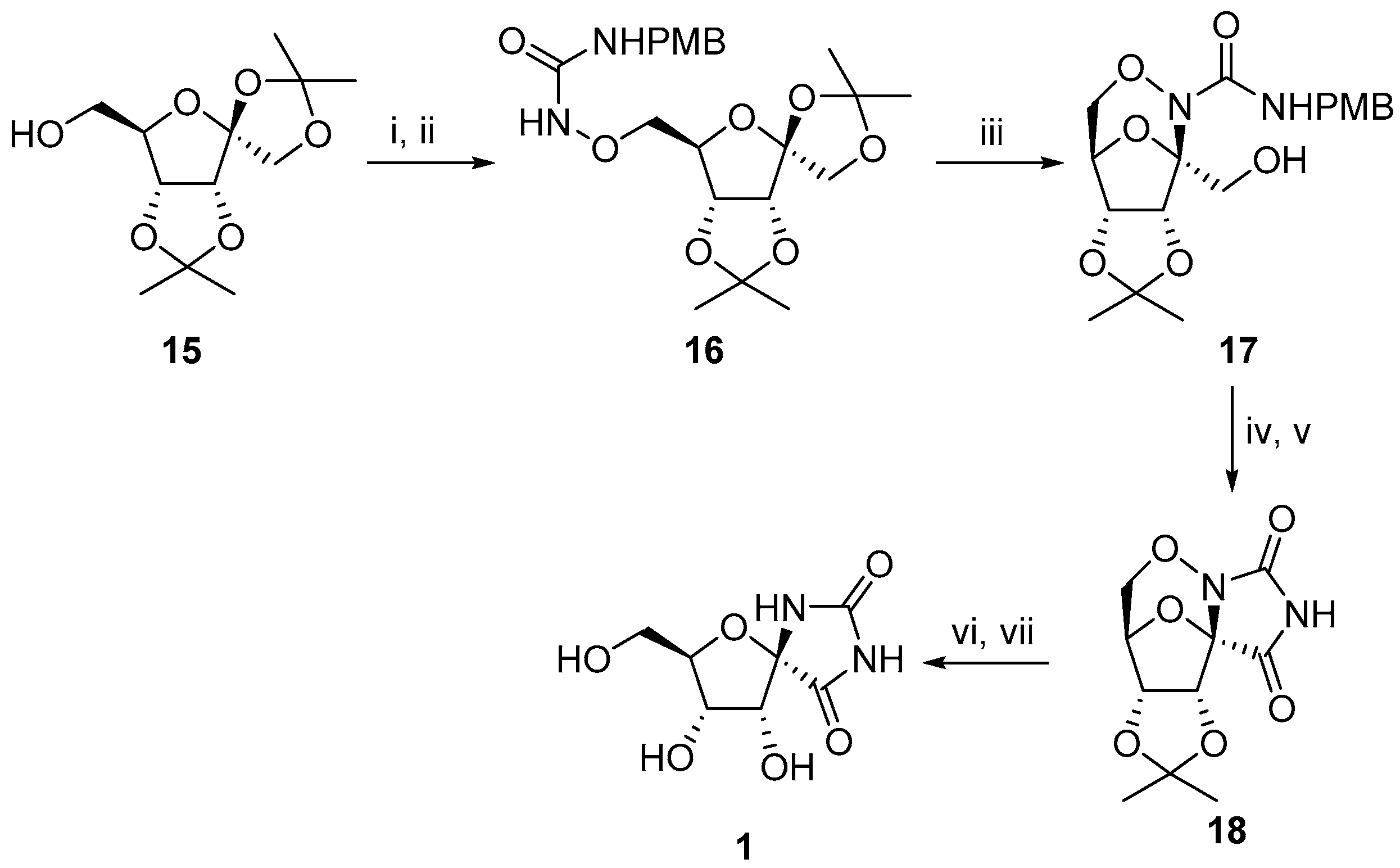
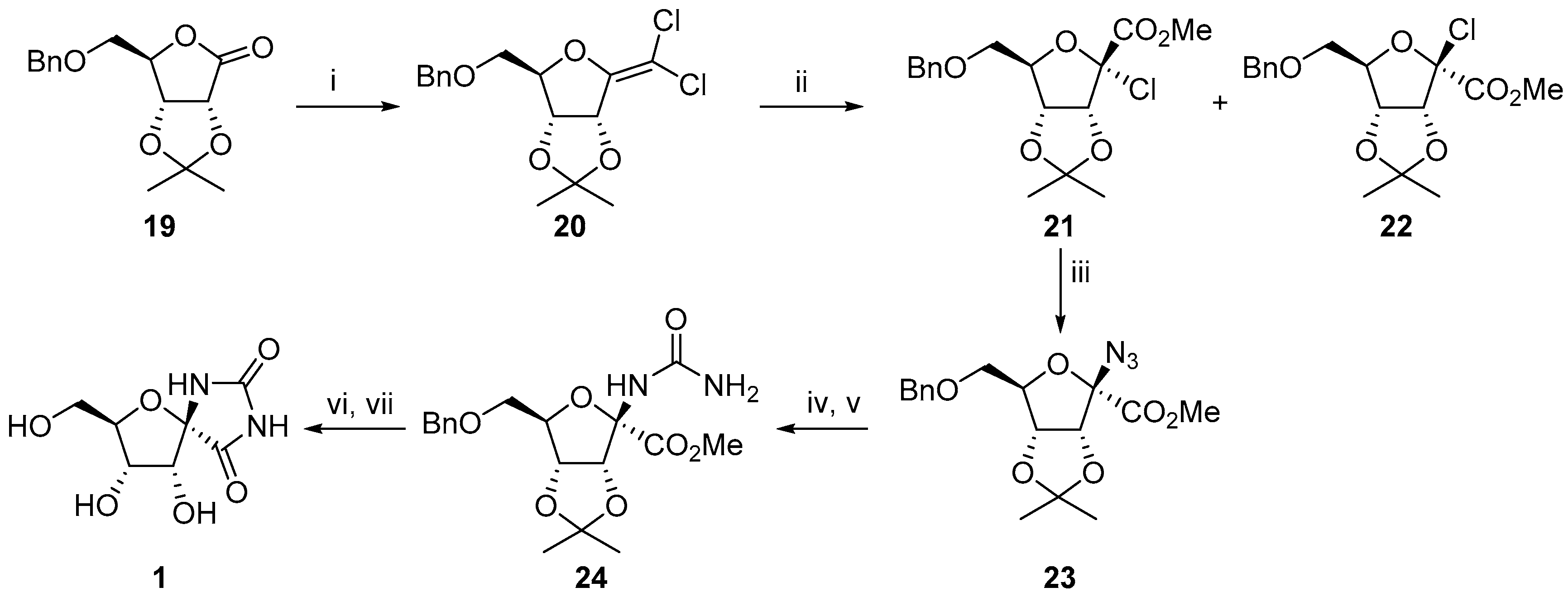
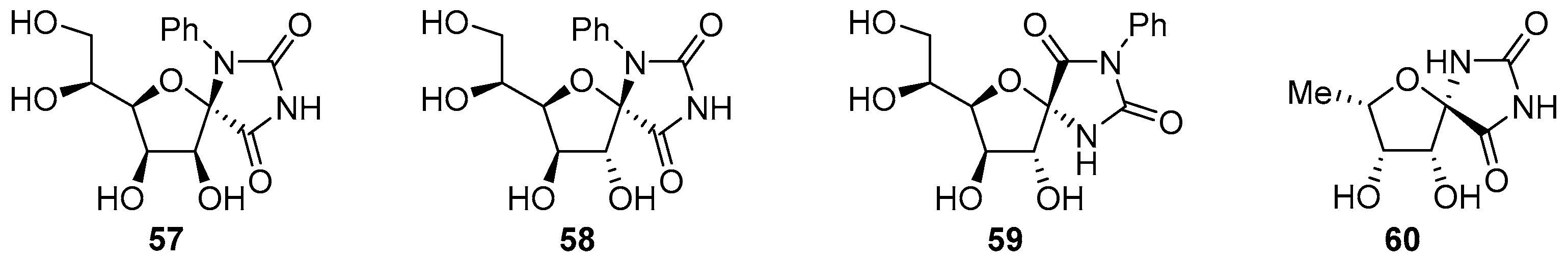
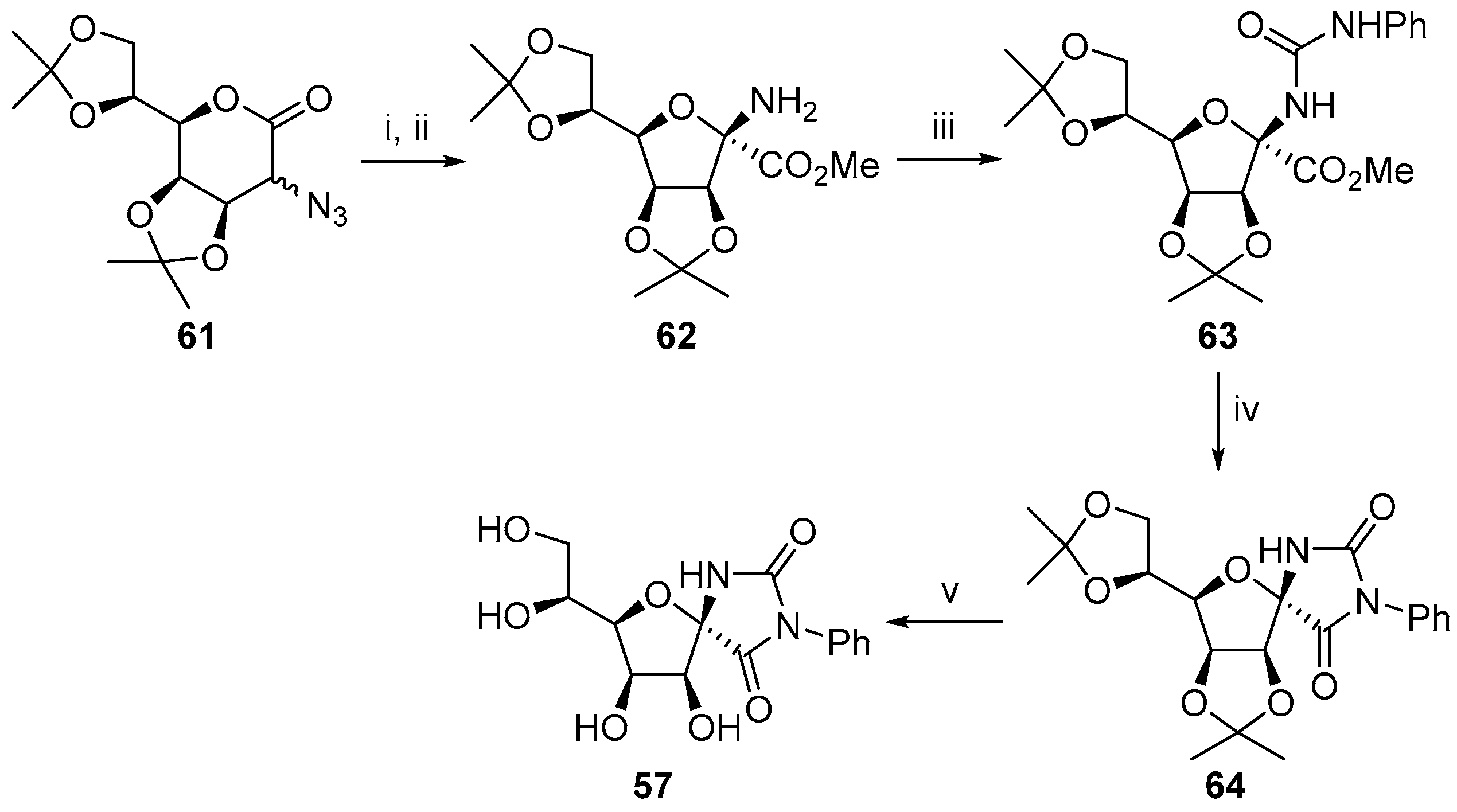
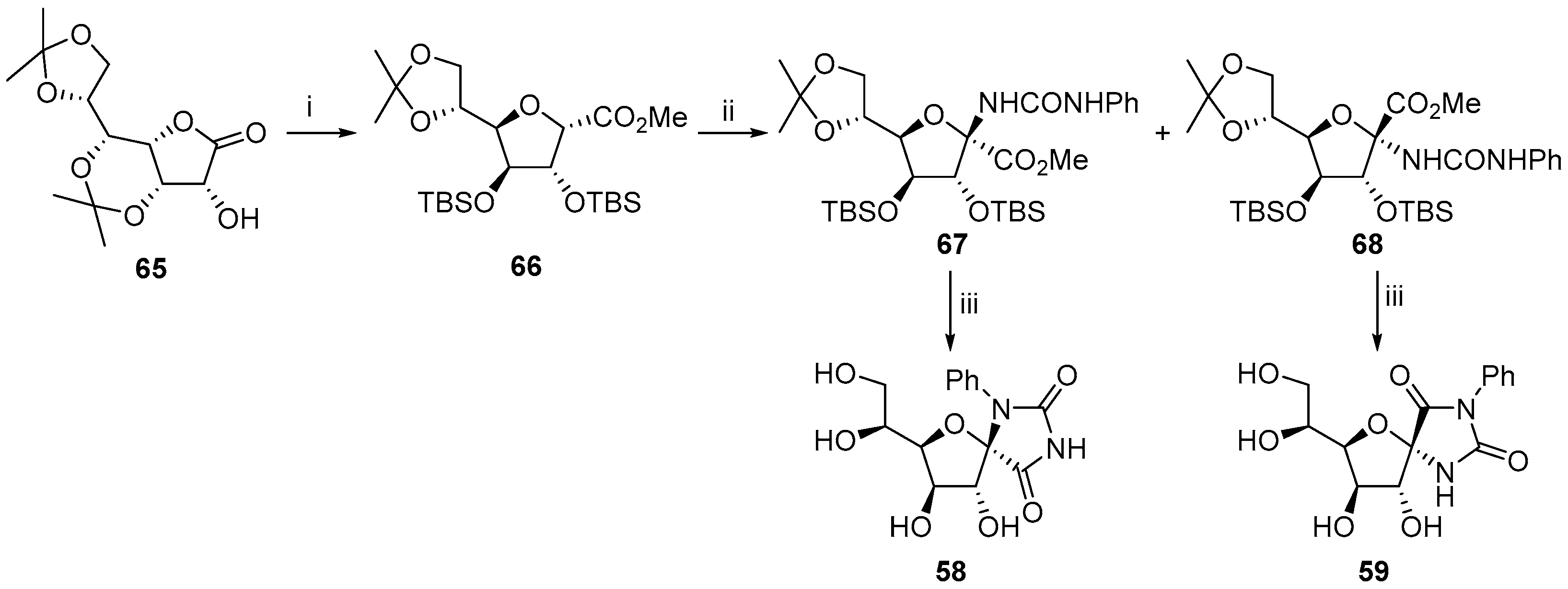
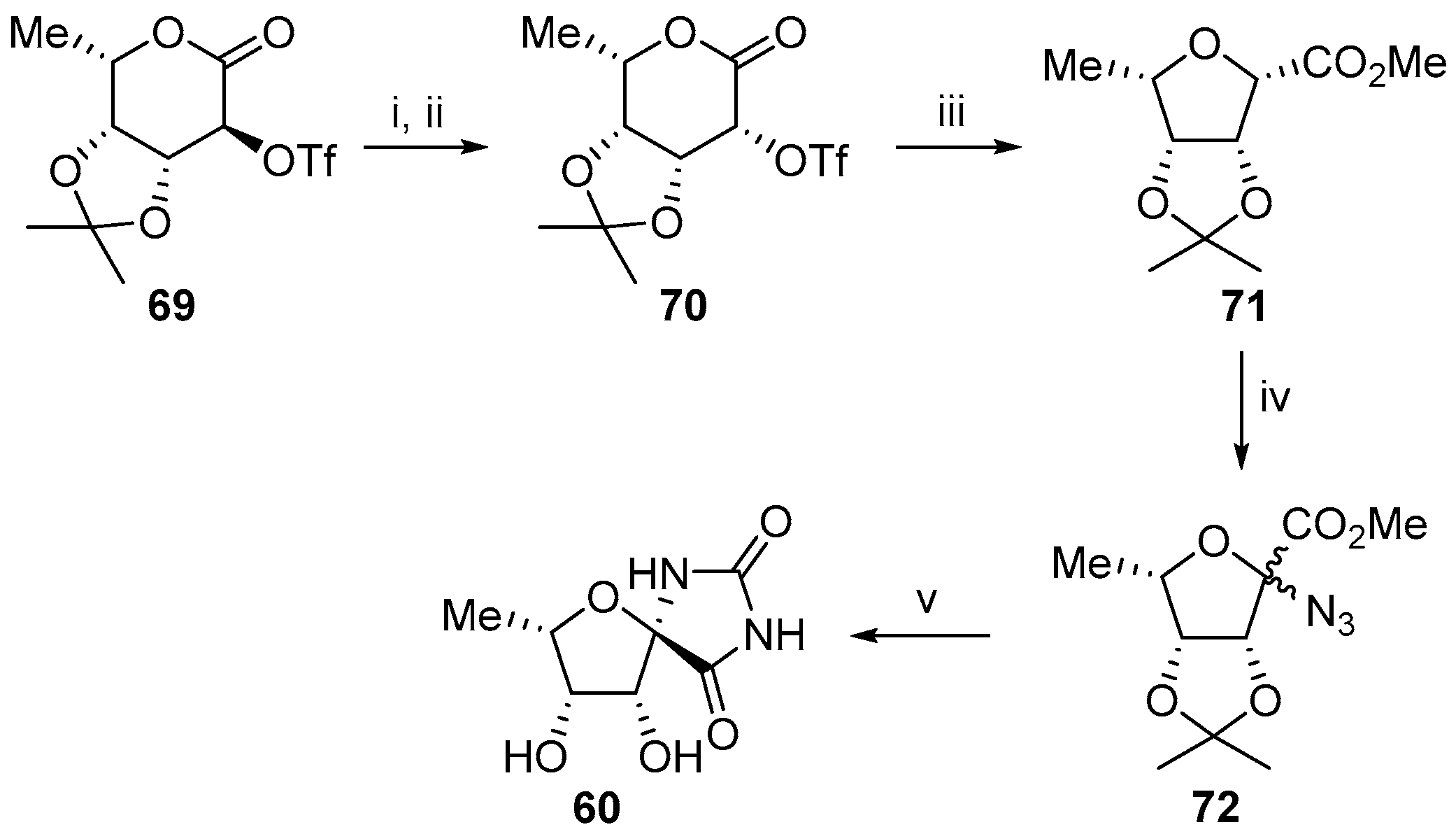
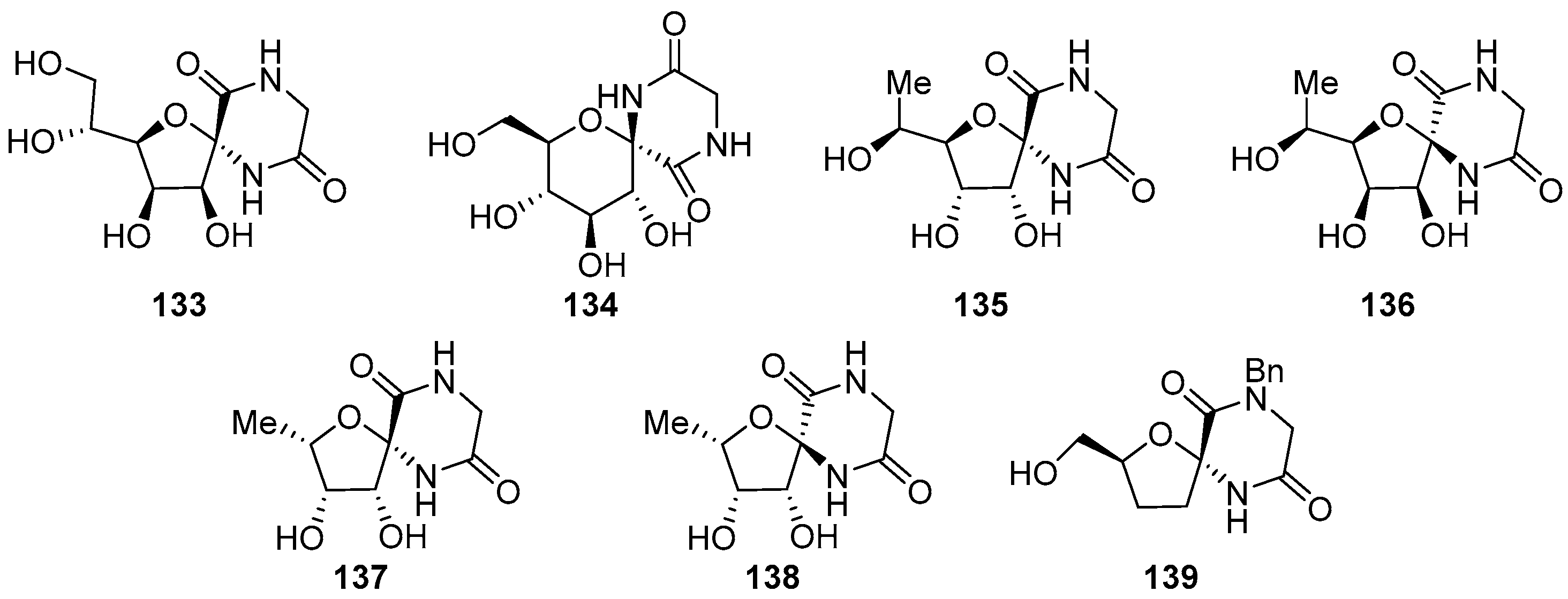
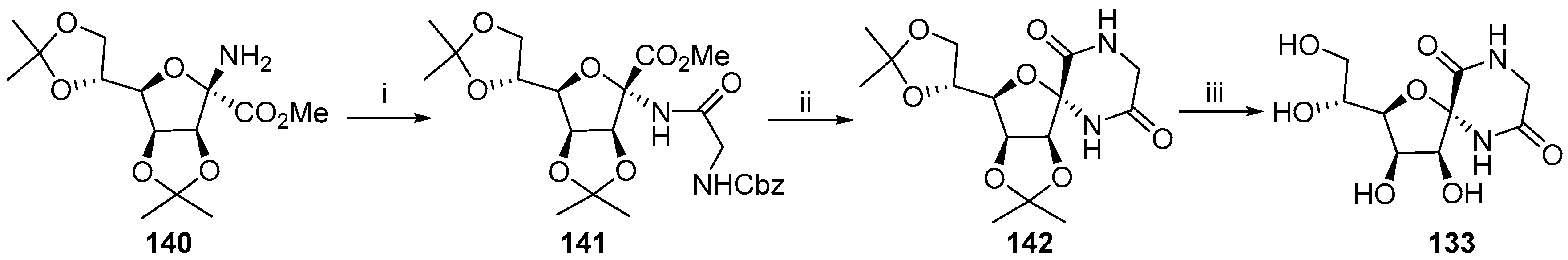
2.1. (+)-Hydantocidin
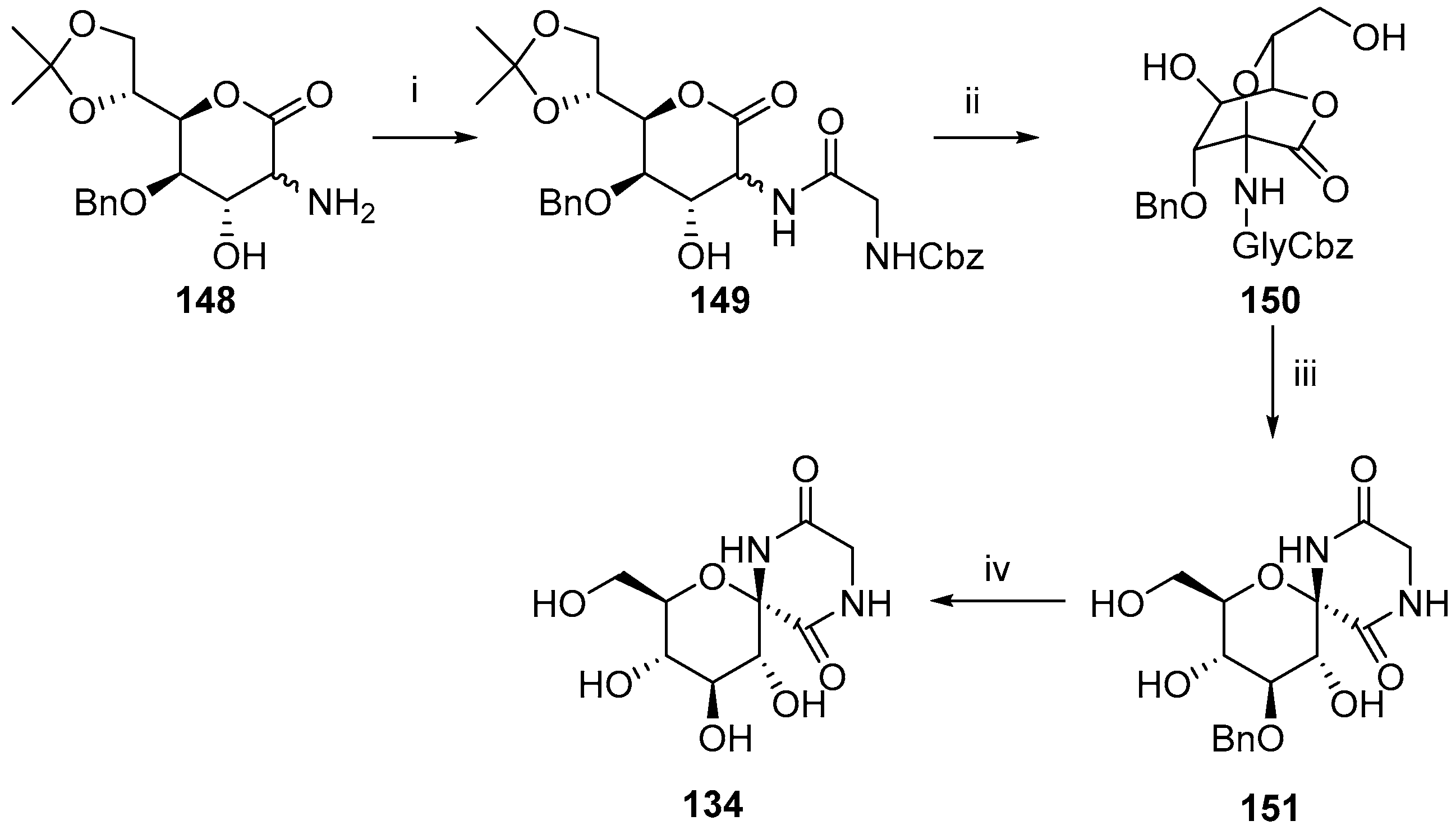
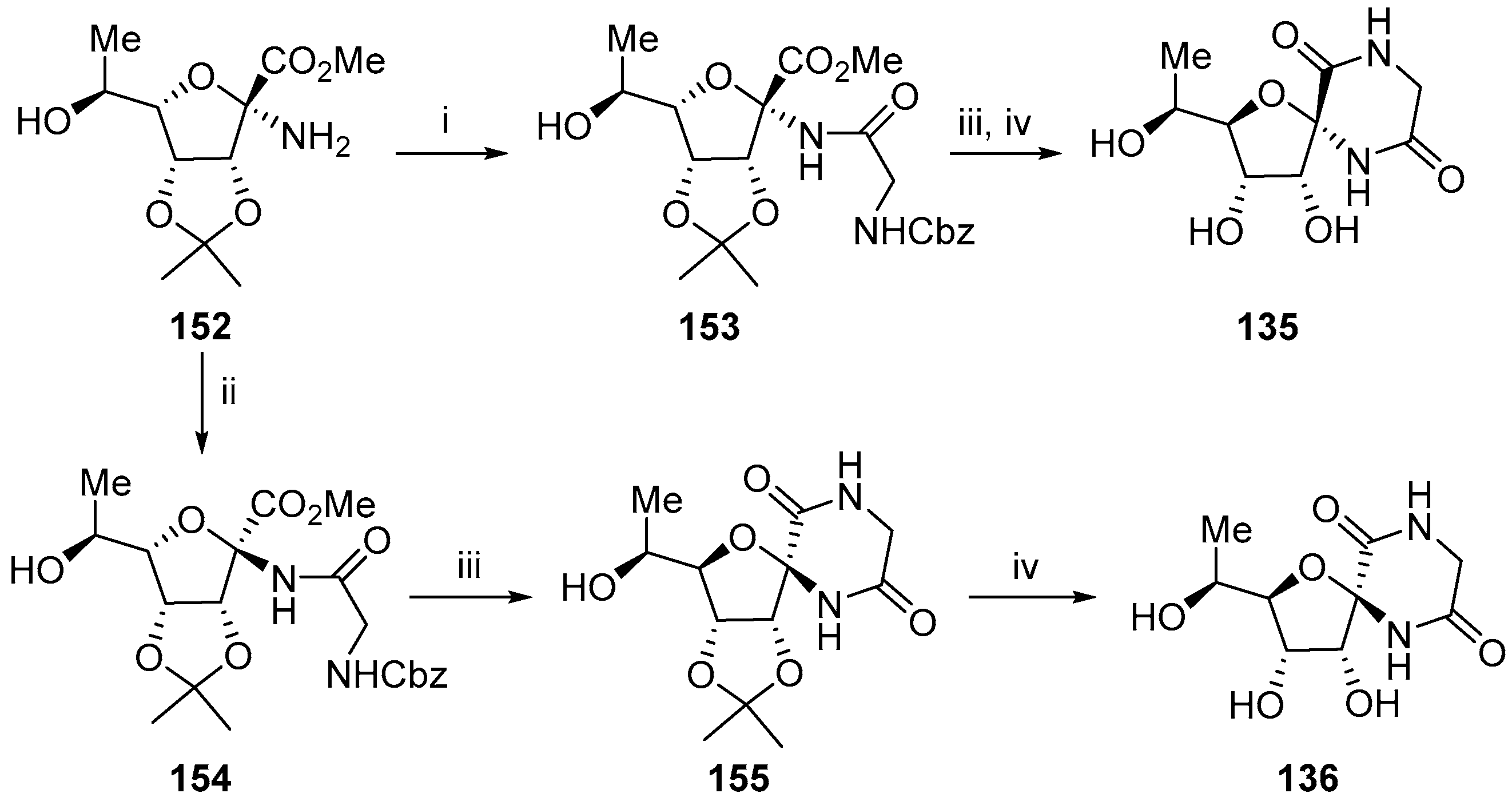
2.2. Modifications of Hydantocidin in the Sugar Ring
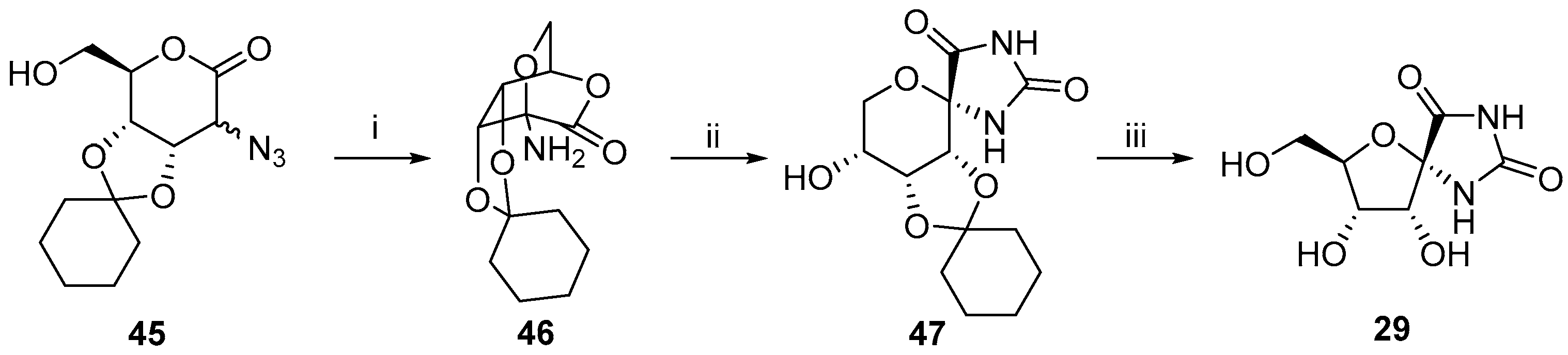
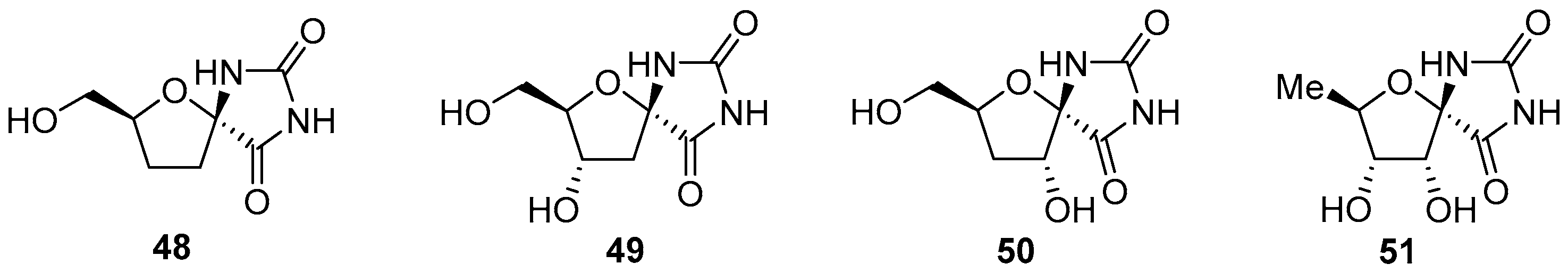
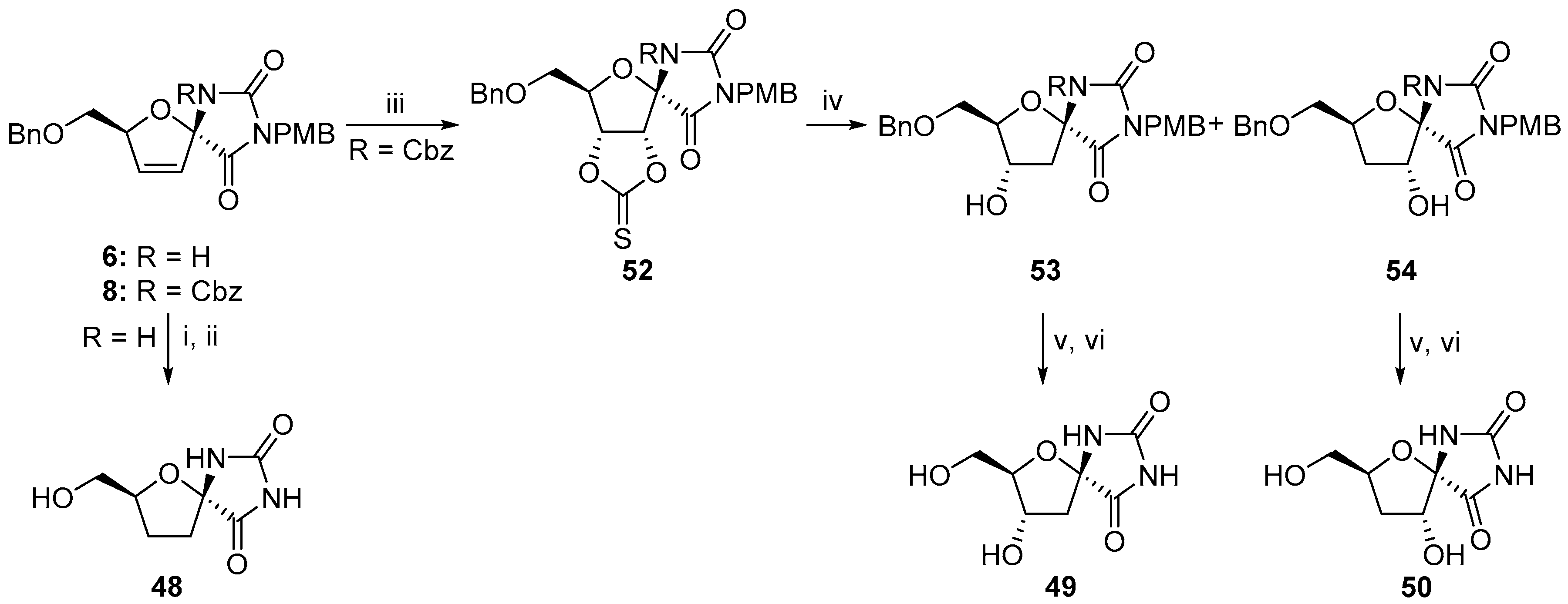
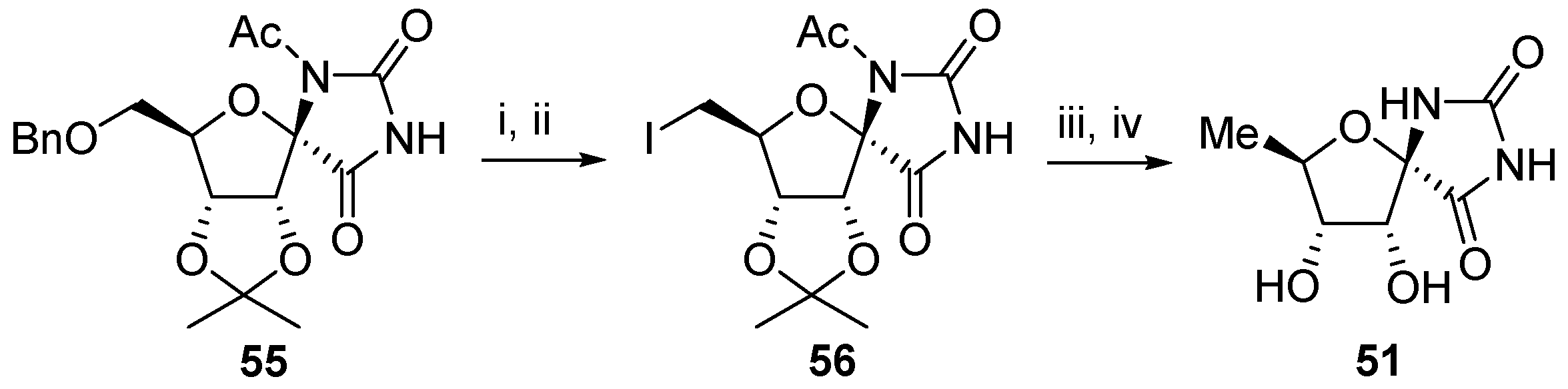
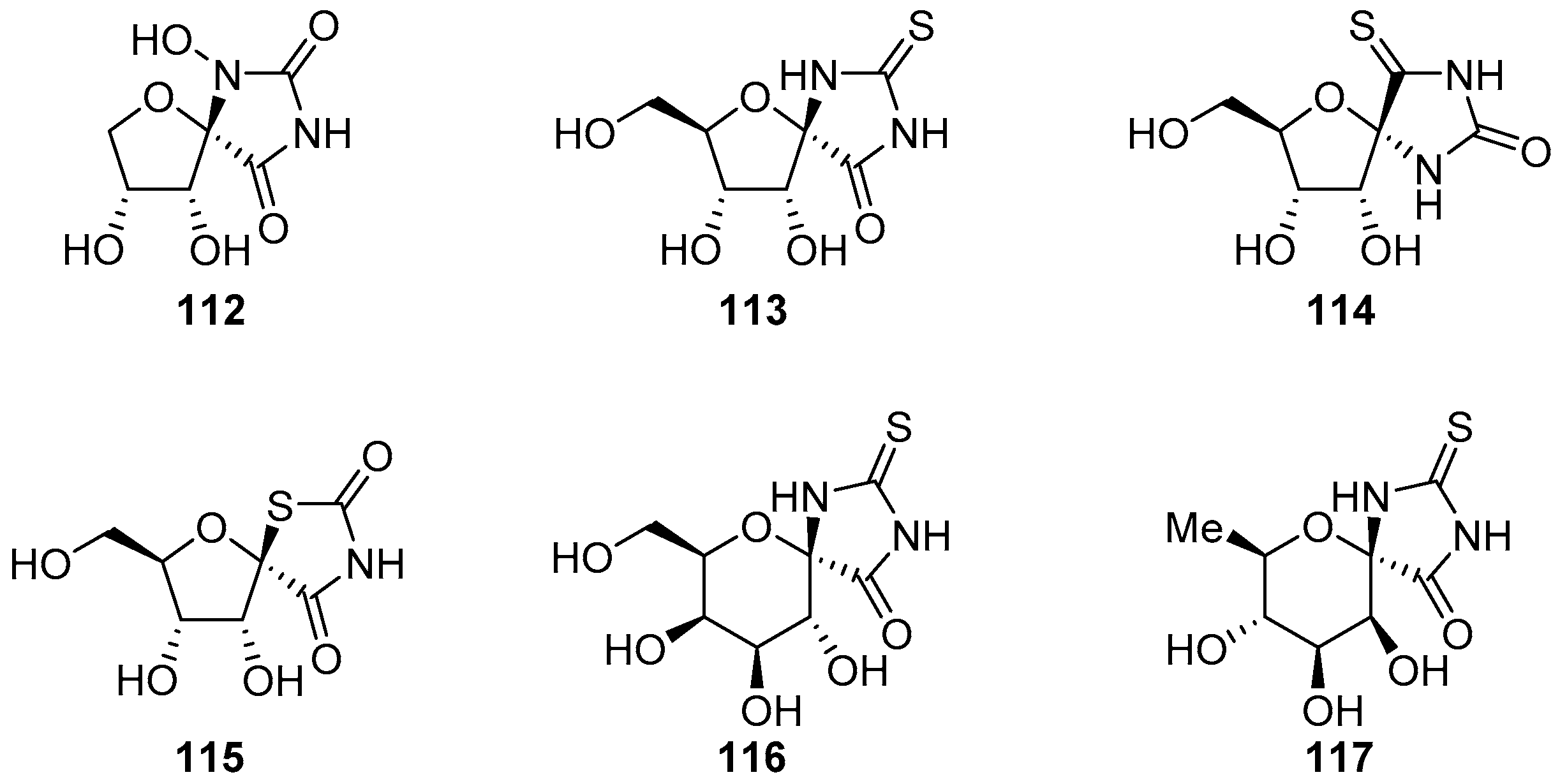
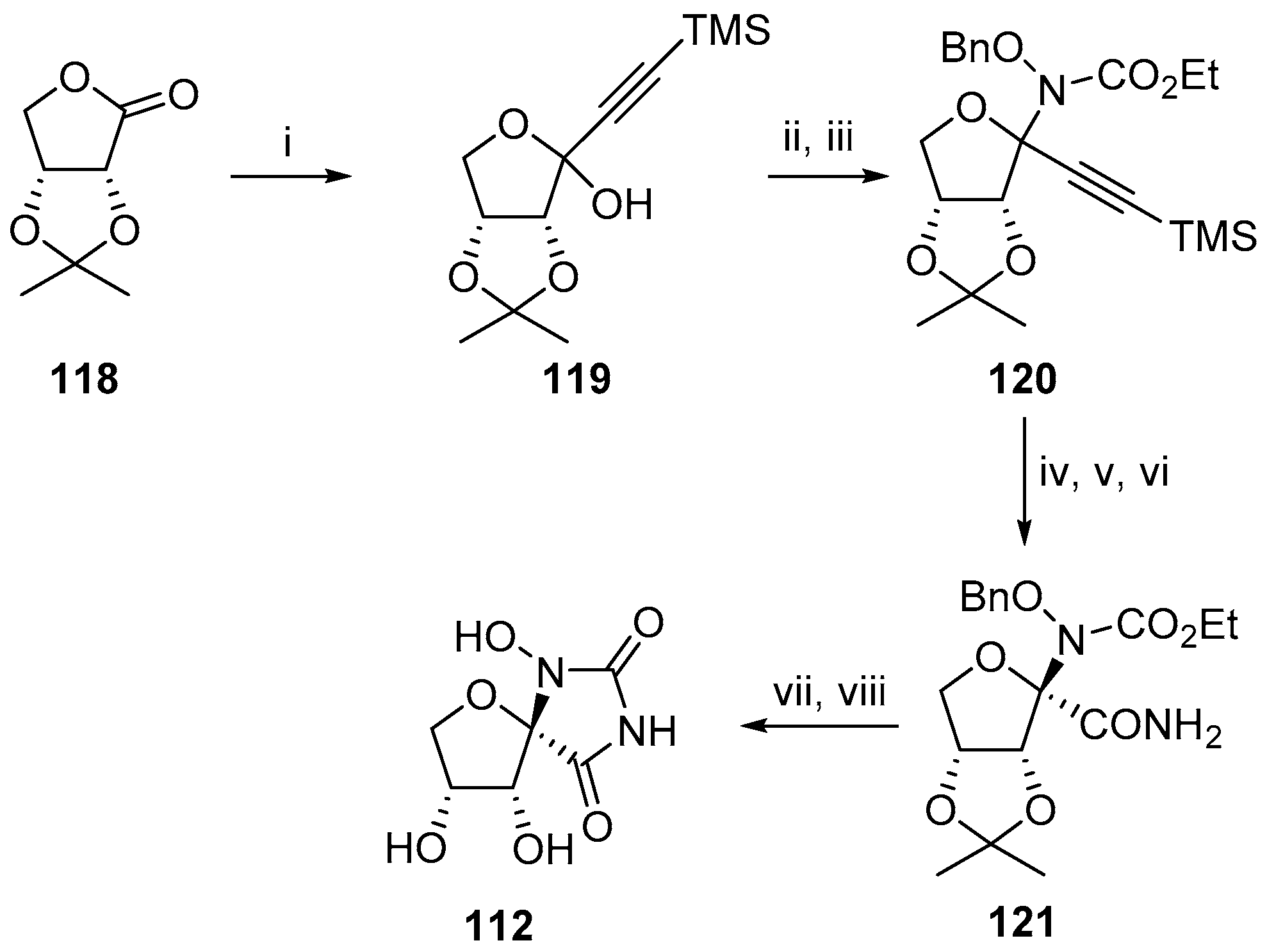
2.2.1. Furanoses
2.2.2. Pyranoses
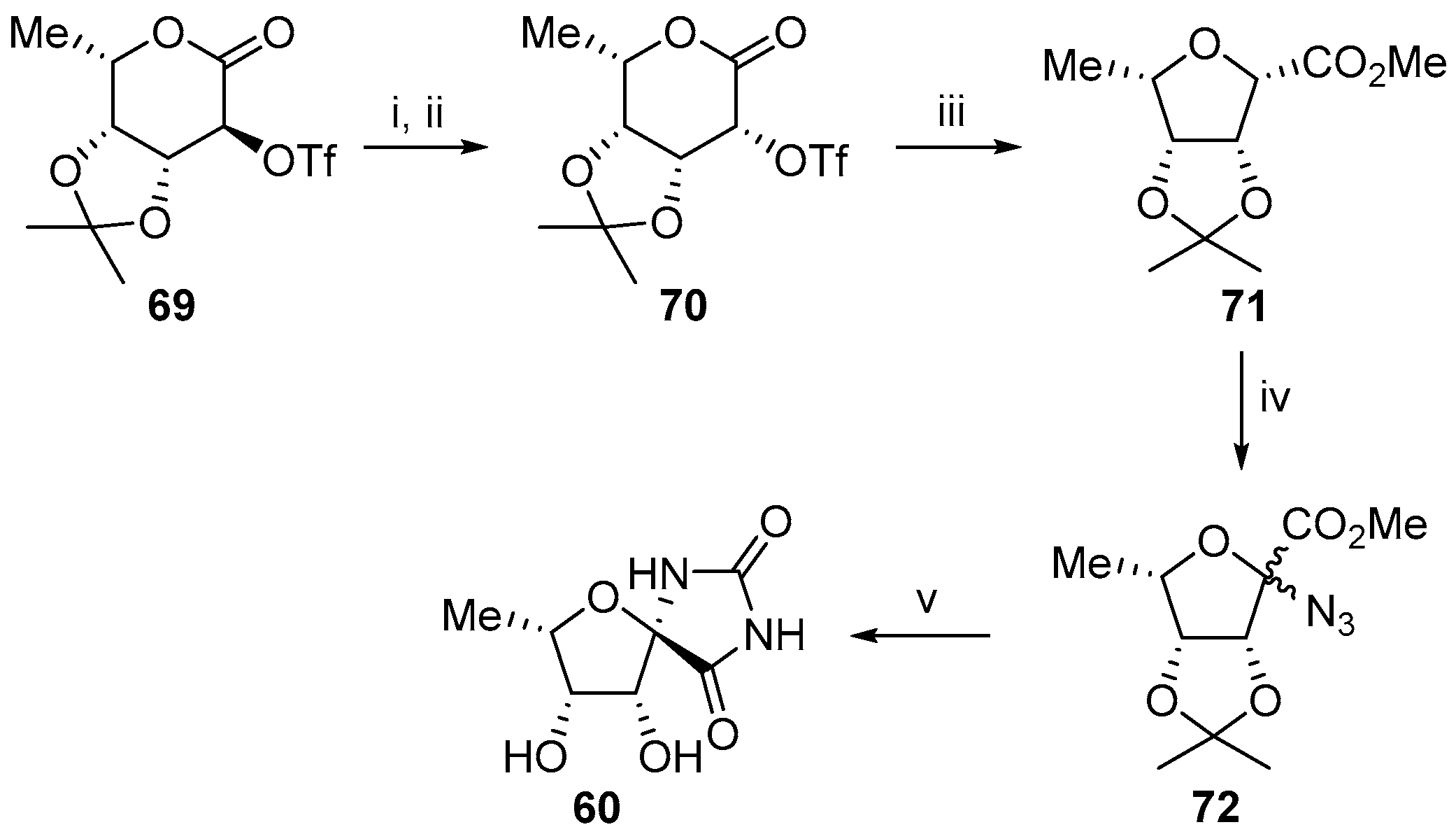
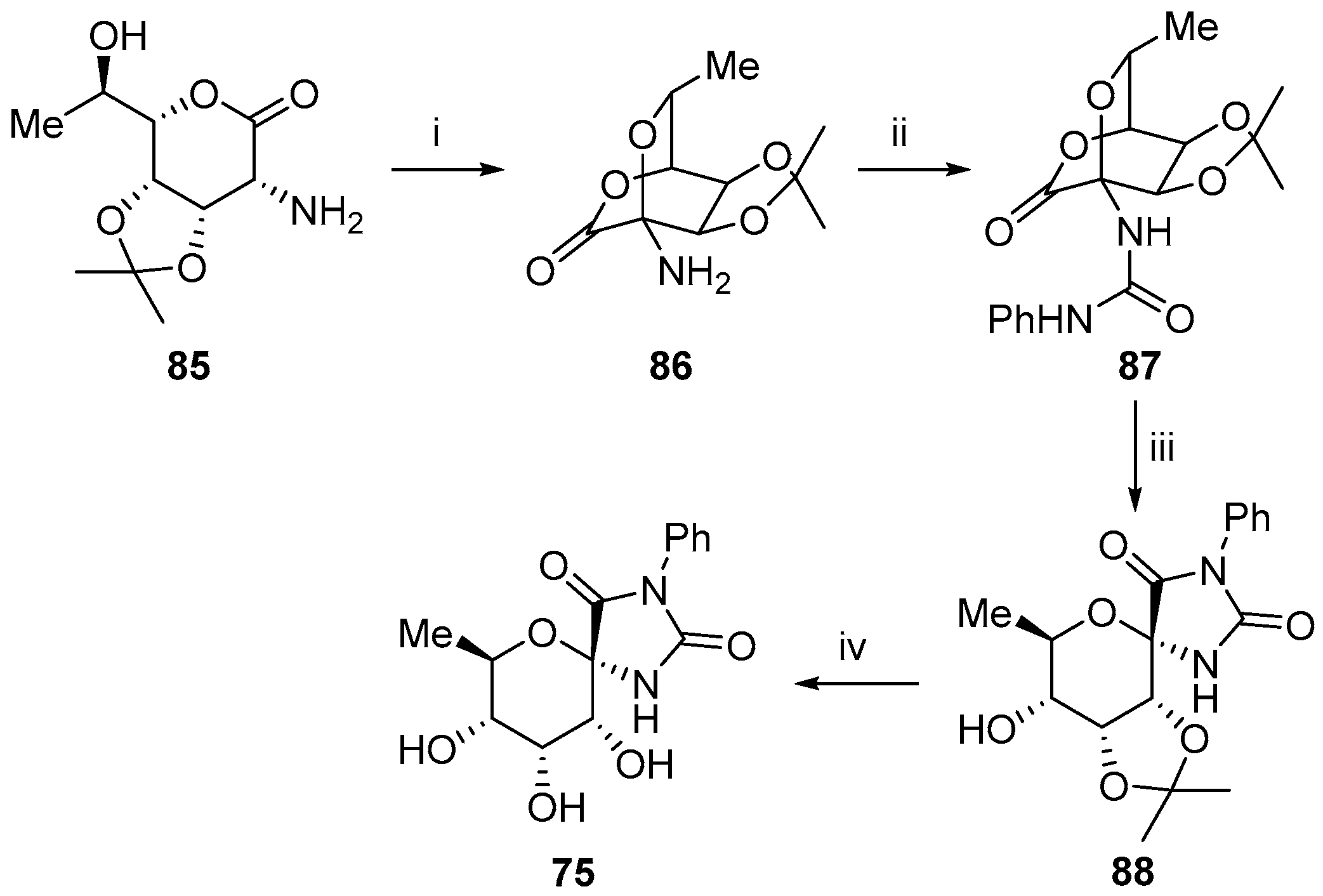
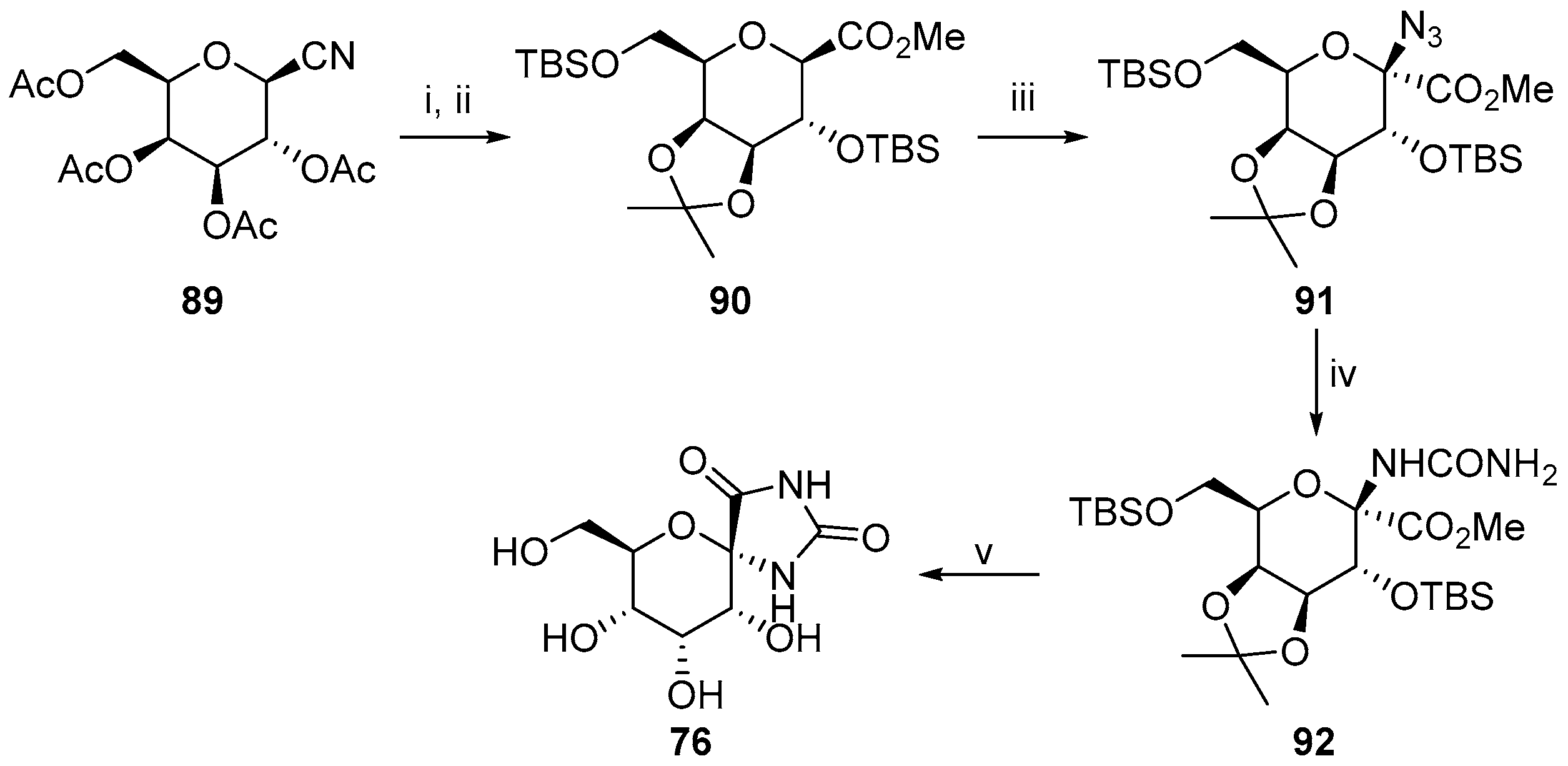
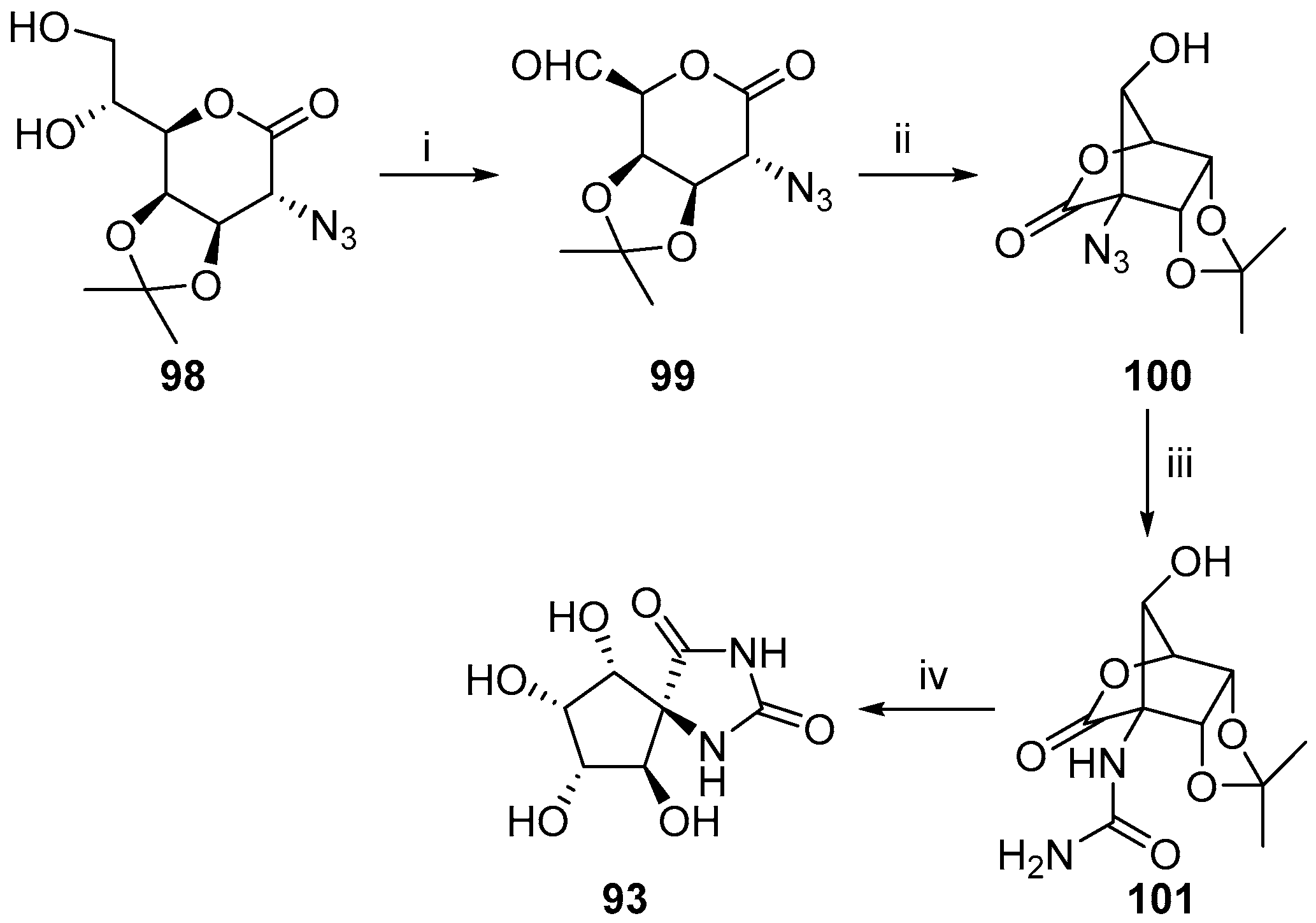
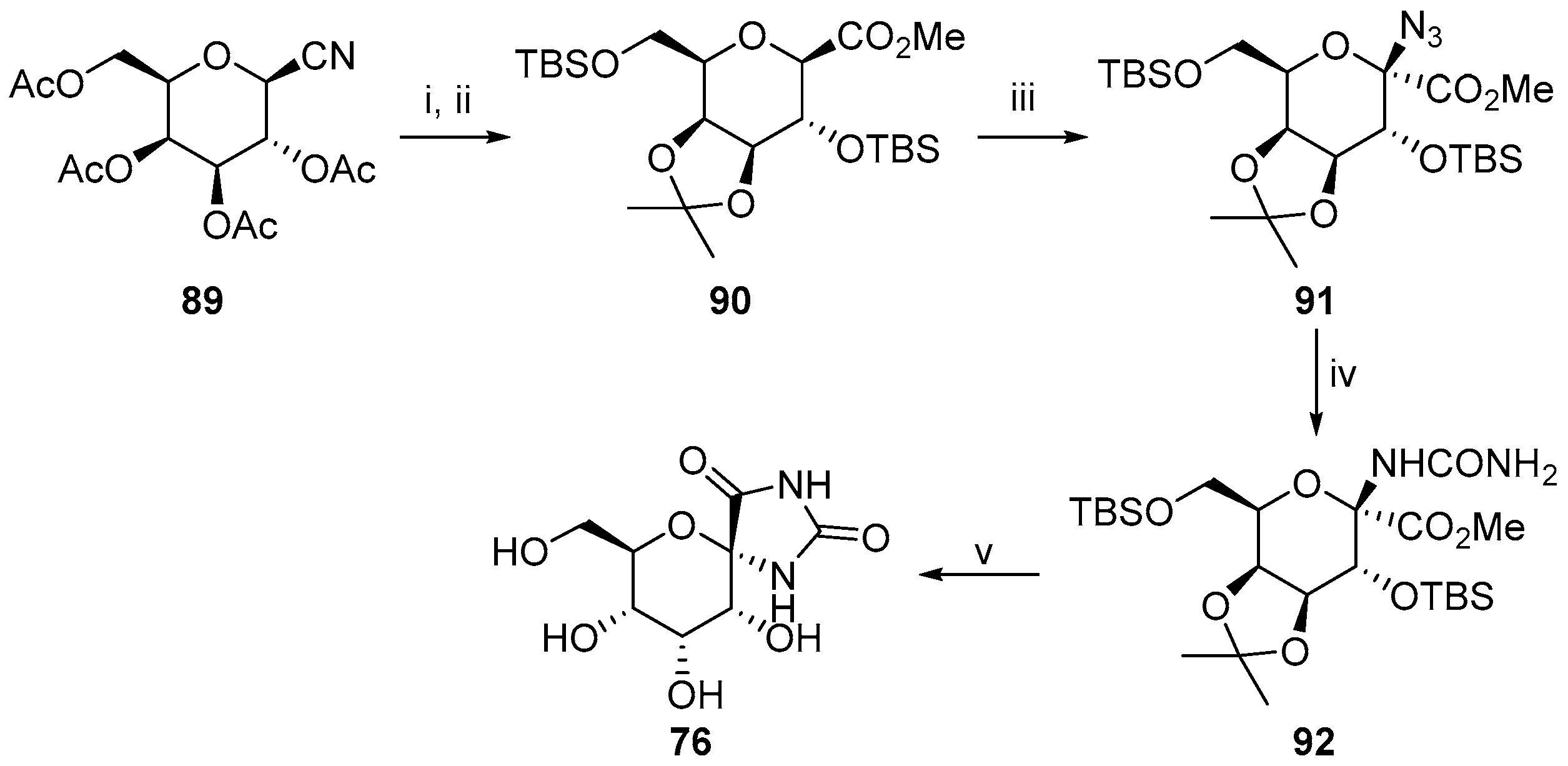
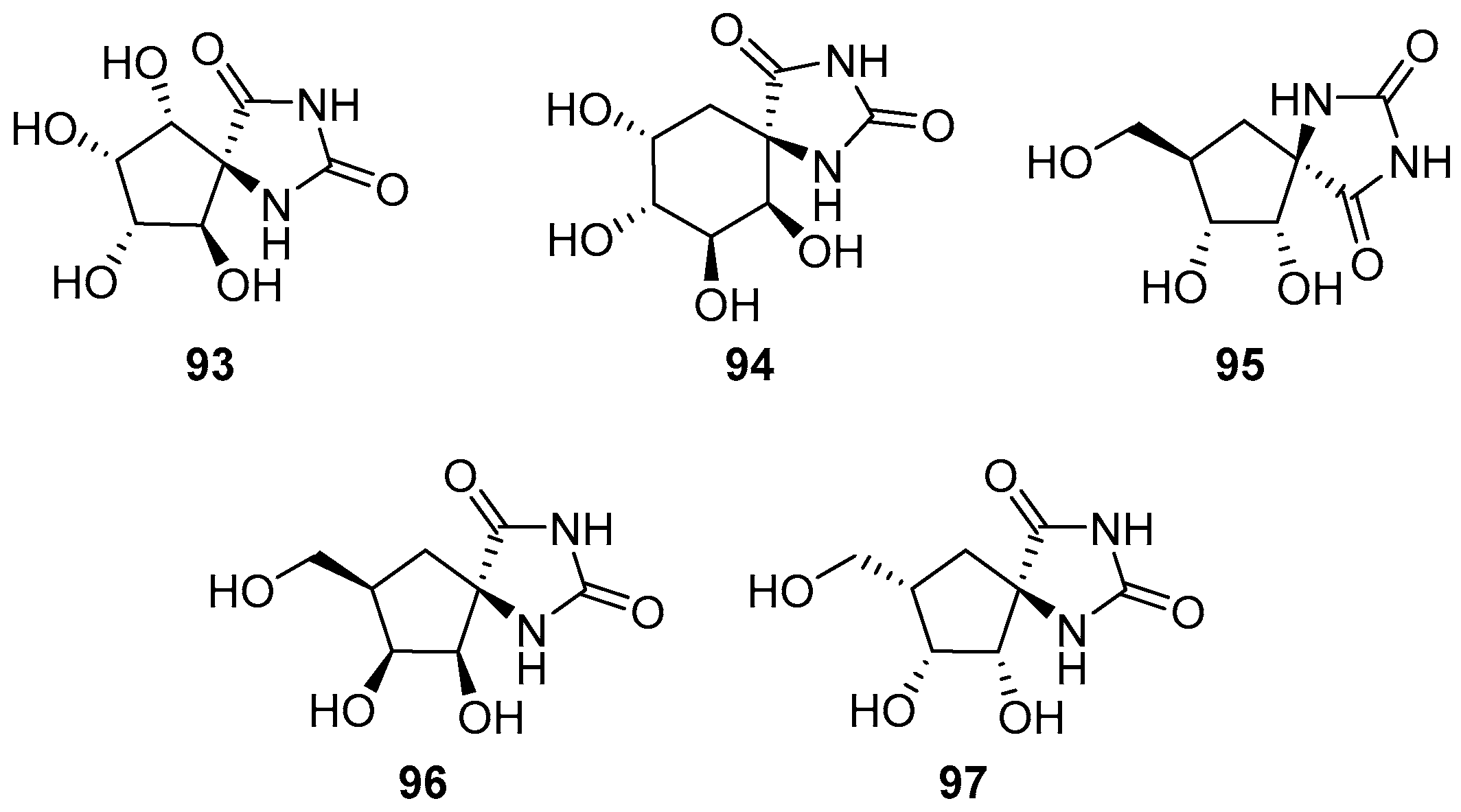
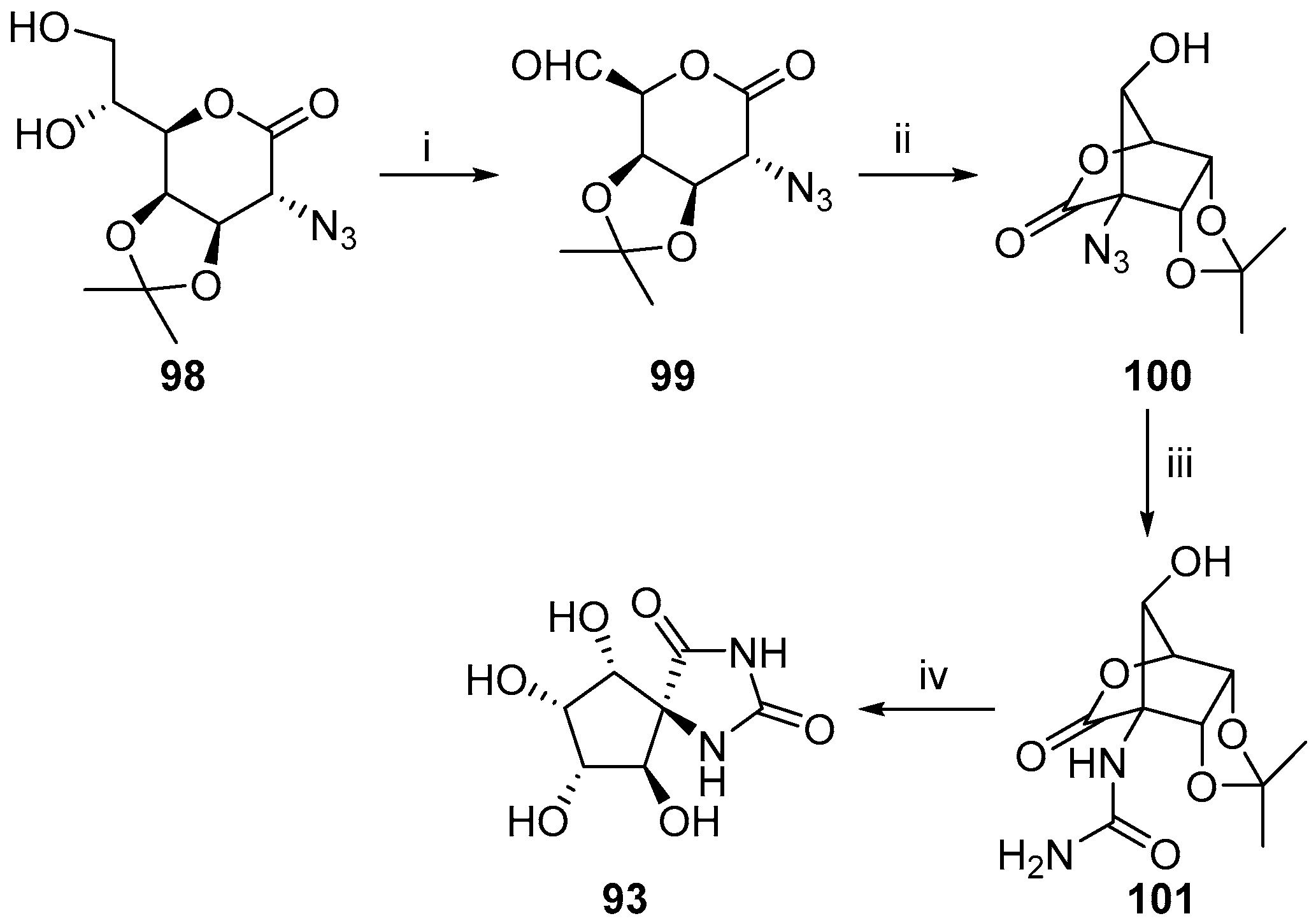
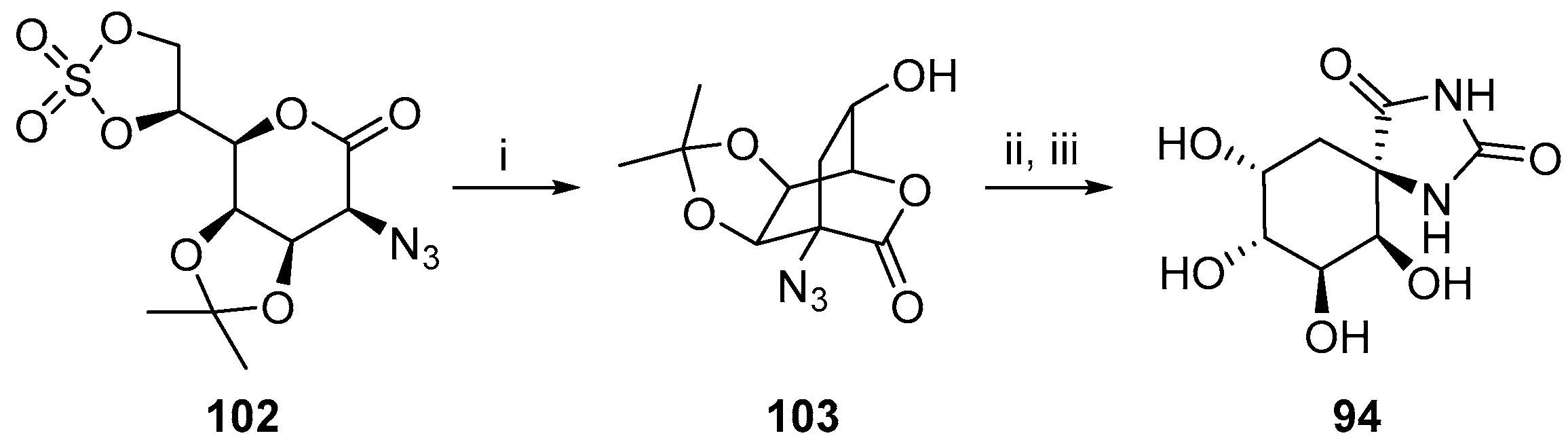
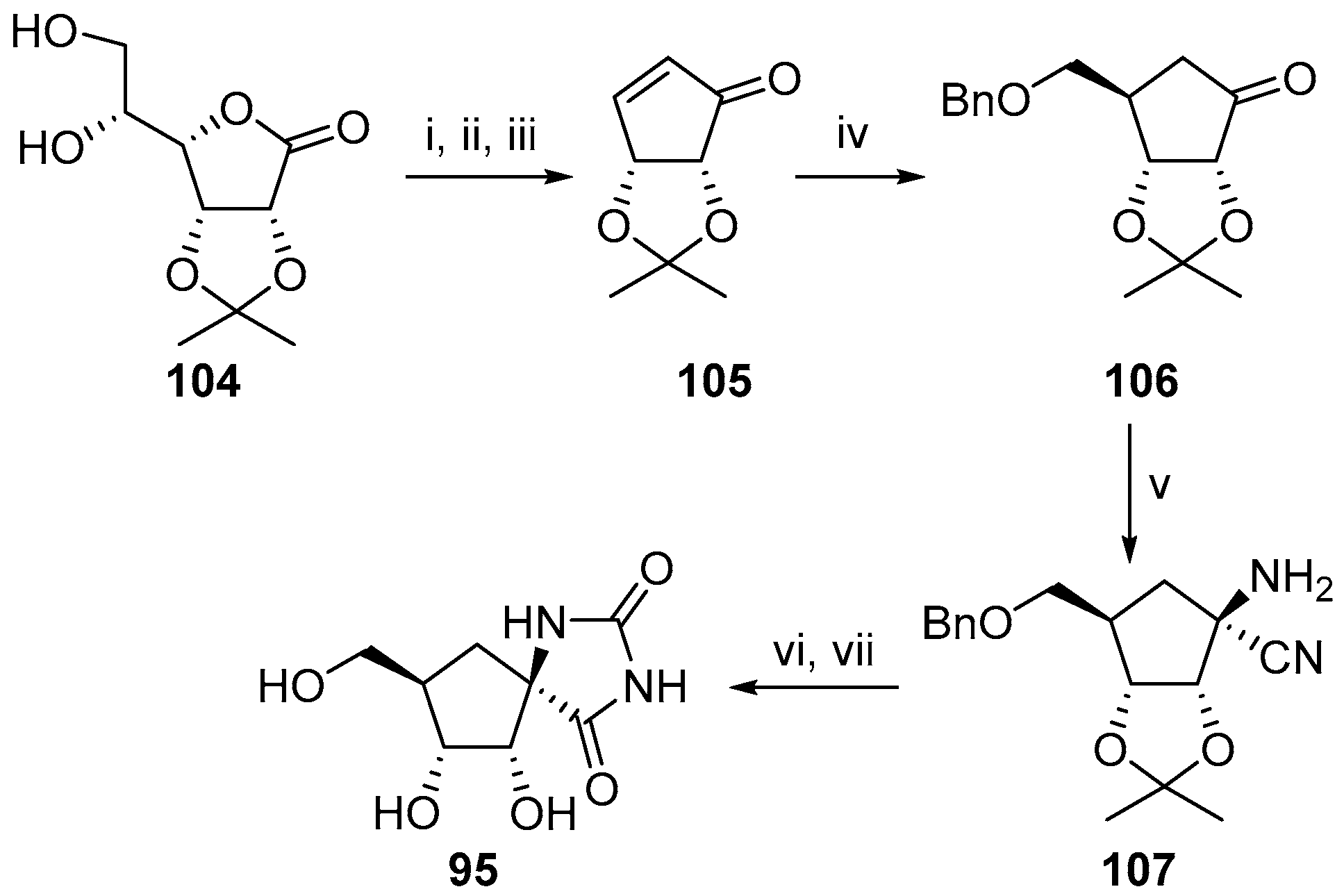
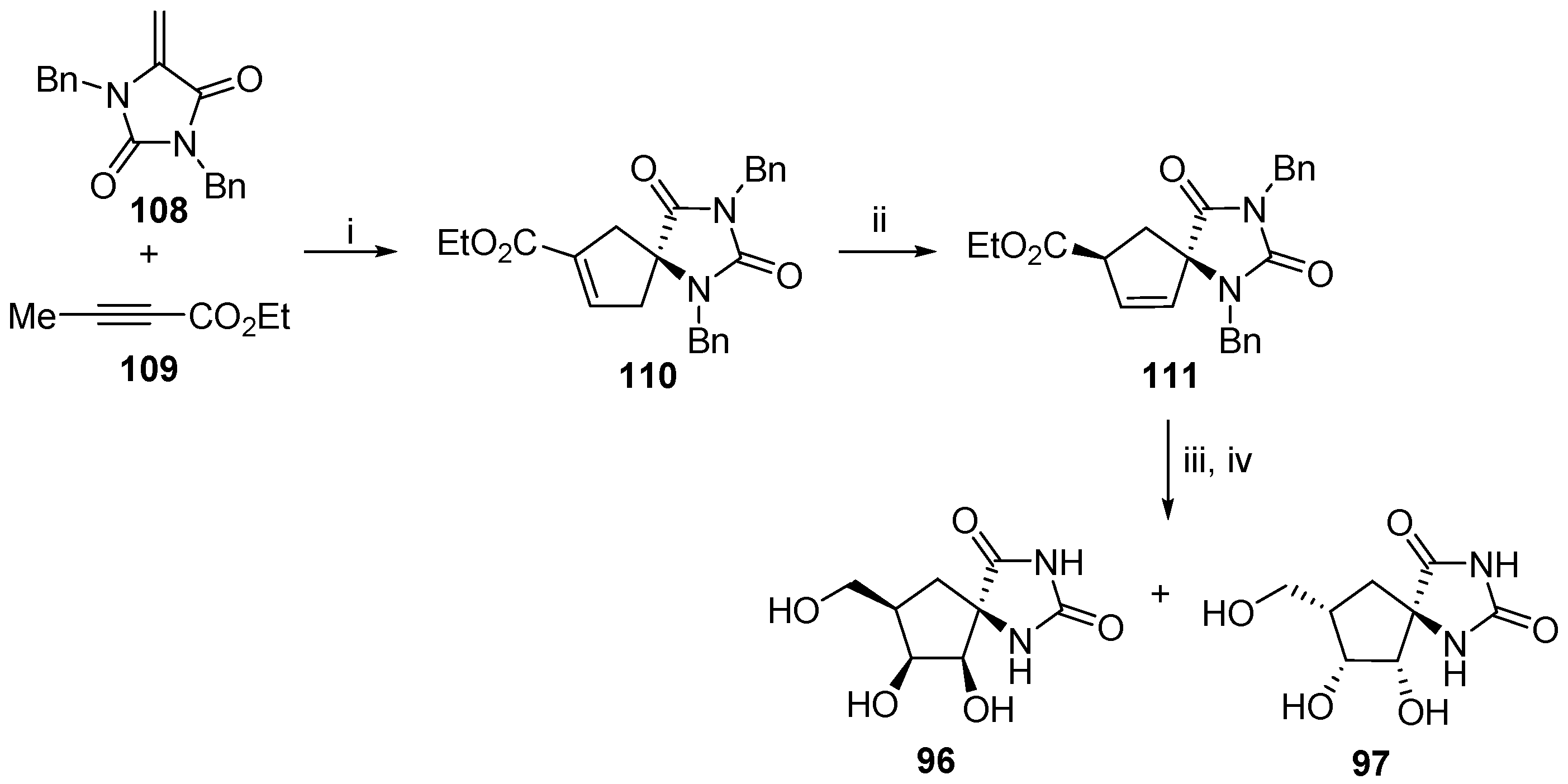
2.2.3. Carbocycles
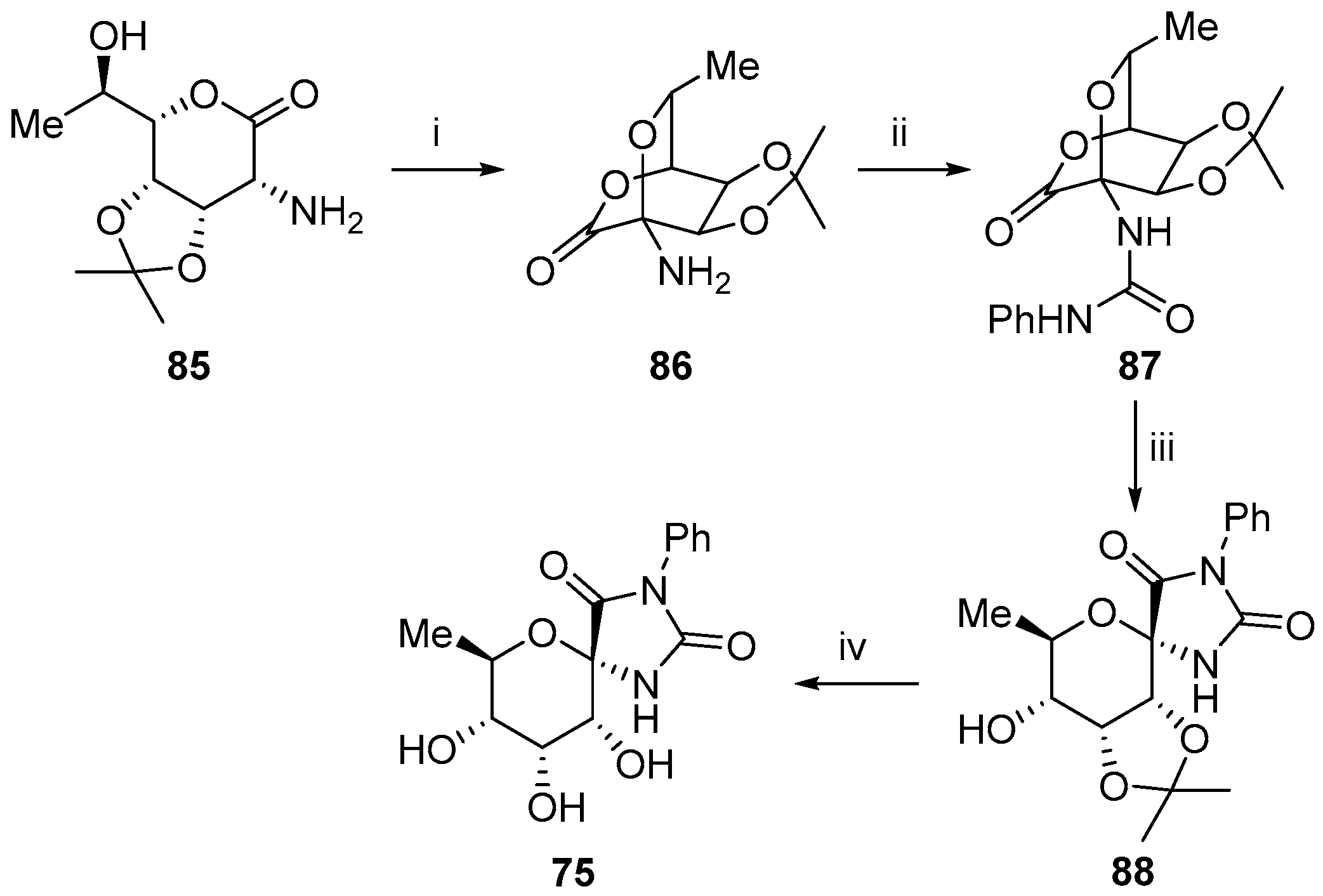
2.3. Modifications of Hydantocidin in the Hydantoin Ring
3. Synthesis of Diketopiperazine Analogues
4. Synthesis of Barbiturate Analogues
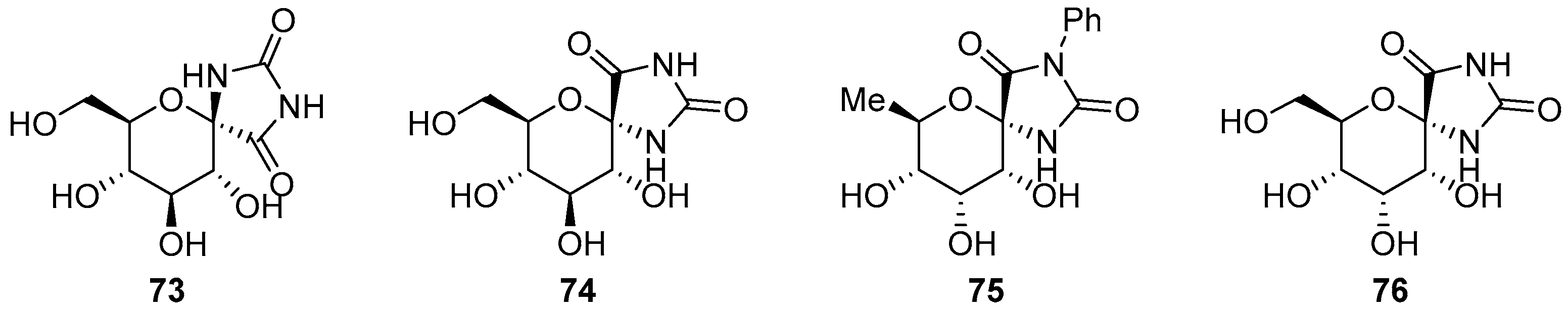
5. Synthesis of Miscellaneous Spiroheterocyclic Units
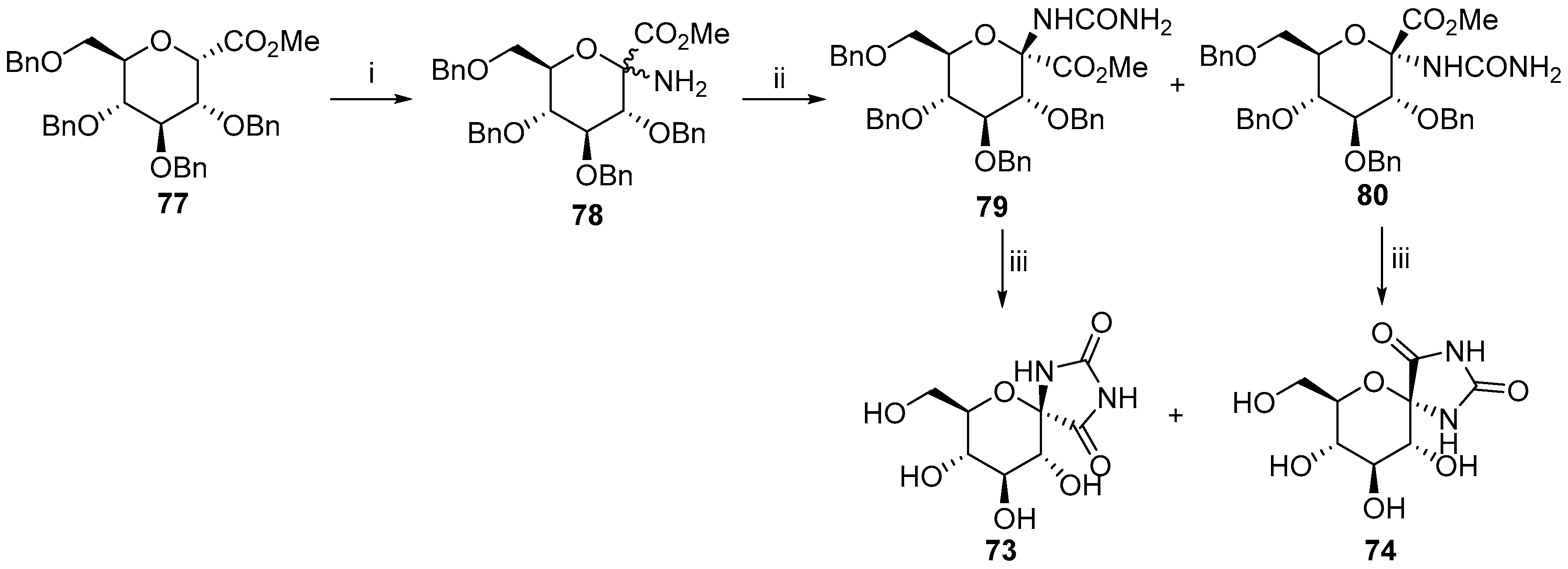
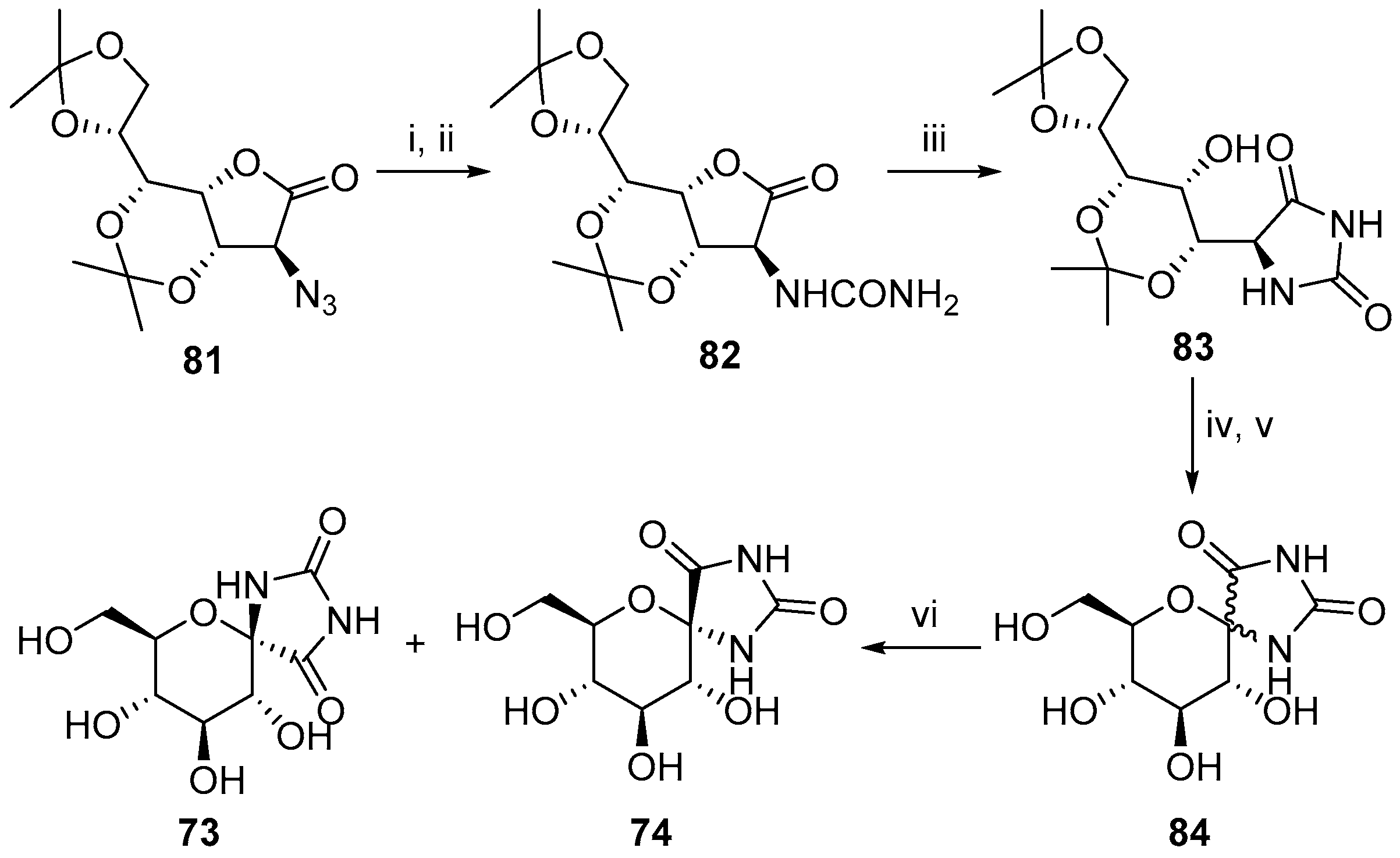
6. Polycyclic Spironucleosides
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. A 40-year journey in search of selective antiviral chemotherapy. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 1–24. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Highlights in the Discovery of Antiviral Drugs: A Personal Retrospective. J. Med. Chem. 2010, 53, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; De Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The history of antiretrovirals: Key discoveries over the past 25 years. Rev. Med. Virol. 2009, 19, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Bonate, P.L.; Arthaud, L.; Cantrell, W.R., Jr.; Stephenson, K.; Secrist, J.A., III; Weitman, S. Discovery and development of clofarabine: A nucleoside analogue for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Elgemeie, G. Thioguanine, mercaptopurine: Their analogs and nucleosides as antimetabolites. Curr. Pharm. Des. 2003, 9, 2627–2642. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Izuta, S. DNA polymerases as targets of anticancer nucleosides. Curr. Drug Targets 2004, 5, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.; Secrist, J.; Waud, W. Purine nucleoside antimetabolites in development for the treatment of cancer. Curr. Opin. Investig. Drugs 2004, 5, 592–596. [Google Scholar] [PubMed]
- Sharma, P.; Nurpeisov, V.; Hernandez-Santiago, B.; Beltran, T.; Schinazi, R. Nucleoside inhibitors of human immunodeficiency virus type 1 reverse transcriptase. Curr. Top. Med. Chem. 2004, 4, 895–919. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. New nucleoside reverse transcriptase inhibitors for the treatment of HIV infections. Curr. Opin. Pharmacol. 2004, 4, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.K. Locked nucleic acid: Tighter is different. Chem. Commun. 2013, 49, 5618–5620. [Google Scholar] [CrossRef] [PubMed]
