Mechanistic Study of Copper-Catalyzed C-H Hydroxylation/C-S Coupling by ESI-HR MS and DFT Calculations

Abstract
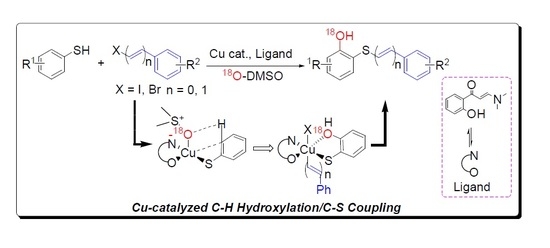
:
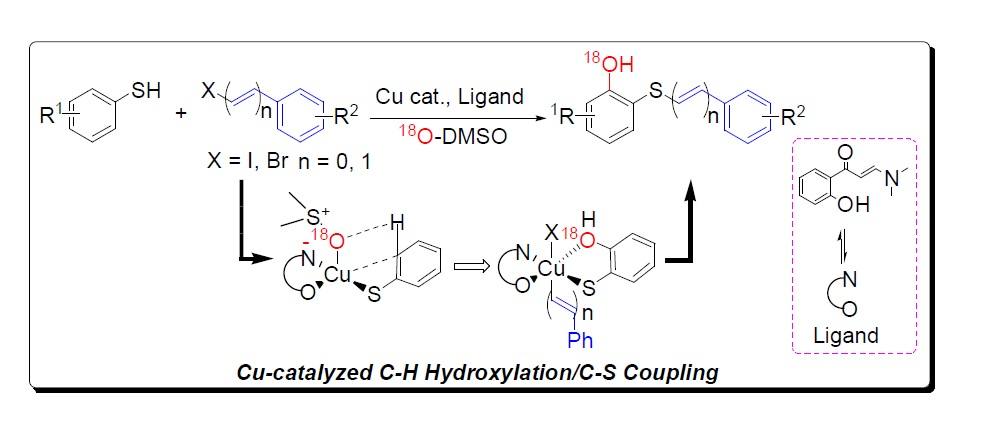