Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides

Abstract
:
1. Introduction
2. Experimental Method
2.1. General Experimental Procedures
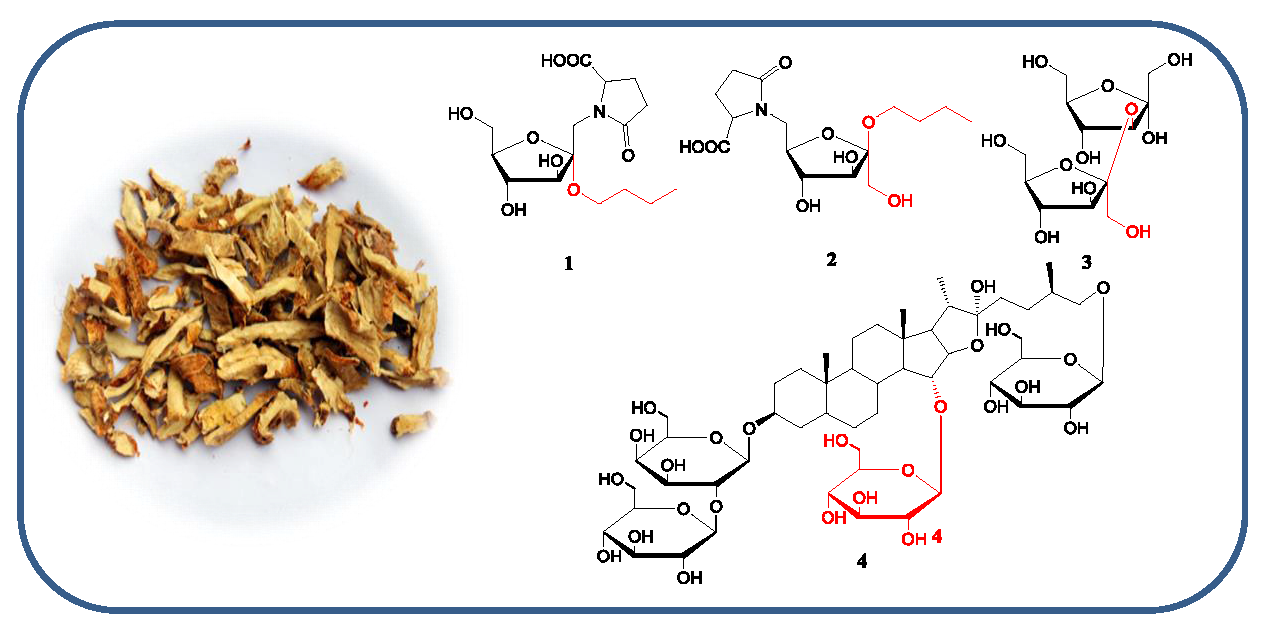
2.2. Plant Material
2.3. Extration and Isolation
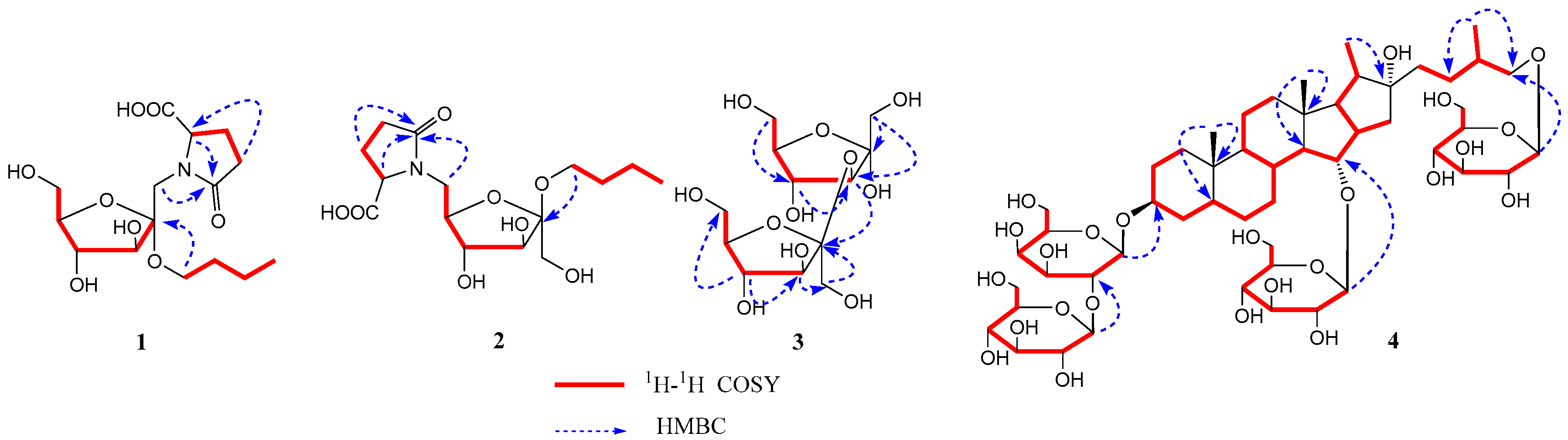
2.3.1. Aneglycoside A—Compound 1
2.3.2. Aneglycoside B—Compound 2
2.3.3. Aneglycoside C—Compound 3
2.3.4. Timosaponin U—Compound 4
2.4. Acid Hydrolysis, GC Analysis, and Opitcal Rotation Test
2.5. Cytotoxicity Assays
3. Results
3.1. Structure Determination
3.2. Cytotoxic Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peng, Y.; Zhang, Y.J.; Ma, Z.Q.; Pan, W.S.; Sun, Y.Q.; Song, S.J. Two new saponins from Anemarrhena asphodeloides Bge. Chin. Chem. Lett. 2007, 18, 171–174. [Google Scholar] [CrossRef]
- Saito, S.; Nagase, S.; Ichinose, K. New steroidal saponins from the Rhizomes of Anemarrhena asphodeloides Bunge (Liliaceae). Chem. Pharm. Bull. 1994, 42, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Xu, S.X.; Huang, S.; Wang, Z.X. A Study on the chemical constituents of Anemarrhena asphodeloides Bge. J. Shenyang Pharm. Univ. 1996, 13, 34–39. [Google Scholar]
- Zhang, C.; Xu, X.Z.; Li, C. Fructosides from Cynomorium songaricum. Phytochemistry 1996, 41, 975–976. [Google Scholar] [CrossRef]
- Wu, S.B.; Meyer, R.S.; Whitaker, B.D.; Litt, A.; Kennelly, E.J. Antioxidant glucosylated caffeoylquinic acid derivatives in the invasive tropica soda apple, Solanum viarum. J. Nat. Prod. 2012, 75, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Guo, R.; Li, T.; Liu, Y.; Wang, C.F.; Shu, Z.P.; Wang, Z.B.; Zhang, J.; Xia, Y.G.; Jiang, H.; et al. Five withanolides from the leaves of Datura metel L. and their inhibitory effects on nitric oxide production. Molecules 2014, 19, 4548–4559. [Google Scholar] [CrossRef] [PubMed]