- Marquez, V.E.; Ezzitouni, A.; Russ, P.; Siddiqui, M.A.; Ford, H.; Feldman, R.J.; Mitsuya, H.; George, C.; Barchi, J.J. HIV-1 Reverse transcriptase can discriminate between two conformationally locked carbocyclic AZT triphosphate analogues. J. Am. Chem. Soc. 1998, 120, 2780–2789. [Google Scholar] [CrossRef]
- Herdewijn, P. Targeting RNA with conformationally restricted oligonucleotides. Liebigs Ann. Chem. 1996, 1996, 1337–1348. [Google Scholar] [CrossRef]
- Kool, E.T. Preorganization of DNA: Design principles for improving nucleic acid recognition by synthetic oligonucleotides. Chem. Rev. 1997, 97, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Wengel, J. Synthesis of 3′-C- and 4′-C-branched oligodeoxynucleotides and the development of locked nucleic acid (LNA). Acc. Chem. Res. 1999, 32, 301–310. [Google Scholar] [CrossRef]
- Soengas, R.G.; Sandrina, S. Spirocyclic nucleosides in medicinal chemistry: An overview. Mini Rev. Med. Chem. 2012, 12, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Haneishi, T.; Nakajima, M.; Torikata, A.; Okazaki, T.; Tohjigamori, M.; Kawakubo, K. Process for the Production of an Imidazoledione Compound by a Strain of Streptomyces hygroscopicus. U.S. Patent 5,064,760, 12 November 1991. [Google Scholar]
- Haneishi, T.; Nakajima, M.; Torikata, A.; Okazaki, T.; Tohjigamori, M.; Kawakubo, K. Imidazoledione Compounds Useful as Herbicides. U.S. Patent 4,952,234, 28 August 1990. [Google Scholar]
- Pachlatko, J.P.; Zaehner, H.P. New Streptomyces hygroscopicus TU-2474 for Production of the Herbicide Hydantocidin and the Bactericide Homomycin. D.E. Patent 4,129,616, 03 December 1992. [Google Scholar]
- Mitsubishi Kasei Corp. Synthesis of Spiroacetal Nucleosides. J.P. Patent 04,222,589, 1990. [Google Scholar]
- Nakajima, M.; Itoi, K.; Takamatsu, Y.; Kinoshita, T.; Okazaki, T.; Kawakubo, K.; Shindo, M.; Honma, T.; Tohjigamori, M.; Haneishi, T. Hydantocidin: A new compound with herbicidal activity from Streptomyces hygroscopicus. J. Antibiot. 1991, 44, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Heim, D.R.; Cseke, C.; Gerwick, B.C.; Murdoch, M.G.; Green, S.B. Hydantocidin: a possible proherbicide inhibiting purine biosynthesis at the site of adenylosuccinate synthetase. Pestic. Biochem. Physiol. 1995, 53, 138–145. [Google Scholar] [CrossRef]
- Smith, J.L. Enzymes of nucleotide synthesis. Curr. Opin. Struct. Biol. 1995, 5, 752–757. [Google Scholar] [CrossRef]
- Gregoriou, M.; Noble, M.E.M.; Watson, K.A.; Garman, E.F.; Krülle, T.M.; de la Fuente, C.; Fleet, G.W.J.; Oikonomakos, N.G.; Johnson, L.N. The structure of a glycogen phosphorylase glucopyranose spirohydantoin complex at 1.8 A resolution and 100 K: The role of the water structure and its contribution to binding. Protein Sci. 1998, 7, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Johnson, L.N. The allosteric transition of glycogen phosphorylase. Nature 1989, 340, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Livanova, N.B.; Kornilaev, B.A. Structure, regulation, and denaturation of rabbit muscle glycogen phosphorylase b. Biochemistry 1996, 61, 1432–1442. [Google Scholar]
- Johnson, L.N.; O’Reilly, M. Control by phosphorylation. Curr. Opin. Struct. Biol. 1996, 6, 762–769. [Google Scholar] [CrossRef]
- Ortmeyer, H.K.; Bodkin, N.L.; Hansen, B.C. Insulin regulates liver glycogen synthase and glycogen phosphorylase activity reciprocally in rhesus monkeys. Am. J. Physiol. 1997, 272, E133–E138. [Google Scholar] [PubMed]