{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
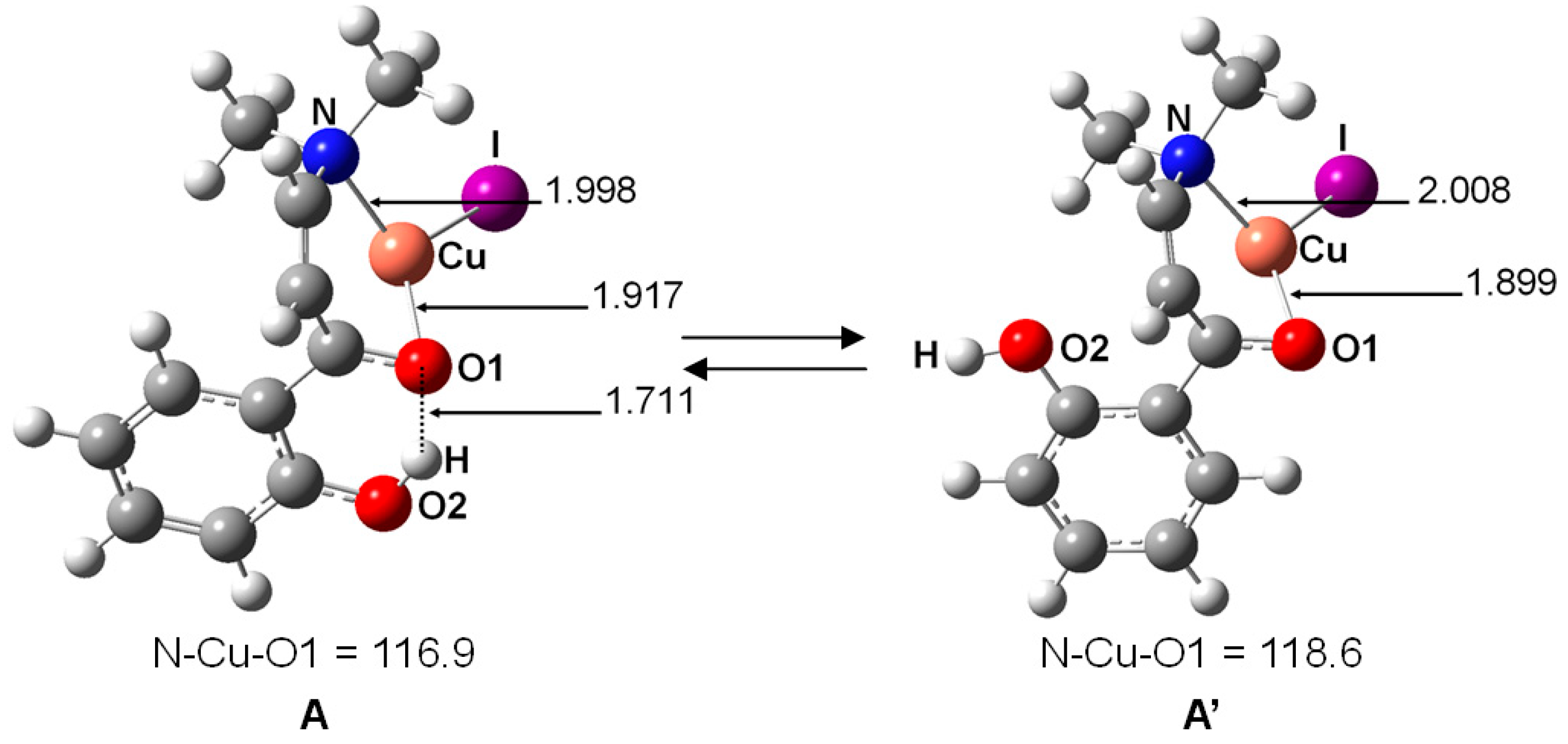
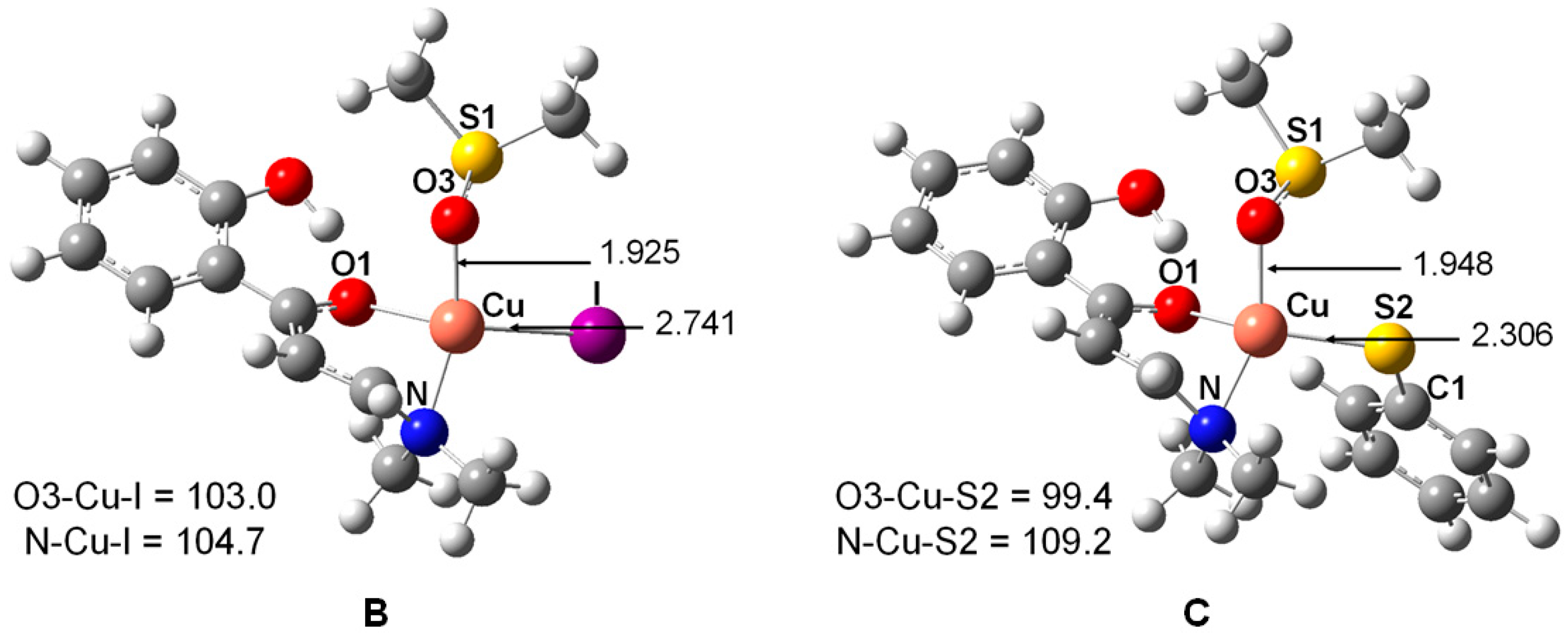
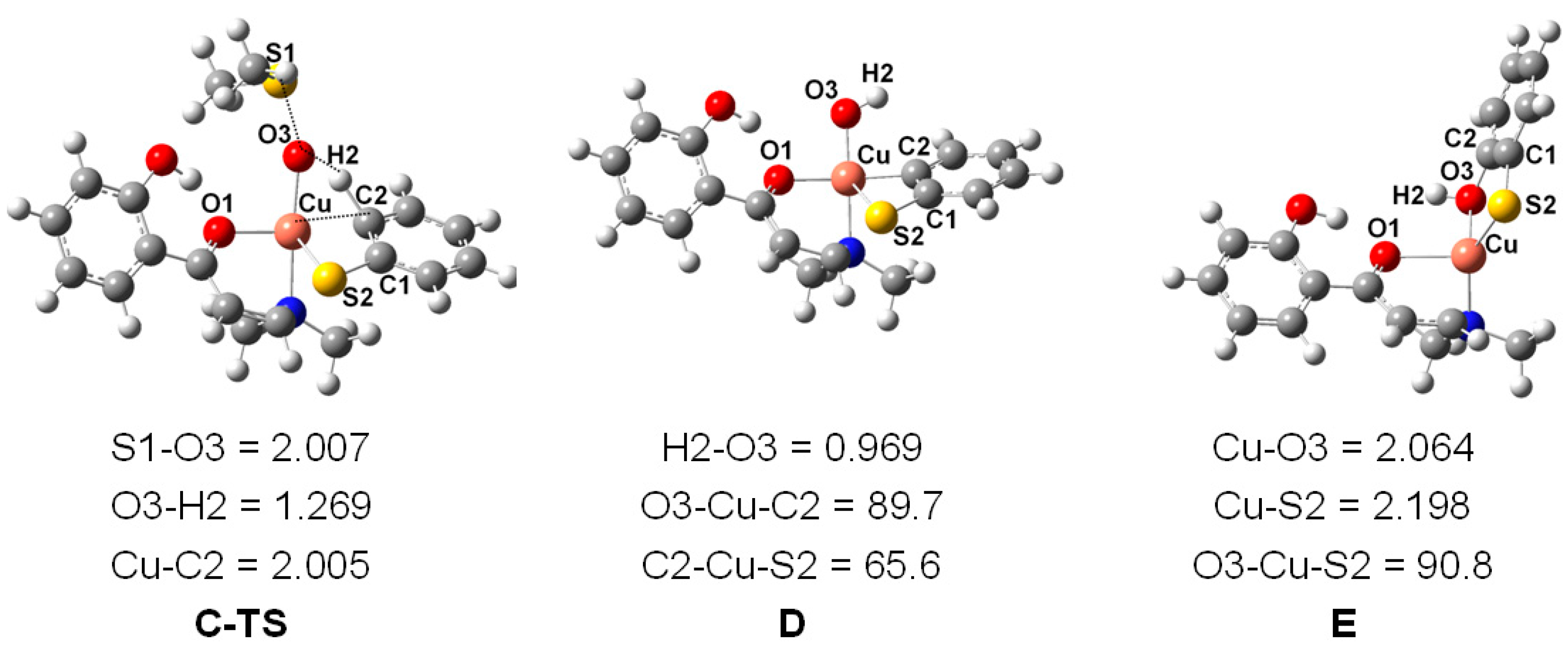