- Greenaway, W.; Whatley, F.R. Enzymic synthesis of l-glutamic acid-15N and 4-aminobutyric acid-15N and the preparation of l-pyroglutamic acio-15N. J. Label. Compd. Radiopharm. 1975, 11, 395–400. [Google Scholar] [CrossRef]
- Hao, X.Y.; Tan, N.H.; Zhou, J. Chemical Constituents of Gastrodia elata Bl. Yunnan Plant Res. 2000, 22, 81–84. [Google Scholar]
- Zhang, Z.; Wang, D.; Zhao, Y.; Gao, H.; Hu, Y.H.; Hu, J.F. Fructose-derived carbohydrates from Alisma orientalis. Nat. Prod. Res. 2009, 23, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Zhang, J.; Liu, Y.; Kuang, H.X. Steroidal saponins from the rhizomes of Anemarrhena asphodeloides. Molecules 2016, 21, 1075. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, A.; Mimaki, Y. Steroidal saponins from the whole plants of Agave utahensis and their cytotoxic activity. Phytochemistry 2009, 70, 807–815. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–4 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
| No. | Compound 1 | Compound 2 | ||
|---|---|---|---|---|
| δC | δH Mult (J, Hz) | δC | δH Mult (J, Hz) | |
| Sugar | ||||
| 1 | 64.0 | 4.35 (d, 11.6) | 61.7 | 3.65 (d, 11.8) |
| 4.24 (d, 11.6) | 3.52 (d, 11.8) | |||
| 2 | 102.9 | 105.6 | ||
| 3 | 80.2 | 3.96 (d, 8.0) | 78.1 | 4.12 (d, 8.0) |
| 4 | 76.8 | 3.91 (d, 8.0) | 77.4 | 3.99 (d, 8.0) |
| 5 | 83.5 | 3.71 overlap | 80.0 | 3.92 overlap |
| 6 | 64.7 | 3.73 overlap | 67.5 | 4.28 overlap |
| 3.56 (m) | ||||
| Pyroglutamic Acid | ||||
| 2′ | 173.4 | 173.8 | ||
| 3′ | 25.8 | 2.49 (m) | 25.9 | 2.49 (m) |
| 2.23 (m) | 2.21 (m) | |||
| 4′ | 30.2 | 2.33 (m) | 30.2 | 2.34 (m) |
| 5′ | 57.1 | 4.32 (dd, 8.0, 4.0) | 57.0 | 4.30 (m) |
| 6′ | 181.2 | 181.1 | ||
| n-butyl | ||||