- Martin, J.L.; Veluraja, K.; Ross, K.; Johnson, L.N.; Fleet, G.W.J.; Ramsden, N.G.; Bruce, I.; Orchard, M.G.; Oikonomakos, N.G.; Papageorgiou, A.C.; et al. Glucose analogue inhibitors of glycogen phosphorylase: The design of potential drugs for diabetes. Biochemistry 1991, 30, 10101–10116. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.A.; Mitchell, E.P.; Johnson, L.N.; Son, J.C.; Bichard, C.J.F.; Orchard, M.G.; Fleet, G.W.J.; Oikonomakos, N.G.; Leonidas, D.D.; Kontou, M.; et al. Design of inhibitors of glycogen phosphorylase: A study of .alpha.- and .beta.-C-glucosides and 1-thio-.beta.-d-glucose compounds. Biochemistry 1994, 33, 5745–5758. [Google Scholar] [CrossRef] [PubMed]
- Board, M.; Bollen, M.; Stalmans, W.; Kim, Y.; Fleet, G.W.J.; Johnson, L.N. Effects of C-1-substituted glucose analogue on the activation states of glycogen synthase and glycogen phosphorylase in rat hepatocytes. Biochem. J. 1995, 311, 845–852. [Google Scholar] [CrossRef]
- Martin, J.L.; Johnson, L.N.; Withers, S.G. Comparison of the binding of glucose and glucose 1-phosphate derivatives to T-state glycogen phosphorylase b. Biochemistry 1990, 29, 10745–10757. [Google Scholar] [CrossRef] [PubMed]
- Osei, K. Diabetes, Clinical Science in Practice; Leslie, R.D.G., Robbins, D.C., Eds.; Cambridge University: Cambridge, UK, 1995; Chapter 12; ISBN 0521450292. [Google Scholar]
- DeFronzo, R.A. The triumvirate: Cell, muscle, liver: A collusion responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef] [PubMed]
- Mio, S.; Ichinose, R.; Goto, K.; Sugai, S. Synthetic studies on (+)-hydantoicin: A total synthesis of (+)-hydantoicin, a new herbicidal metabolite from microorganism. Tetrahedron 1991, 47, 2111–2120. [Google Scholar] [CrossRef]
- Mio, S.; Kumagawa, Y.; Sugai, S. Synthetic studies on (+)-hydantocidin (3): A new synthetic method for construction of the spiro-hydantoin ring at the anomeric position of d-ribofuranose. Tetrahedron 1991, 47, 2133–2144. [Google Scholar] [CrossRef]
- Prisbe, E.J.; Smejkal, J.; Verheyder, J.P.H.; Moffat, L.J.G. Halo sugar nucleosides. V. Synthesis of angustmycin A and some base analogues. J. Org. Chem. 1976, 41, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Chemla, P. Stereoselective Synthesis of (+)-Hydantocidin. Tetrahedron Lett. 1993, 34, 7391–7394. [Google Scholar] [CrossRef]
- Shiozaki, M. Syntheses of hydantocidin and C-2-thioxohydantocidin. Carbohydr. Res. 2002, 337, 2077–2088. [Google Scholar] [CrossRef]
- Mio, S.; Shiraishi, M.; Sugai, S. Synthetic studies on (+)-hydantocidin (2): Aldol addition approaches towards the stereoisomers of (+)-hydantocidin. Tetrahedron 1991, 47, 2121–2132. [Google Scholar] [CrossRef]
- Mio, S.; Ueda, M.; Hamura, M.; Kitakawa, J.; Sugai, S. Synthetic studies on (+)-hydantocidin (4): Synthesis of stereoisomers of (+)-hydantocidin. Tetrahedron 1991, 47, 2145–2154. [Google Scholar] [CrossRef]
- Fairbanks, A.J.; Fleet, G.W.J. Synthesis of 5-epihydantocidin from d-Ribose. Tetrahedron 1995, 51, 3881–3894. [Google Scholar] [CrossRef]
- Hsia, K.Y.; Ward, P.; Lamont, R.B.; Lilley, P.M.; De, Q.; Watkin, D.J.; Fleet, G.W.J. Formation of highly substituted cyclopentanes from radical and anionic Michael cyclisations of α-iodo-γ- and -δ-lactones. Tetrahedron Lett. 1994, 35, 4823–4826. [Google Scholar] [CrossRef]