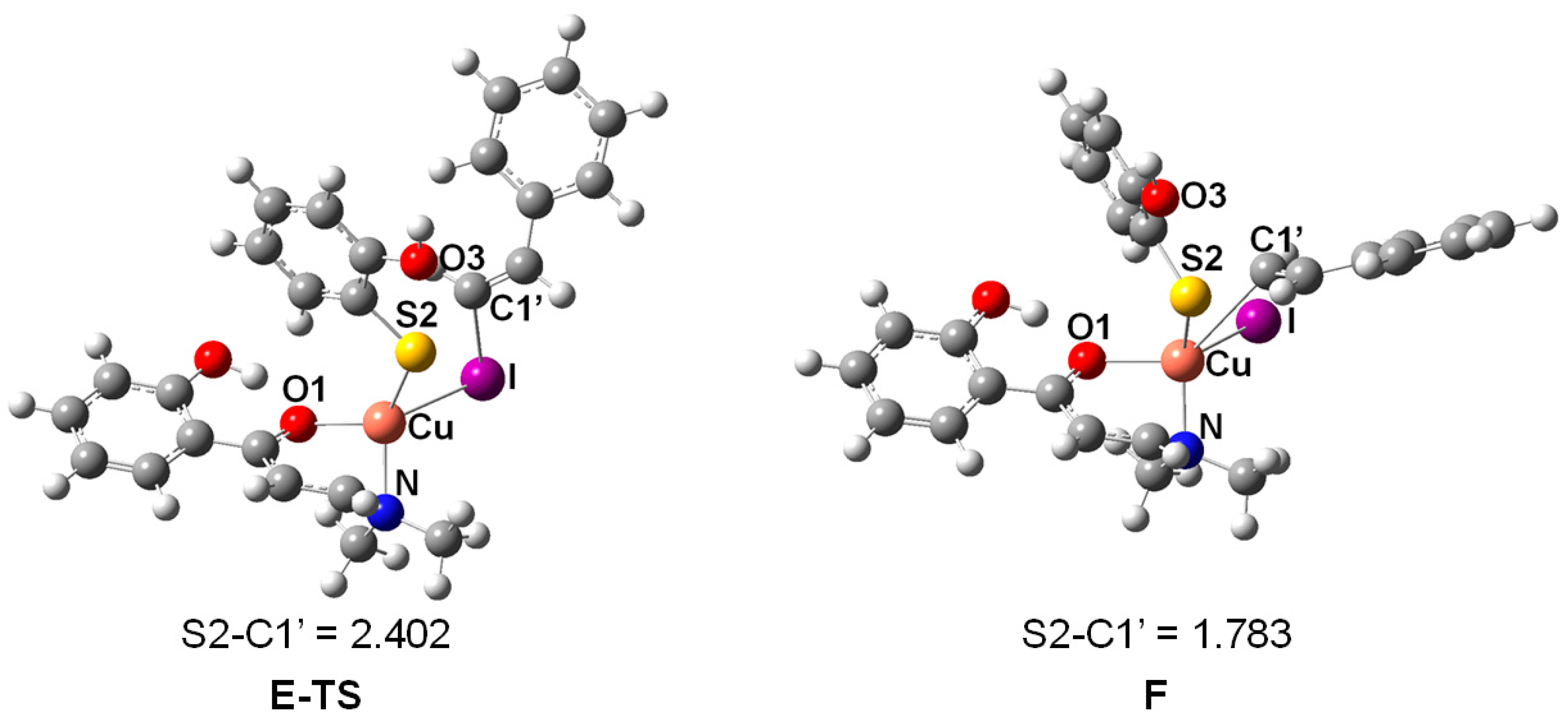
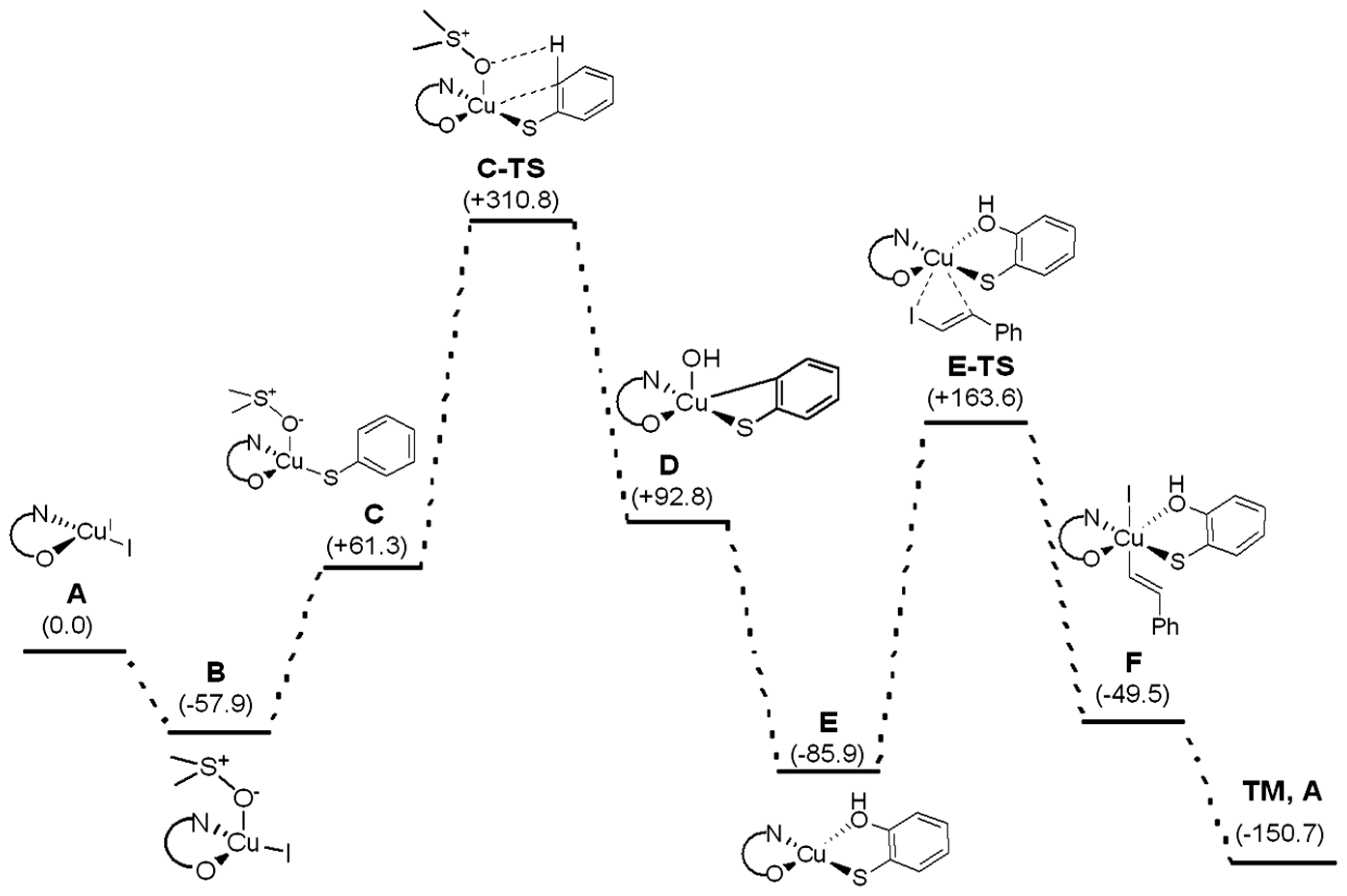