| 1″ | 62.3 | 3.74 (m) | 62.3 | 3.61 (m) |
| 3.52 (m) | 3.45 (m) | |||
| 2″ | 33.2 | 1.53 (m) | 33.4 | 1.52 (m) |
| 3″ | 20.3 | 1.37 (m) | 20.3 | 1.37 (m) |
| 4″ | 14.3 | 0.92 (3H, t, 7.2) | 14.4 | 0.92 (3H, t, 7.2) |
| No. | δC | δH Mult (J, Hz) | No. | δC | δH Mult (J, Hz) |
|---|---|---|---|---|---|
| α-Fruf | β-Fruf | ||||
| 1 | 98.8 | 3.60 (d, 12.3) | 1′ | 62.5 | 3.52 (d, 12.3) |
| 4.13 (d, 12.3) | 4.06 (d, 12.3) | ||||
| 2 | 81.8 | 2′ | 102.5 | ||
| 3 | 77.7 | 3.94 (d, 2.4) | 3′ | 76.9 | 3.67 (d, 7.8) |
| 4 | 83.5 | 3.86 (dd, 2.4, 5.7) | 4′ | 74.5 | 4.02 (d, 7.8) |
| 5 | 61.2 | 3.92 (m) | 5′ | 81.2 | 3.83 (m) |
| 6 | 61.7 | 3.68 (d, 12.3) | 6′ | 62.6 | 3.58 (d, 12.4) |
| 3.77 (d, 12.3) | 3.77 (d, 12.4) |
| No. | δC | δH Mult (J, Hz) | No. | δC | δH Mult (J, Hz) |
|---|---|---|---|---|---|
| 1 | 31.2 | 1.86 (m) | 3-gal | ||
| 1.48 (m) | 1′ | 102.4 | 4.89 (d, 7.6) | ||
| 2 | 27.2 | 1.84 (m) | 2′ | 81.6 | 4.65 (m) |
| 1.29 (m) | 3′ | 76.9 | 4.03 (m) | ||
| 3 | 75.5 | 4.32 (m) | 4′ | 69.9 | 4.54 (m) |
| 4 | 31.0 | 1.48 (m) | 5′ | 76.6 | 4.07 (m) |
| 1.85 (m) | 6′ | 62.2 | 2.06 (m) | ||
| 5 | 37.0 | 2.15 (m) | 4.42 (m) | ||
| 6 | 27.1 | 1.92 (m) | glc | ||
| 1.52 (m) | 1″ | 106.0 | 5.27 (d, 7.6) | ||
| 7 | 27.0 | 1.87 (m) | 2″ | 75.3 | 4.37 (m) |
| 1.28 (m) | 3″ | 78.1 | 4.18 (m) | ||
| 8 | 36.5 | 1.85 (m) | 4″ | 71.8 | 4.27 (m) |
| 9 | 40.9 | 1.21 (m) | 5″ | 78.4 | 3.83 (m) |
| 10 | 35.5 | 6″ | 62.8 | 4.52 (m) | |
| 11 | 21.3 | 1.26 (m) | 4.32 (m) | ||
| 1.37 (m) | 15-glc | ||||
| 12 | 41.5 | 1.21 (m) | 1′′′ | 105.1 | 4.80 (d, 7.8) |
| 1.69 (m) | 2′′′ | 75.3 | 4.27 (m) | ||
| 13 | 41.4 | 3′′′ | 78.5 | 3.82 (m) | |
| 14 | 61.0 | 1.62 (m) | 4′′′ | 71.8 | 4.18 (m) |
| 15 | 79.2 | 4.38 (m) | 5′′′ | 78.6 | 3.95 (m) |
| 16 | 91.4 | 5.06 (dd, 3.6, 8.7) | 6′′′ | 62.8 | 4.52 (m) |