- Mio, S.; Sane, H.; Shindou, M.; Honma, T.; Sugai, S. Synthesis and herbicidal activity of deoxy derivatives of (+)-hydantocidin. Agric. Biol. Chem. 1991, 55, 1105–1109. [Google Scholar]
- Burton, J.W.; Son, J.C.; Fairbanks, A.J.; Choi, S.S.; Taylor, H.; Watkin, D.J.; Winchester, B.G.; Fleet, G.W.J. Anomeric spirohydantoins of mannofuranose: Approaches to novel anomeric amino acids by oxidative ring contraction. Tetrahedron Lett. 1993, 34, 6119–6122. [Google Scholar] [CrossRef]
- Brandstetter, T.W.; Kim, Y.; Son, J.C.; Taylor, H.M.; Lilley, P.M.D.Q.; Watkin, D.J.; Johnson, L.N.; Oikonomakos, N.G.; Fleet, G.W.J. Spirohydantoins of glucofuranose: Analogues of hydantocidin. Tetrahedron Lett. 1995, 36, 2149–2152. [Google Scholar] [CrossRef]
- Blériot, Y.; Simone, M.I.; Wormald, M.R.; Dwek, R.A.; Watkin, D.J.; Fleet, G.W.J. Sugar amino acids at the anomeric position of carbohydrates: Synthesis of spirocyclic amino acids of 6-deoxy-l-lyxofuranose. Tetrahedron: Asymm. 2006, 17, 2276–2286. [Google Scholar] [CrossRef]
- Szewczyk, K. Aldono-1,5-lactones: Preparation, nucleophilic substitution and deoxygenation reactions. Pol. J. Chem. 2000, 74, 1275–1282. [Google Scholar]
- Bichard, C.J.F.; Mitchell, E.P.; Wormald, M.R.; Watson, K.A.; Johnson, L.N.; Zographos, S.E.; Koutra, D.D.; Oikonomakos, N.G.; Fleet, G.W.J. Potent inhibition of glycogen phosphorylase by a spirohydantoin of glucopyranose: First pyranose analogues of hydantocidin. Tetrahedron Lett. 1995, 36, 2145–2148. [Google Scholar] [CrossRef]
- Bichard, C.J.F.; Wheatley, J.R.; Fleet, G.W.J. Acetonides of heptonolactones: Kiliani ascension of 3-O-benzyl-d-glucose and 3-O-benzyl-d-allose. Tetrahedron Asymm. 1994, 5, 431–440. [Google Scholar] [CrossRef]
- Fuente, C.; Krülle, T.M.; Watson, K.A.; Gregoriou, M.; Johnson, L.N.; Zographos, S.E.; Oikonomakos, N.G.; Fleet, G.W.J. Glucopyranose analogues of spirohydantoins: Specific inhibitors of glycogen phosphorylase. Synlett 1997, 485–487. [Google Scholar] [CrossRef]
- Wheatley, J.R.; Beacham, A.R.; Lilley, P.M.D.Q.; Watkin, D.J.; Fleet, G.W.J. Ketals of l-rhamnoheptonolactones: Potential mimics of l-rhamnose. Tetrahedron Asymm. 1994, 5, 2523–2534. [Google Scholar] [CrossRef]
- Estevez, J.C.; Smith, M.D.; Lane, A.L.; Crook, S.; Watkin, D.J.; Besra, G.S.; Brennan, P.J.; Nash, R.J.; Fleet, G.W.J. Mimics of l-rhamnose: Analogues of rhamnopyranose containing a constituent amino acid at the anomeric position. A rhamnopyranose analogue of hydantocidin. Tetrahedron Asymm. 1996, 7, 391–394. [Google Scholar] [CrossRef]
- Brandstetter, T.W.; Wormald, M.R.; Dwek, R.A.; Butters, T.D.; Platt, F.M.; Tsitsanou, K.E.; Zographos, S.E.; Oikonomakos, N.G.; Fleet, G.W.J. A galactopyranose analogue of hydantocidin. Tetrahedron Asymm. 1996, 7, 157–170. [Google Scholar] [CrossRef]
- Köll, P.; Frötsch, A. Ein neuer effizienter weg zur darstellung von glycopyranosylcyaniden (2,6-anhydroaldononitrilen) ohne nachbargruppenbeteiligung. Reduktion von 2,6-anhydro-1-desoxy-1-nitroalditolen mit phosphortrichlorid. Carbohydr. Res. 1987, 171, 301–315. [Google Scholar] [CrossRef]
- Elliott, R.P.; Hui, A.; Fairbanks, A.J.; Nash, R.J.; Winchester, B.G.; Way, G.; Smith, C.; Lamont, B.; Storer, R.; Fleet, G.W.J. Highly substituted cis-β-cyclopentane amino acids: An approach to the synthesis of trehazolin analogues. Tetrahedron Lett. 1993, 34, 7949–7952. [Google Scholar] [CrossRef]