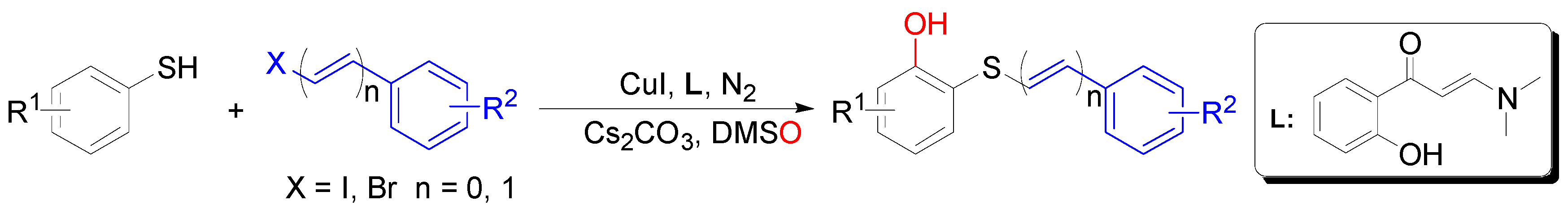
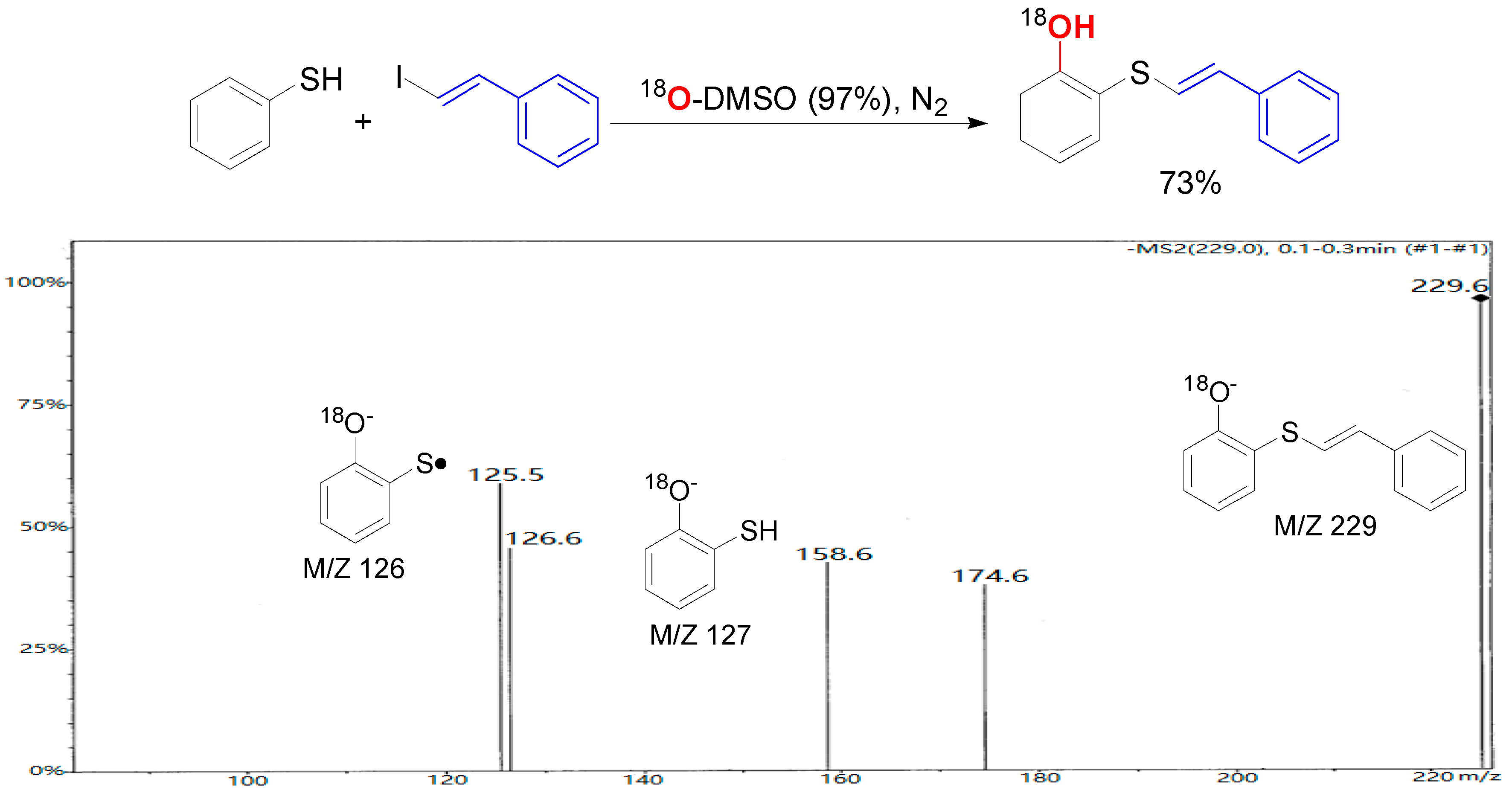