| 17 | 61.5 | 2.25 (m) | 4.32 (m) | ||
| 18 | 18.2 | 0.95 (3H, s) | 26-glc | ||
| 19 | 24.3 | 0.89 (3H, s) | 1′′′′ | 105.2 | 4.83 (d, 7.8) |
| 20 | 40.5 | 1.42 (m) | 2′′′′ | 75.3 | 4.27 (m) |
| 21 | 16.6 | 1.32 (3H, d, 6.8) | 3′′′′ | 78.7 | 4.19 (m) |
| 22 | 110.5 | 4′′′′ | 71.8 | 4.23 (m) | |
| 23 | 37.2 | 1.98 (m) | 5′′′′ | 78.6 | 3.95 (m) |
| 2.12 (m) | 6′′′′ | 62.9 | 4.53 (m) | ||
| 24 | 28.5 | 1.72 (m) | 4.36 (m) | ||
| 2.09 (m) | |||||
| 25 | 34.5 | 1.92 (m) | |||
| 26 | 75.1 | 3.55 (m) | |||
| 4.00 (m) | |||||
| 27 | 17.6 | 1.03 (3H, d, 6.7) |
| Compound | IC50 (μM) | Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| HepG2 | Hela | SGC7901 | HepG2 | Hela | SGC7901 | ||
| 1 | 38.4 ± 2.4 | 29.7 ± 1.9 | >100 | 4 | 61.8 ± 4.1 | 39.7 ± 3.7 | 44.5 ± 2.0 |
| 2 | 41.8 ± 3.5 | 34.2 ± 3.6 | >100 | doxorubicin | 8.4 ± 2.2 | 9.0 ± 1.4 | 6.7 ± 1.8 |
| 3 | >100 | >100 | >100 | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.-Y.; Bi, X.-Y.; Liu, Y.; Li, G.-Y.; Yin, X.; Kuang, H.-X. Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides. Molecules 2017, 22, 1995. https://doi.org/10.3390/molecules22111995
Yang B-Y, Bi X-Y, Liu Y, Li G-Y, Yin X, Kuang H-X. Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides. Molecules. 2017; 22(11):1995. https://doi.org/10.3390/molecules22111995
Chicago/Turabian StyleYang, Bing-You, Xue-Yan Bi, Yan Liu, Guo-Yu Li, Xin Yin, and Hai-Xue Kuang. 2017. "Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides" Molecules 22, no. 11: 1995. https://doi.org/10.3390/molecules22111995
APA StyleYang, B.-Y., Bi, X.-Y., Liu, Y., Li, G.-Y., Yin, X., & Kuang, H.-X. (2017). Four New Glycosides from the Rhizoma of Anemarrhena asphodeloides. Molecules, 22(11), 1995. https://doi.org/10.3390/molecules22111995