- Fairbanks, A.J.; Elliott, R.P.; Smith, C.; Hui, A.; Way, G.; Storer, R.; Taylor, H.; Watkin, D.J.; Winchester, B.G.; Fleet, G.W.J. Synthesis of cyclopentane spirohydantoins by aldol cyclisations: An approach to highly substituted α-cyclopentane amino acids. Tetrahedron Lett. 1993, 34, 7953–7956. [Google Scholar] [CrossRef]
- Skead, B.M.; Fleet, G.W.J.; Saunders, J.R.B.; Lament, R.B. Cyclic sulphates of δ-lactones in the synthesis of tetrahydrofurans, tetrahydropyrans and cyclohexanes. Tetrahedron Lett. 1993, 34, 6115–6118. [Google Scholar] [CrossRef]
- Fairbanks, A.J.; Hui, A.; Skead, B.M.; Lilley, P.M.; Lamont, R.B.; Storer, R.; Saunders, J.; Watkin, D.J.; Fleet, G.W.J. Polyhydroxylated cyclohexane and cyclopentane α-amino acids from cyclisations of an azidolactone. Tetrahedron Lett. 1994, 35, 8891–8894. [Google Scholar] [CrossRef]
- Sano, H.; Sugai, S. Synthesis of (±)-carbocyclic analogue of spirohydantoin nucleoside. Tetrahedron 1995, 51, 4635–4646. [Google Scholar] [CrossRef]
- Sano, H.; Sugai, S. Synthesis of an optically active carbocyclic derivative of (+)-hydantocidin. Tetrahedron Asymm. 1995, 6, 1143–1150. [Google Scholar] [CrossRef]
- Pham, T.Q.; Pyne, S.G.; Skelton, B.W.; White, A.H. Synthesis of carbocyclic hydantocidins via regioselective and diastereoselective phosphinecatalyzed [3 + 2]-cycloadditions to 5-methylenehydantoins. J. Org. Chem. 2005, 70, 6369–6377. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Sancéau, J.-Y.; Chemla, P. Synthesis of surrogate structures related to the herbicidal agent hydantocidin. Tetrahedron 1995, 51, 6669–6678. [Google Scholar] [CrossRef]
- Sano, H.; Mio, S.; Kitagawa, J.; Shindou, M.; Honma, T.; Sugai, S. Synthesis of spirothiohydantoin analogues of hydantocidin. Tetrahedron 1995, 51, 12561–12572. [Google Scholar] [CrossRef]
- Lamberth, C.; Blarer, S. Concise approach to 1-thia-hydantocidin. Synth. Commun. 1996, 26, 75–81. [Google Scholar] [CrossRef]
- Utimoto, K.; Wakabayashi, Y.; Horiie, T.; Inoue, M.; Shishiyama, Y.; Obayashi, M.; Nozaki, H. Cyanotrimethylsilane as a versatile reagent for introducing cyanide functionality. Tetrahedron 1983, 39, 967–973. [Google Scholar] [CrossRef]
- Osz, E.; Somsák, L.; Sziládgy, L.; Dinya, Z. A Straightforward route to hydantocidin analogues with pyranose ring structure. Tetrahedron 1997, 53, 5813–5824. [Google Scholar] [CrossRef]
- Osz, E.; Somsák, L.; Sziládgy, L.; Kováks, L.; Docsa, T.; Tóth, B.; Gergely, P. Efficient inhibition of muscle and liver glycogen phosphorylases by a new glucopyranosylidene-spiro-thiohydantoin. Bioorg. Med. Chem. Lett. 1999, 9, 1385–1390. [Google Scholar] [CrossRef]
- Somsák, L.; Nagy, V.; Docsa, T.; Tóth, B.; Gergely, P. Gram-scale synthesis of a glucopyranosylidene-spirothiohydantoin and its effect on hepatic glycogen metabolism studied in vitro and in vivo. Tetrahedron Asymm. 2000, 11, 405–408. [Google Scholar] [CrossRef]
- Somsák, L.; Nagy, V. A new, scalable preparation of a glucopyranosylidene-spiro-thiohydantoin: One of the best inhibitors of glycogen phosphorylases. Tetrahedron Asymm. 2000, 11, 1719–1727. [Google Scholar] [CrossRef]
- Somsák, L.; Batta, G.; Farkas, I. Preparation of acetylated C-(1-bromo-d-glycosyl) heterocycles and 1-bromo-d-glycosyl cyanides. Carbohydr. Res. 1983, 124, 43–51. [Google Scholar] [CrossRef]
- Ghoneim, A.A. Synthesis of some nucleosides derivatives from l-rhamnose with expected biological activity. Chem. Cent. J. 2011, 5, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lockhoff, O. Acetale als anomere zentren von kohlenhydraten (Hal/O- und O/O-Acetale). In Methoden der Organischen Chemie (Houben-Weyl); Hagemann, H., Klamann, D., Eds.; Thieme: Stuttgart, Germany, 1992; Volume E14a/3, p. 708. ISBN 1588900223. [Google Scholar]
- Adamczeski, M.; Reed, A.R.; Crews, P. New and known diketopiperazines from the caribbean sponge, calyx cf. podatypa. J. Nat. Prod. 1995, 58, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, Y.; Horiguchi, T.; Iinuma, S.; Tanida, S.; Hamda, S. TAN-1496 A, C and E, diketopiperazine antibiotics with inhibitory activity against mammalian DNA topoisomerase I. J. Antibiot. 1994, 47, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.I.; Musza, L.L.; Cooper, R. Structure-activity studies of the natural product substance P antagonist win 64821. Bioorg. Med. Chem. Lett. 1995, 5, 377–380. [Google Scholar] [CrossRef]
- Estevez, J.C.; Ardron, H.; Wormald, M.R.; Brown, D.; Fleet, G.W.J. Spirocyclic peptides at the anomeric position of mannofuranose. Tetrahedron Lett. 1994, 35, 8889–8890. [Google Scholar] [CrossRef]
- Estevez, J.C.; Burton, J.W.; Estevez, R.J.; Ardron, H.; Wormald, M.R.; Dwek, R.A.; Brown, D.; Fleet, G.W.J. Spirodiketopiperazines of mannofuranose: Carbopeptoid α-amino acid esters at the anomeric position of mannofuranose. Tetrahedron Asymm. 1998, 9, 2137–2154. [Google Scholar] [CrossRef]
- Estévez, J.C.; Estévez, R.J.; Ardron, H.; Wormald, M.R.; Brown, D.; Fleet, G.W.J. Tri- and tetra-peptides incorporating an α-amino acid at the anomeric position of mannofuranose. Tetrahedron Lett. 1994, 35, 8885–8888. [Google Scholar] [CrossRef]
- Long, D.D.; Tennant-Eyles, R.J.; Estevez, J.C.; Wormald, M.R.; Dwek, R.A.; Smith, M.D.; Fleet, G.W.J. Carbopeptoids: Peptides and diketopiperazines incorporating the anomeric centre of mannopyranose. J. Chem. Soc. Perkin Trans. 1 2001, 8, 807–813. [Google Scholar] [CrossRef]
- Krülle, T.M.; Watson, K.A.; Gregoriou, M.; Johnson, L.N.; Crook, S.; Watkin, D.J.; Griffiths, R.C.; Nash, R.J.; Tsitsanou, K.E.; Zographos, S.E.; et al. Specific inhibition of glycogen phosphorylase by a spirodiketopiperazine at the anomeric position of glucopyranose. Tetrahedron Lett. 1995, 36, 8291–8294. [Google Scholar] [CrossRef]
- Estevez, J.C.; Saunders, J.; Besra, G.S.; Brennan, P.J.; Nash, R.J.; Fleet, G.W.J. Mimics of l-rhamnose: Synthesis of C-glycosides of l-rhamnofuranose and an α-azidoester as divergent intermediates for combinatorial generation of rhamnofuranose libraries. Tetrahedron Asymm. 1996, 7, 383–386. [Google Scholar] [CrossRef]
- Paquette, L.A.; Brand, S.; Behrens, C. An enantioselective ring expansion route leading to furanose and pyranose nucleosides featuring spirodiketopiperazines at the anomeric position. J. Org. Chem. 1999, 64, 2010–2025. [Google Scholar] [CrossRef] [PubMed]
- Kyogoku, Y.; Lord, R.C.; Rich, A. Specific hydrogen bonding of barbiturates to adenine derivatives. Nature 1968, 218, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Suzuki, T.; Wellman, S.E.; Ho, I.K. Pharmacology of barbiturate tolerance/dependence: GABAA receptors and molecular aspects. Life Sci. 1996, 59, 169–195. [Google Scholar] [CrossRef]
- Renard, A.; Lhomme, J.; Kotera, M. Synthesis and properties of Spiro nucleosides containing the barbituric acid moiety. J. Org. Chem. 2002, 67, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Mio, S.; Kitakawa, J.; Sugai, S. Stereocontrolled synthesis of spirodihydrouracil nucleoside. Tetrahedron Asymm. 1994, 5, 2233–2240. [Google Scholar] [CrossRef]
- Sano, H.; Mio, S.; Hamura, M.; Kitagawa, J.; Shindou, M.; Honma, T.; Sugai, S. Synthesis and herbicidal activity of hydantocidin analogues: Modification of the carbonyl groups in spirohydantoin. Biosci. Biotechnol. Biochem. 1995, 59, 2247–2250. [Google Scholar] [CrossRef]
- Fuentes, J.; Salameh, B.A.B.; Pradera, M.A.; Fernandez de Cordoba, F.J.; Gasch, C. Stereocontrolled synthesis of thiohydantoin spiro-nucleosides from sugar spiroacetals. Tetrahedron 2006, 62, 97–111. [Google Scholar] [CrossRef]
- Gasch, C.; Pradera, M.A.; Salameh, B.A.B.; Molina, J.L.; Fuentes, J. Isothiocyanato derivatives of sugars in the stereoselective synthesis of spironucleosides and spiro-C-glycosides. Tetrahedron Asymm. 2001, 12, 1267–1277. [Google Scholar] [CrossRef]
- Vangala, M.; Shinde, G.P. Synthesis of d-fructose-derived spirocyclic 2-substituted-2-oxazoline ribosides. Beilstein J. Org. Chem. 2015, 11, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.; Golkowski, M.; Ziegler, T. Spiro-fused carbohydrate oxazoline ligands: Synthesis and application as enantio-discrimination agents in asymmetric allylic alkylation. Beilstein J. Org. Chem. 2016, 12, 166–171. [Google Scholar] [CrossRef] [PubMed]
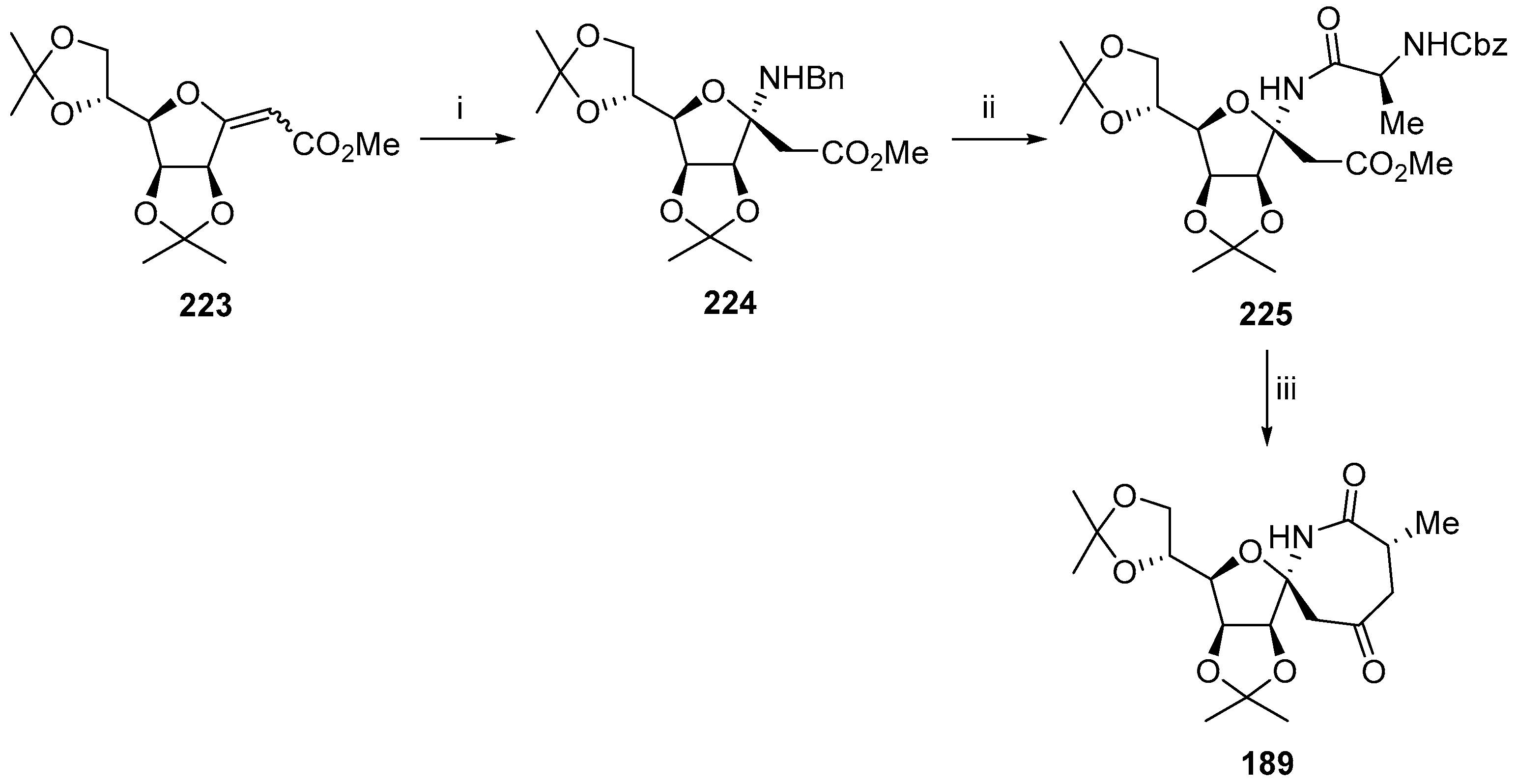
- Soengas, R.G. A straightforward route to novel α,α-disubstituted tetrahydrofuran β-amino acids and spirodiketopiperazines from sugar lactones. Synlett 2010, 2549–2552. [Google Scholar] [CrossRef]
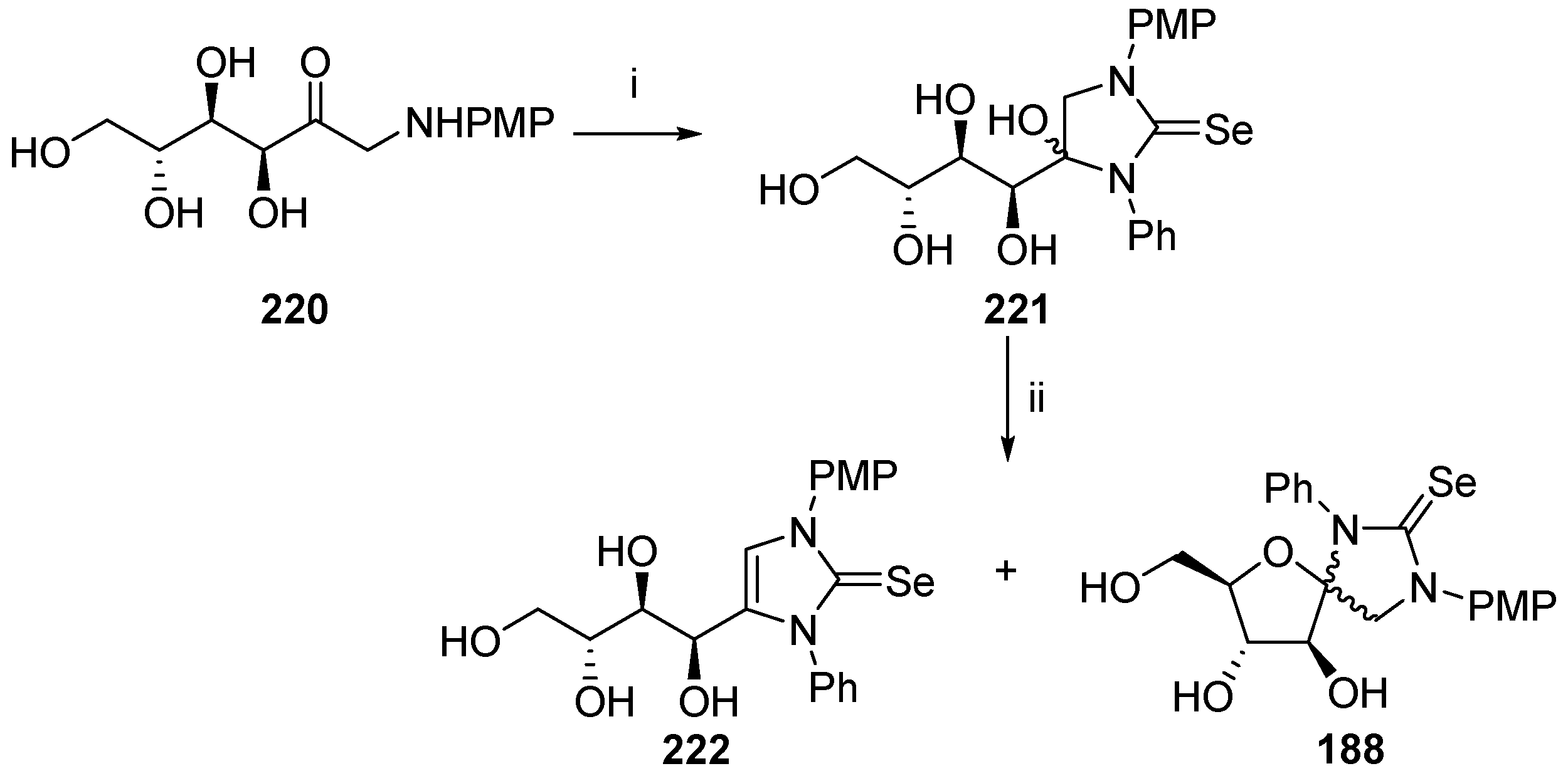
- Maza, S.; López, O.; Martos, S.; Maya, I.; Fernández-Bolaños, J.G. Synthesis of the first selenium-containing acyclic nucleosides and anomeric spironucleosides from carbohydrate precursors. Eur. J. Org. Chem. 2009, 5239–5246. [Google Scholar] [CrossRef]
- Taillefumier, C.; Thielges, S.; Chapleur, Y. Anomeric spiro-annelated 1,4-diazepine 2,5-diones from furano exo-glycals: Towards a new class of spiro-nucleosides. Tetrahedron 2004, 60, 2213–2224. [Google Scholar] [CrossRef]
- Lakhrissi, M.; Chapleur, Y. Wittig Olefination of Lactones. Angew. Chem. Int. Ed. Engl. 1996, 35, 750–752. [Google Scholar] [CrossRef]
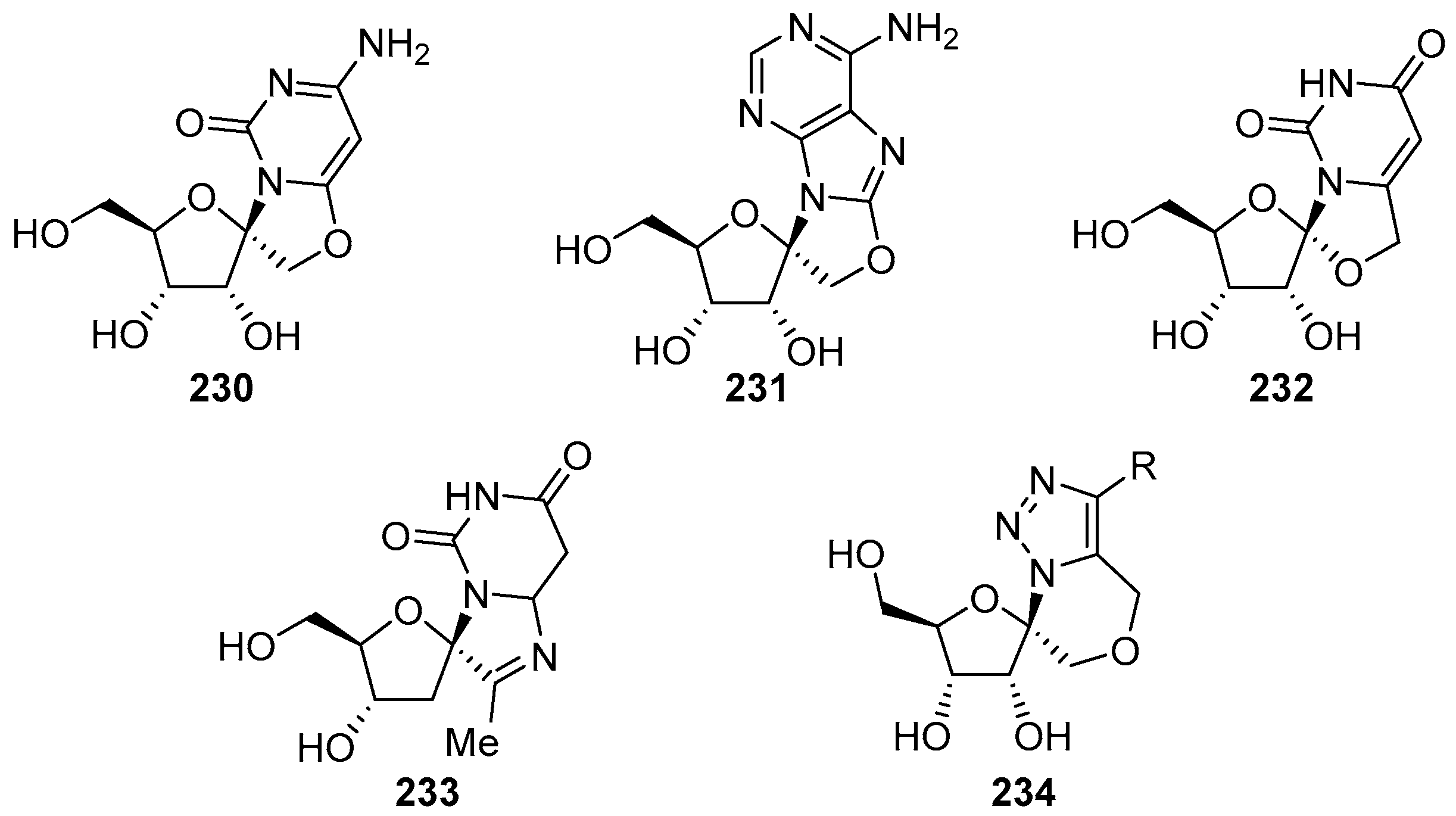
- Nakahara, T.; Okamoto, N.; Suzuki, K.; Kanie, O. Synthetic studies towards a new scaffold, spirobicycloimidazoline. Carbohydr. Res. 2008, 343, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Perez-Martin, I.; Suarez, E. Intramolecular hydrogen abstraction promoted by amidyl radicals. Evidence for electronic factors in the nucleophilic cyclization of ambident amides to oxocarbenium ions. Org. Lett. 2005, 7, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
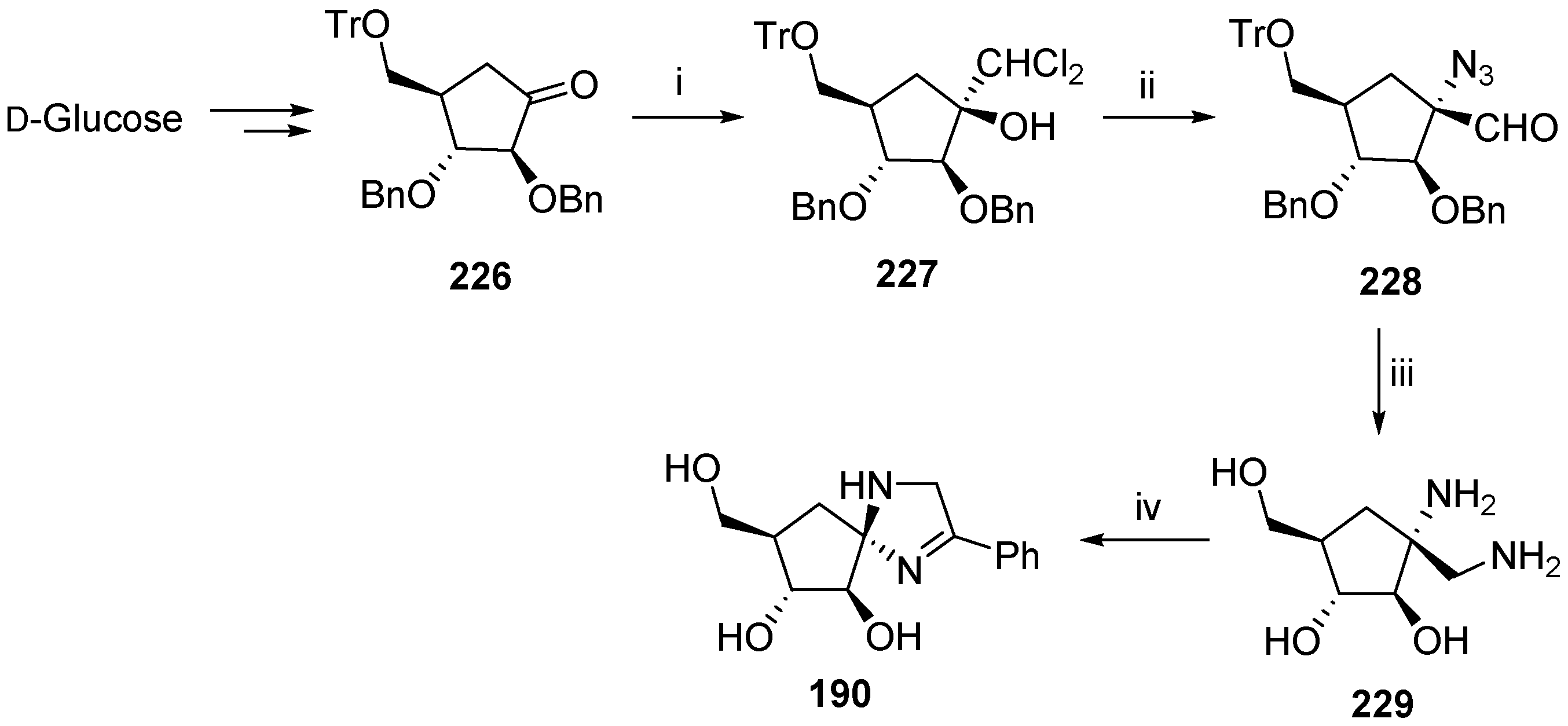
- Pal, A.P.J.; Kadigachalam, P.; Mallick, A.; Doddi, V.R.; Vankar, Y.D. Synthesis of sugar-derived spiroaminals via lactamization and metathesis reactions. Org. Biomol. Chem. 2011, 9, 809–819. [Google Scholar]
- Pal, A.P.J.; Vankar, Y.D. Azidation of anomeric nitro sugars: Application in the synthesis of spiroaminals as glycosidase inhibitors. Tetrahedron Lett. 2010, 51, 2519–2524. [Google Scholar]
- Martin, A.; Perez-Martin, I.; Suarez, E. Synthesis of oxa-aza spirobicycles by intramolecular hydrogen atom transfer promoted by N-radicals in carbohydrate systems. Tetrahedron 2009, 65, 6147–6155. [Google Scholar] [CrossRef]
- Benltifa, M.; De Kiss, M.; Garcia-Moreno, M.I.; Mellet, C.O.; Gueyrard, D.; Wadouachi, A. Regioselective synthesis and biological evaluation of spiro-sulfamidate glycosides from exo-glycals. Tetrahedron Asymm. 2009, 20, 1817–1823. [Google Scholar] [CrossRef]
- Zavgorodny, S.G. A novel type of anhydronucleosides to model syn-conformers of natural nucleosides. Tetrahedron Lett. 1981, 22, 3003–3006. [Google Scholar] [CrossRef]
- Farkaii, J.; Sorm, F. Nucleic acid components and their analogues. XXX. The synthesis of psicofuranine. Collect. Czechoslov. Chem. Commun. 1963, 28, 882–886. [Google Scholar] [CrossRef]
- Gimisis, T.; Castellari, C.; Chatgilialoglu, C. A new class of anomeric spironucleosides. Chem. Commun. 1997, 0, 2089–2090. [Google Scholar] [CrossRef]
- Groziak, M.P.; Koohang, A.; Stevens, W.C.; Robinson, P.D. A new class of nucleosides possessing unusual physical properties: Syntheses, hydration, and structural equilibria of 1-(.beta.-d-glycofuranosyl)uracil-6-carboxaldehydes. J. Org. Chem. 1993, 58, 4054–4060. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Gimisis, T. Anionically induced formation of anomeric spironucleosides from l′-C-Cyano-2′-deoxyuridine. Tetrahedron Lett. 1999, 40, 2837–2840. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Gimisis, T.; Roberti, M.; Balzarini, J.; De Clercq, E. Synthesis and biological evaluation of novel branched and spironucleoside analogues. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1565–1581. [Google Scholar] [CrossRef] [PubMed]
- Dell’Isola, A.; McLachlan, M.M.W.; Neuman, B.W.; Al-Mullah, H.M.N.; Binks, A.W.D.; Elvidge, W.; Shankland, K.; Cobb, A.J.A. Synthesis and antiviral properties of spirocyclic [1,2,3]-triazolooxazine nucleosides. Chem. Eur. J. 2014, 20, 11685–11689. [Google Scholar] [CrossRef] [PubMed]
- Perali, R.S.; Mandava, S.; Bandi, R. A convenient synthesis of l-ribose from d-fructose. Tetrahedron 2011, 67, 4031–4035. [Google Scholar] [CrossRef]
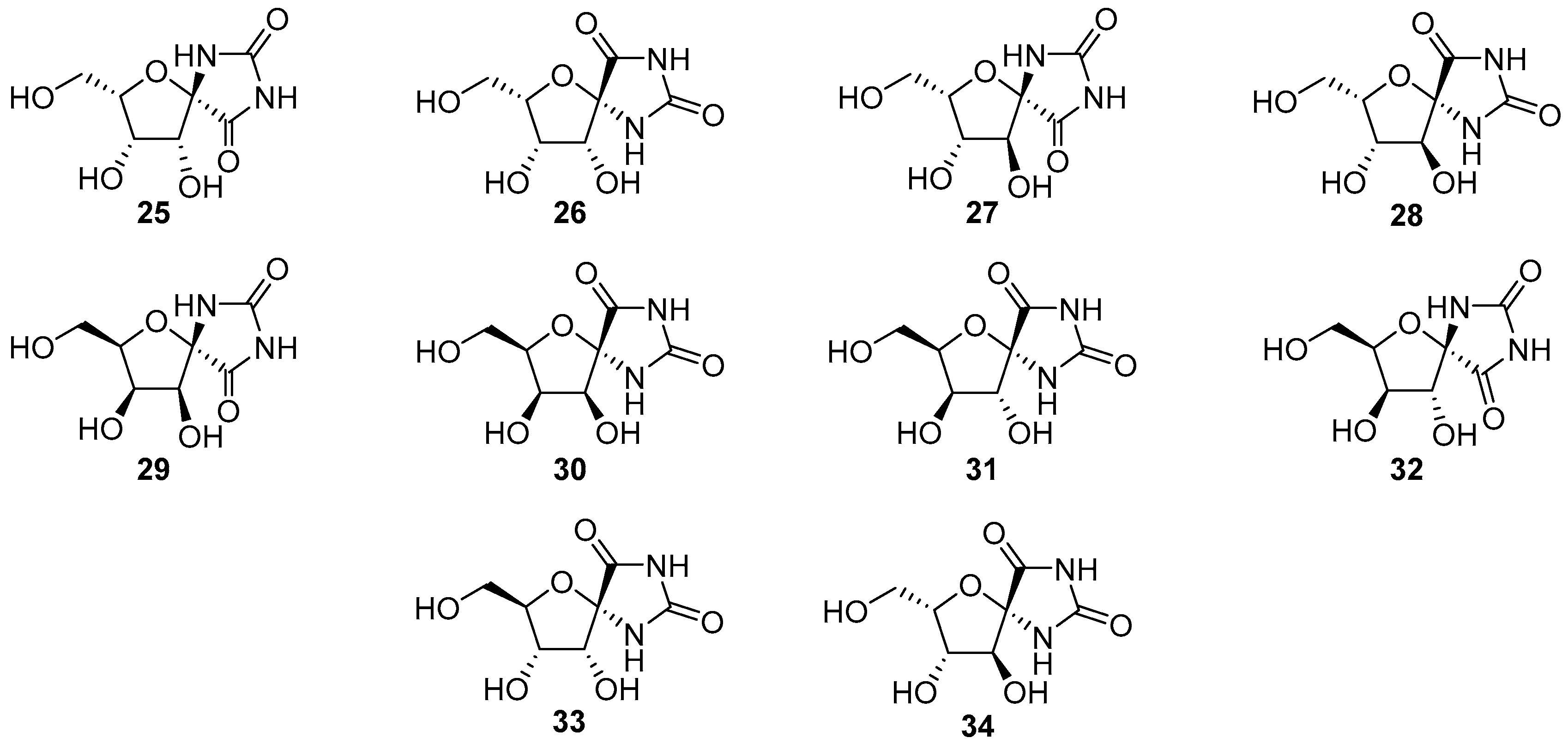
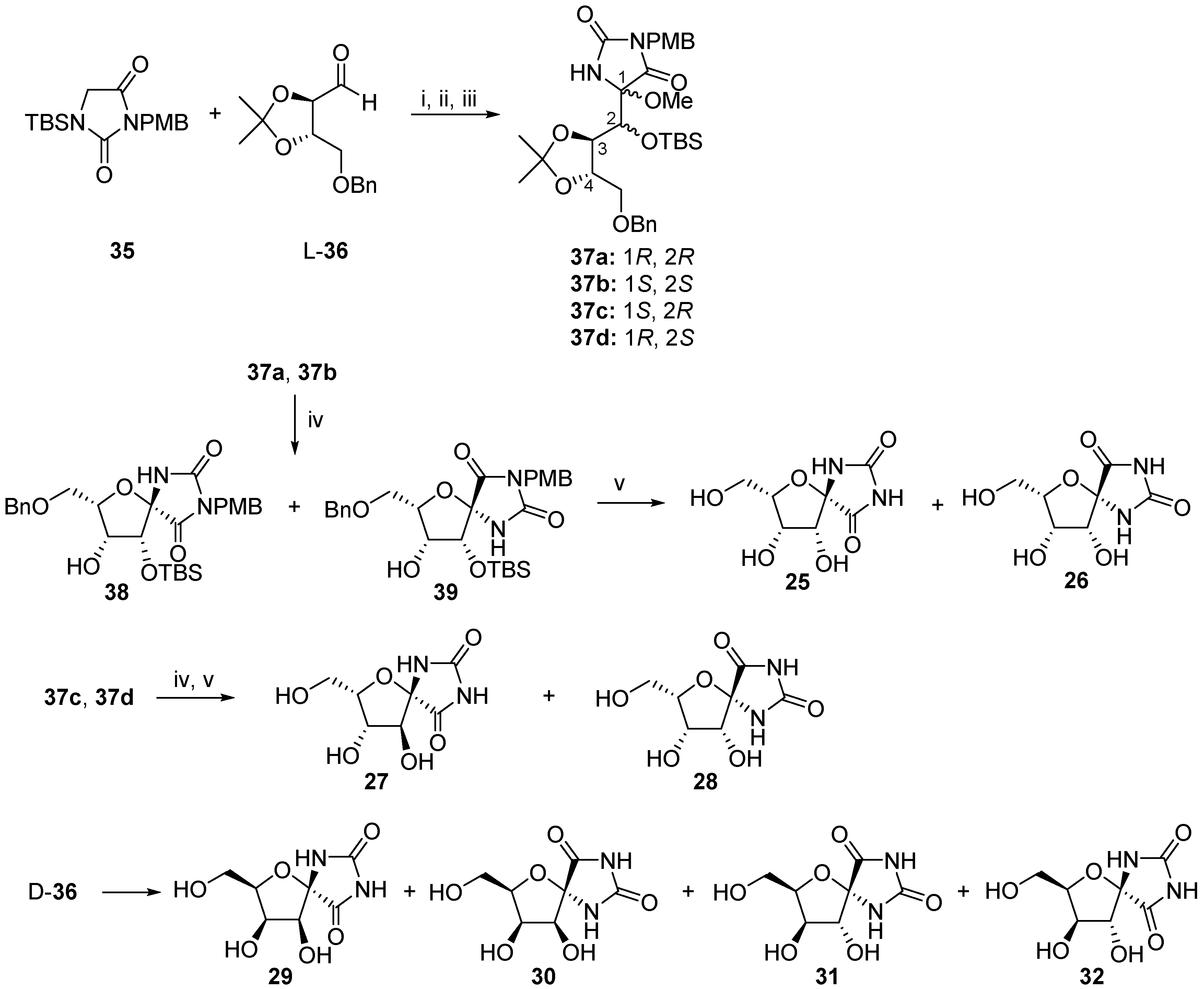
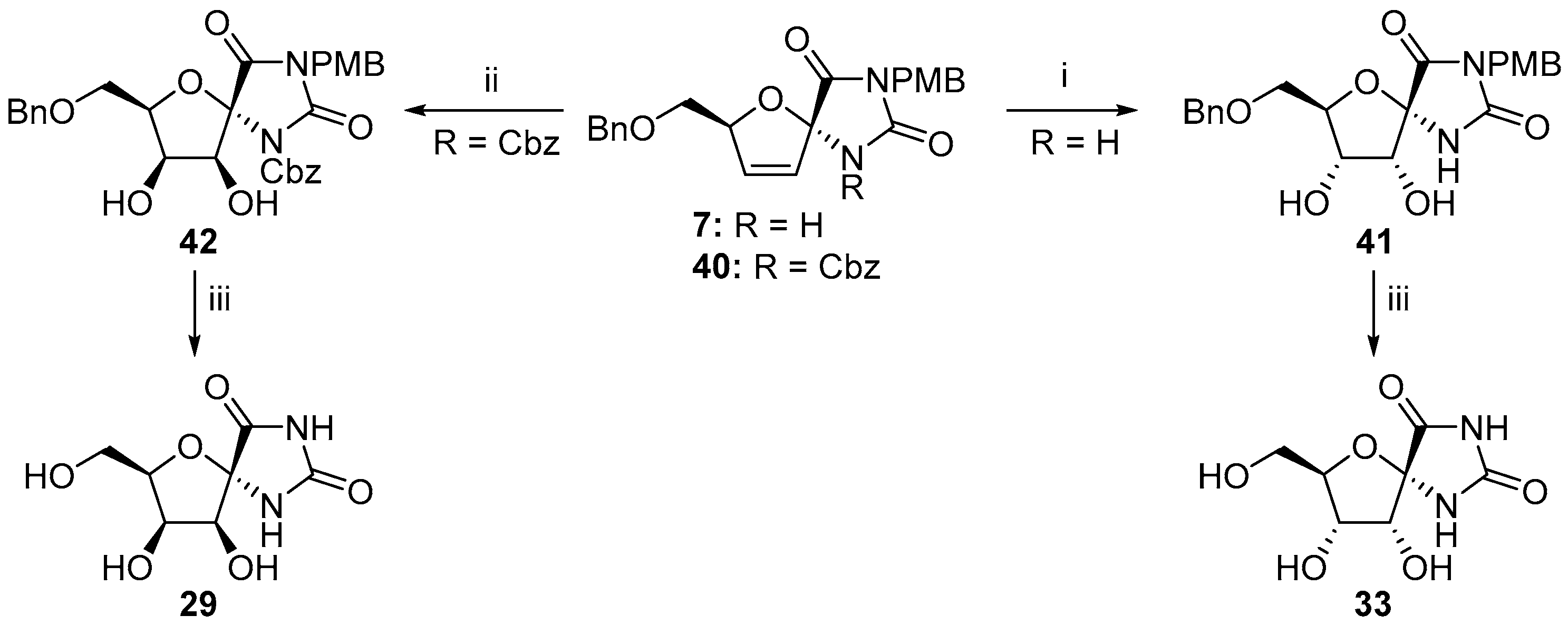
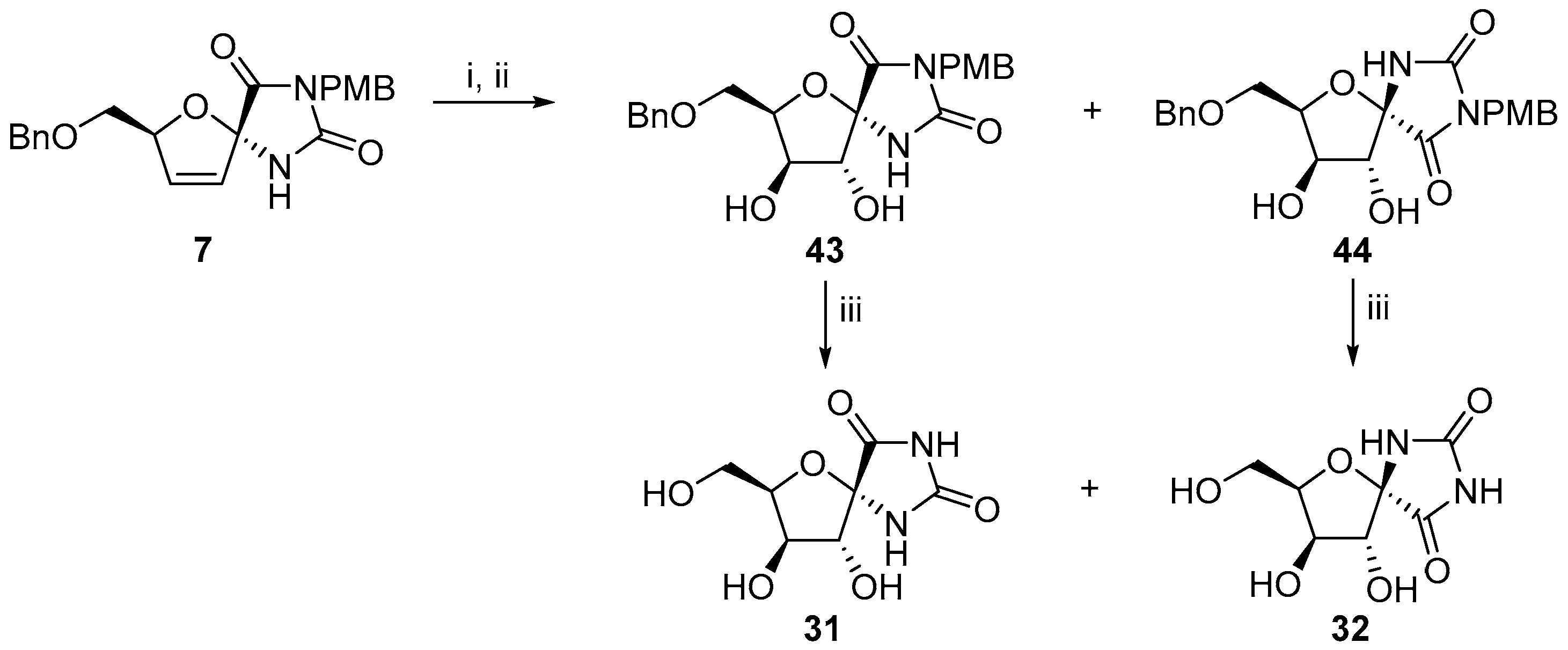

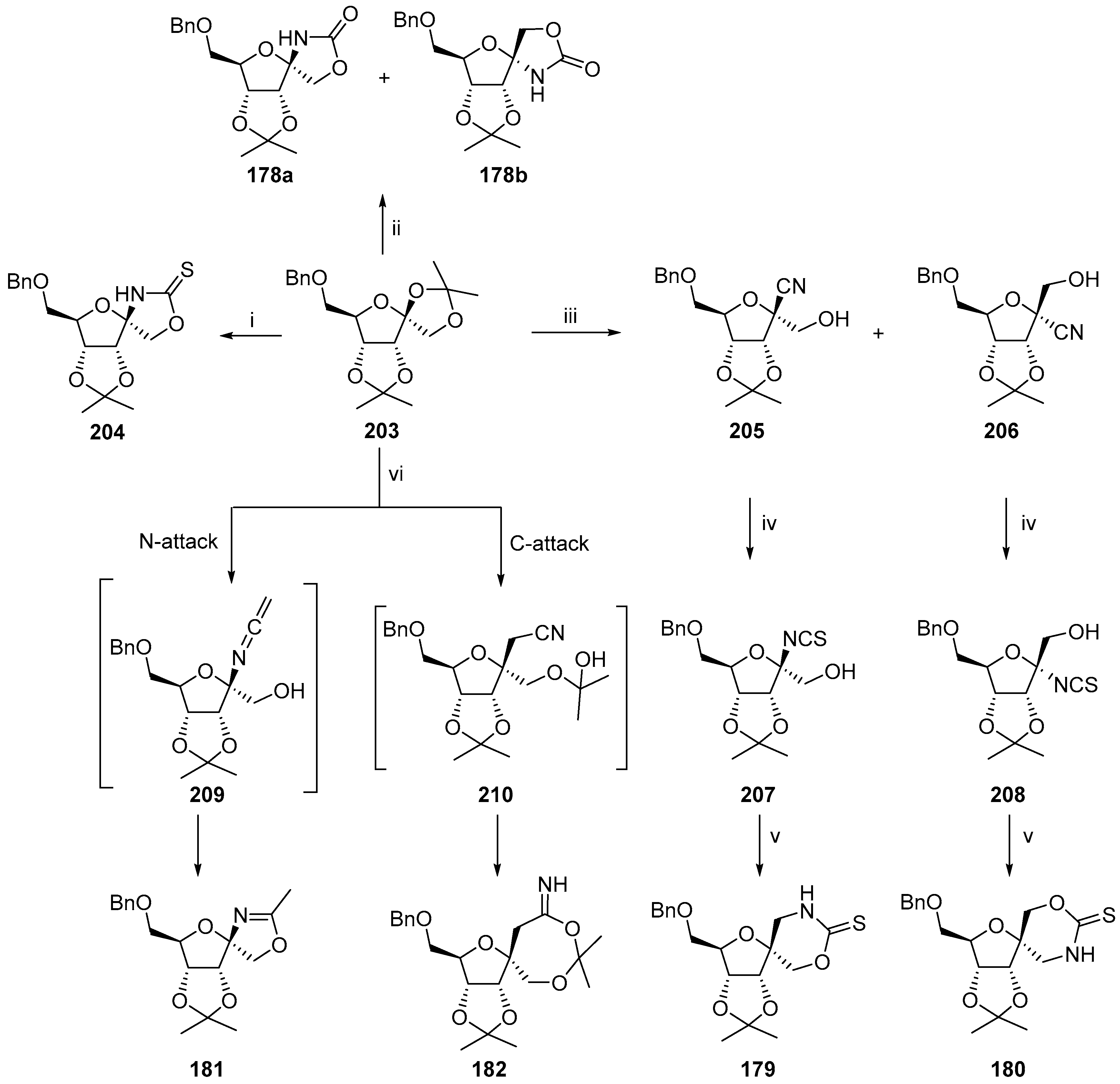
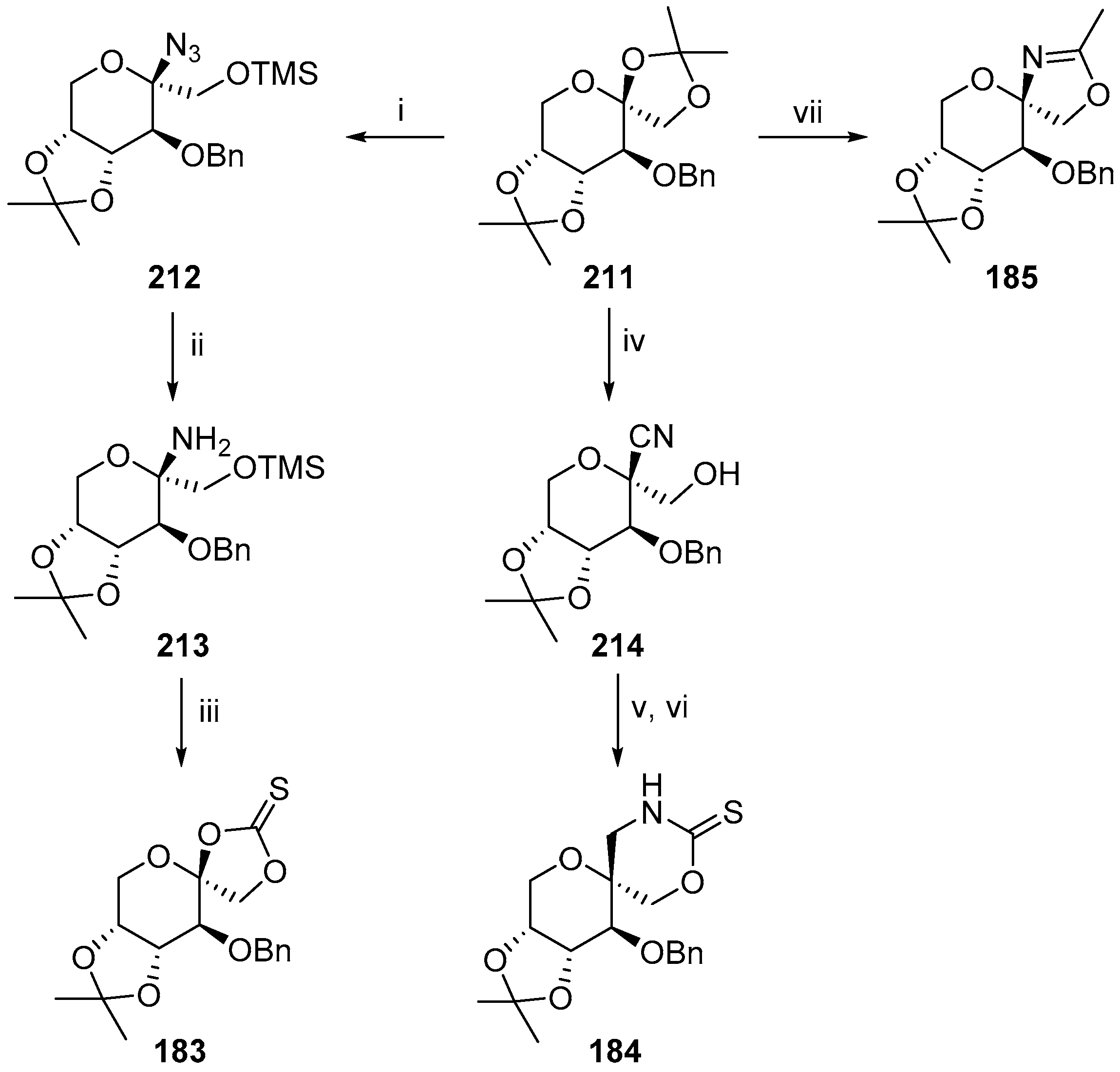
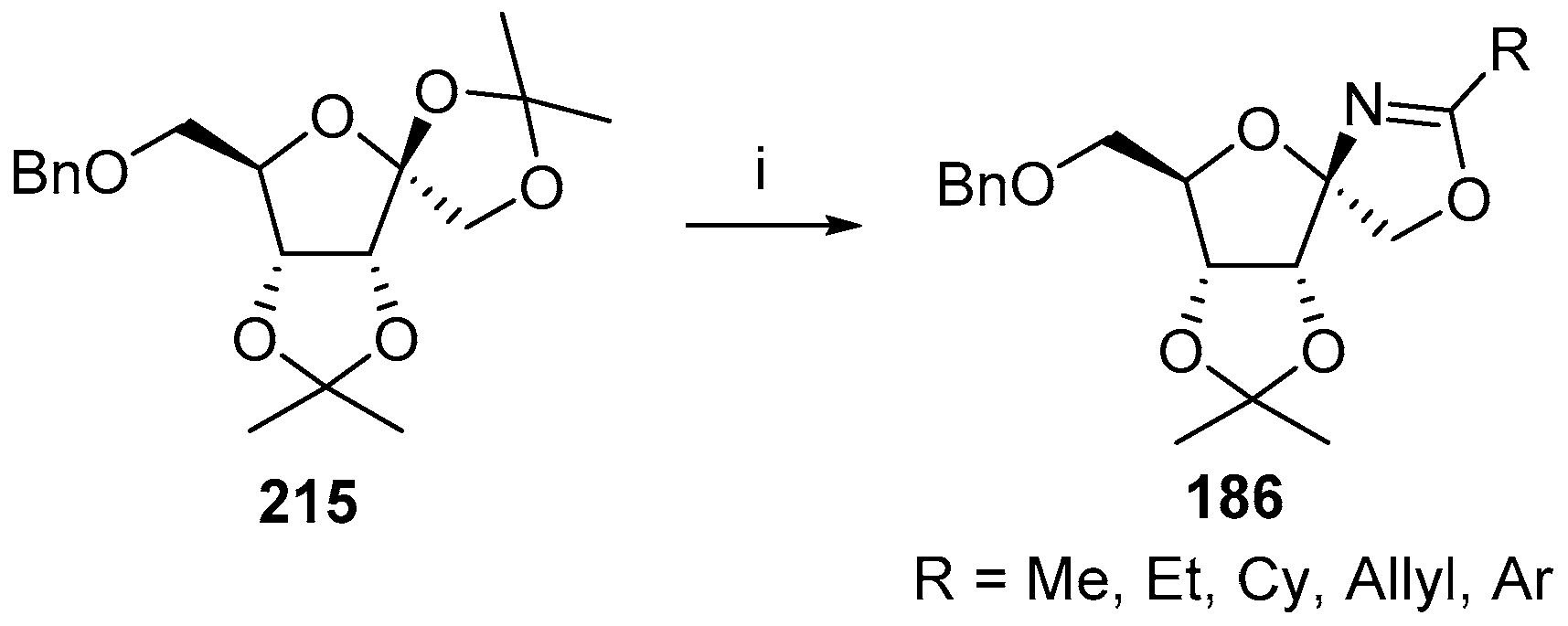
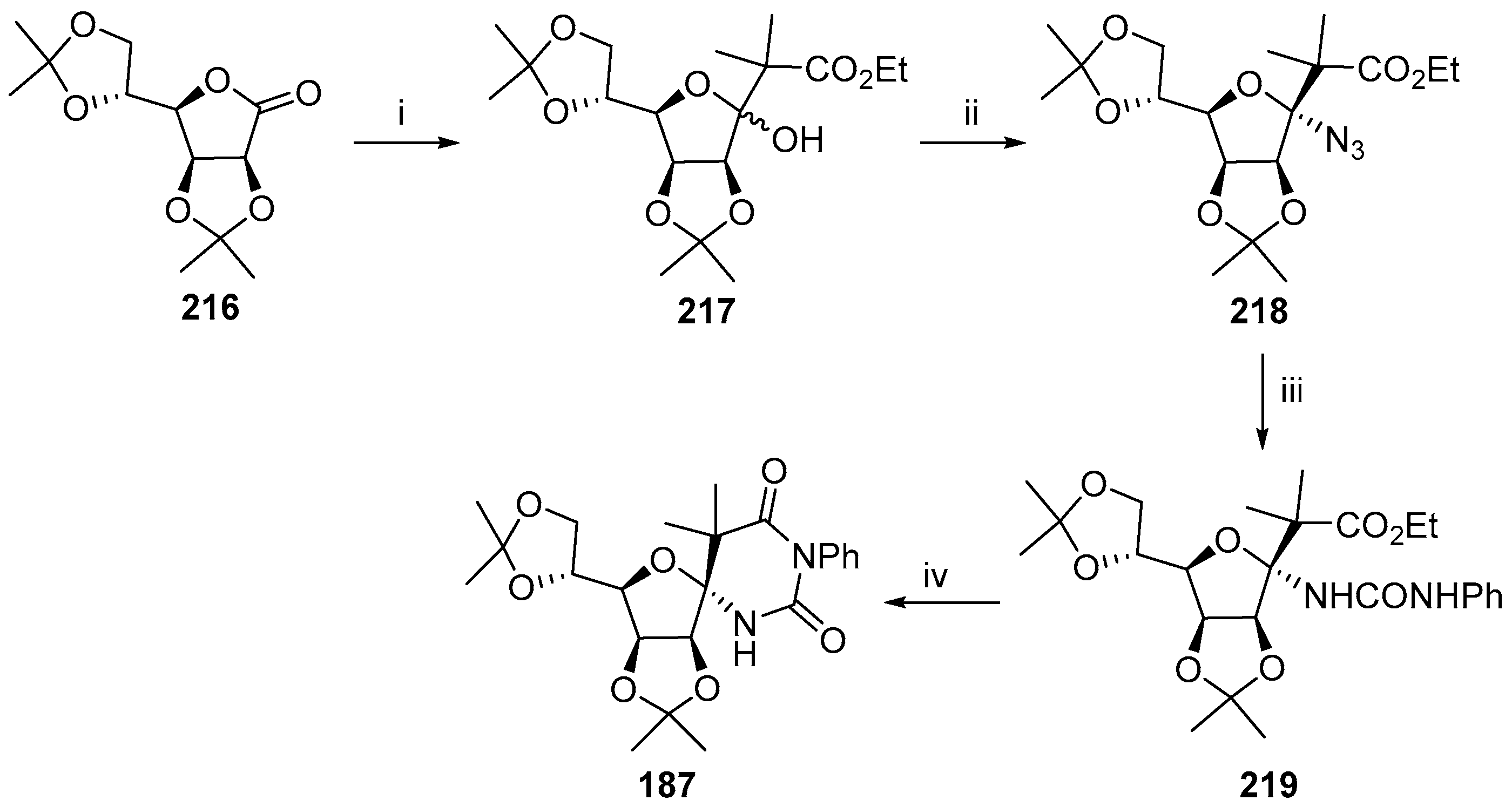
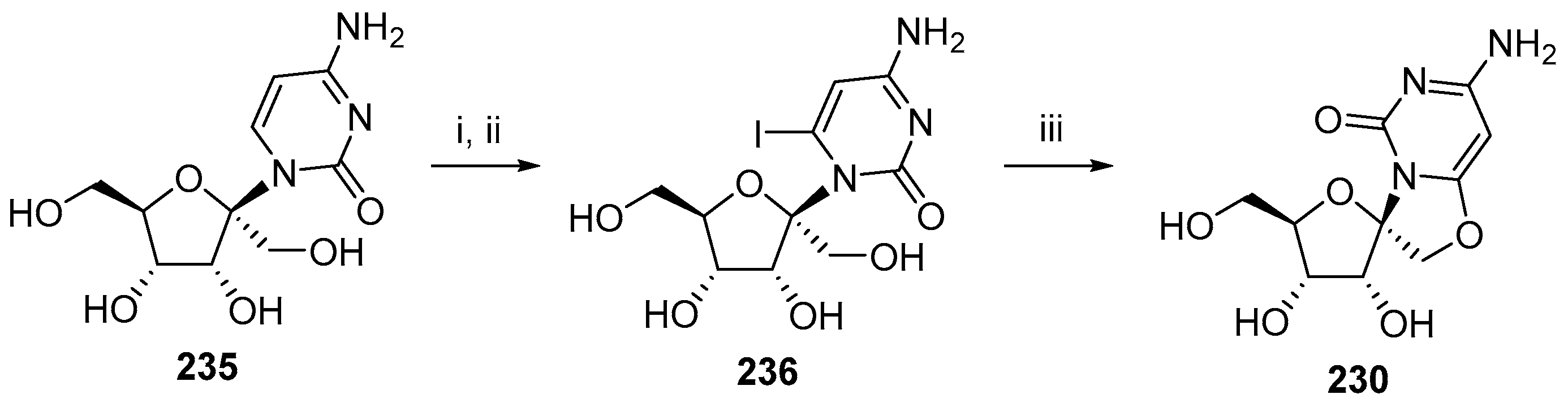

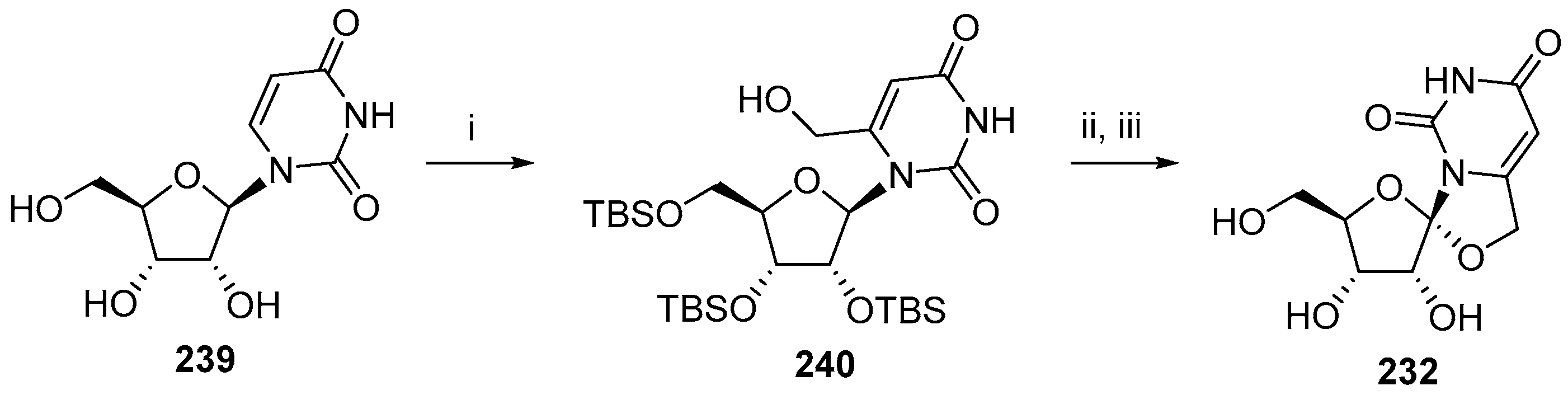
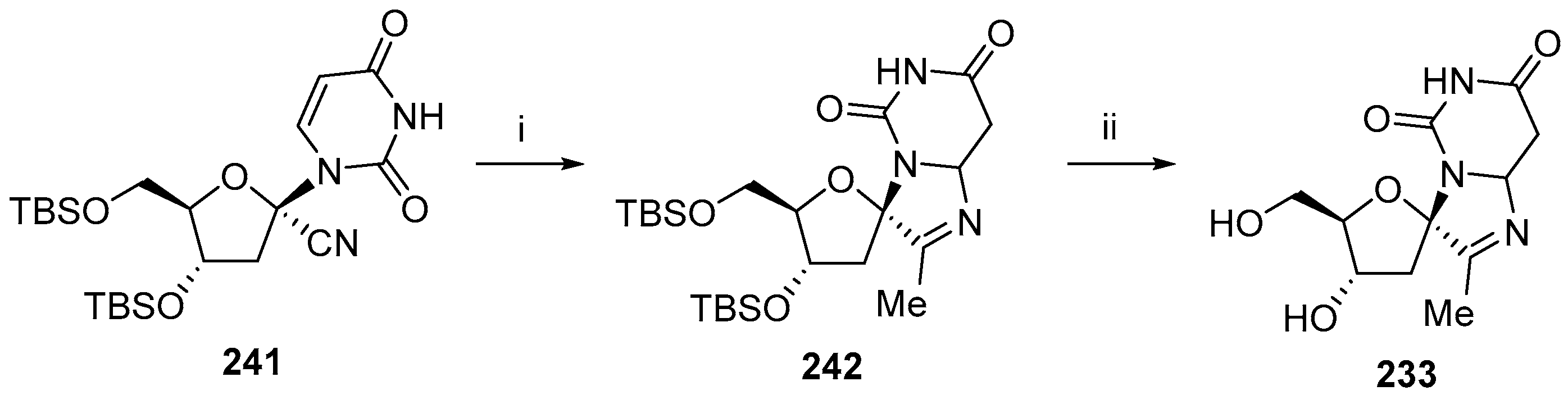
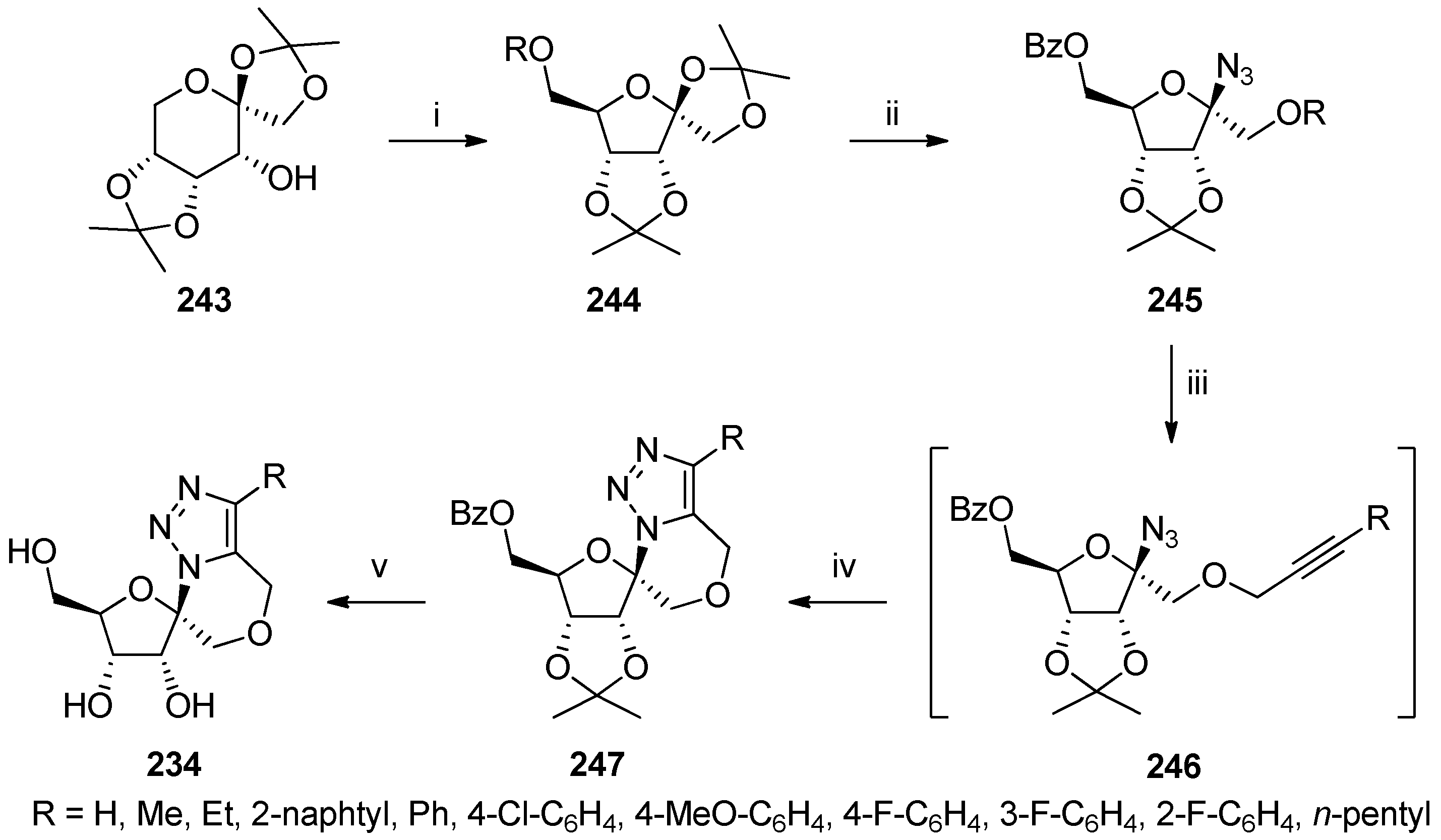
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soengas, R.G.; Da Silva, G.; Estévez, J.C. Synthesis of Spironucleosides: Past and Future Perspectives. Molecules 2017, 22, 2028. https://doi.org/10.3390/molecules22112028
Soengas RG, Da Silva G, Estévez JC. Synthesis of Spironucleosides: Past and Future Perspectives. Molecules. 2017; 22(11):2028. https://doi.org/10.3390/molecules22112028
Chicago/Turabian StyleSoengas, Raquel G., Gustavo Da Silva, and Juan Carlos Estévez. 2017. "Synthesis of Spironucleosides: Past and Future Perspectives" Molecules 22, no. 11: 2028. https://doi.org/10.3390/molecules22112028
APA StyleSoengas, R. G., Da Silva, G., & Estévez, J. C. (2017). Synthesis of Spironucleosides: Past and Future Perspectives. Molecules, 22(11), 2028. https://doi.org/10.3390/molecules22112028