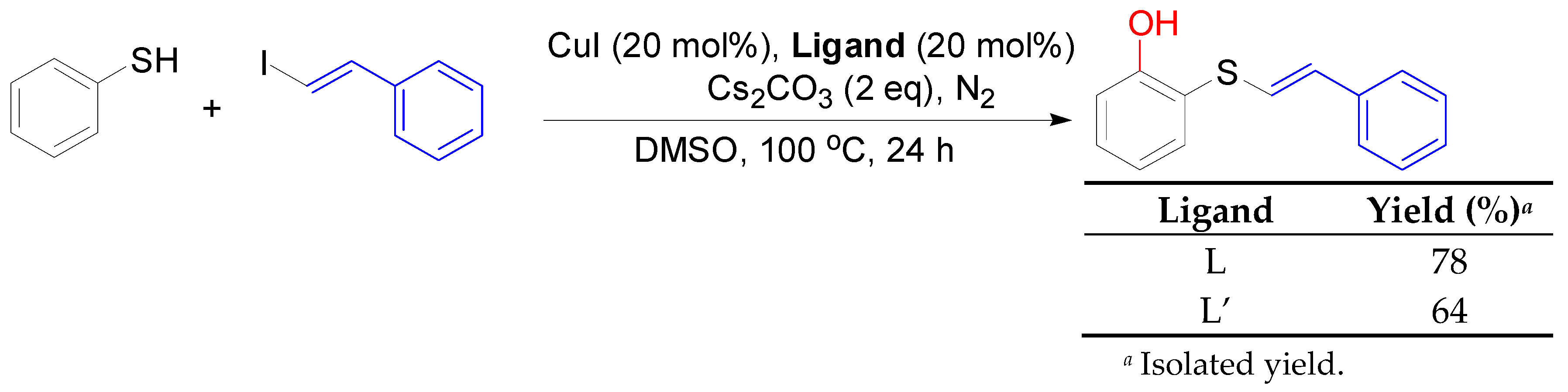
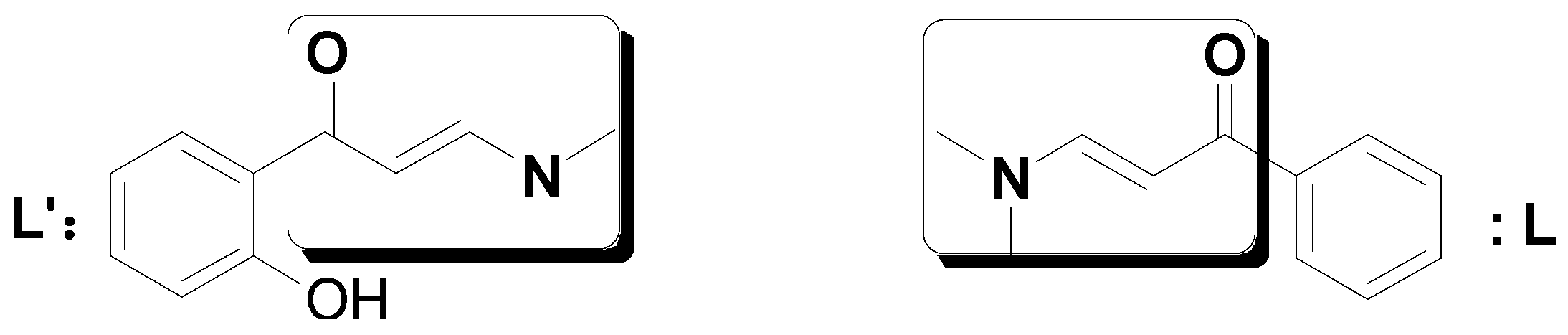
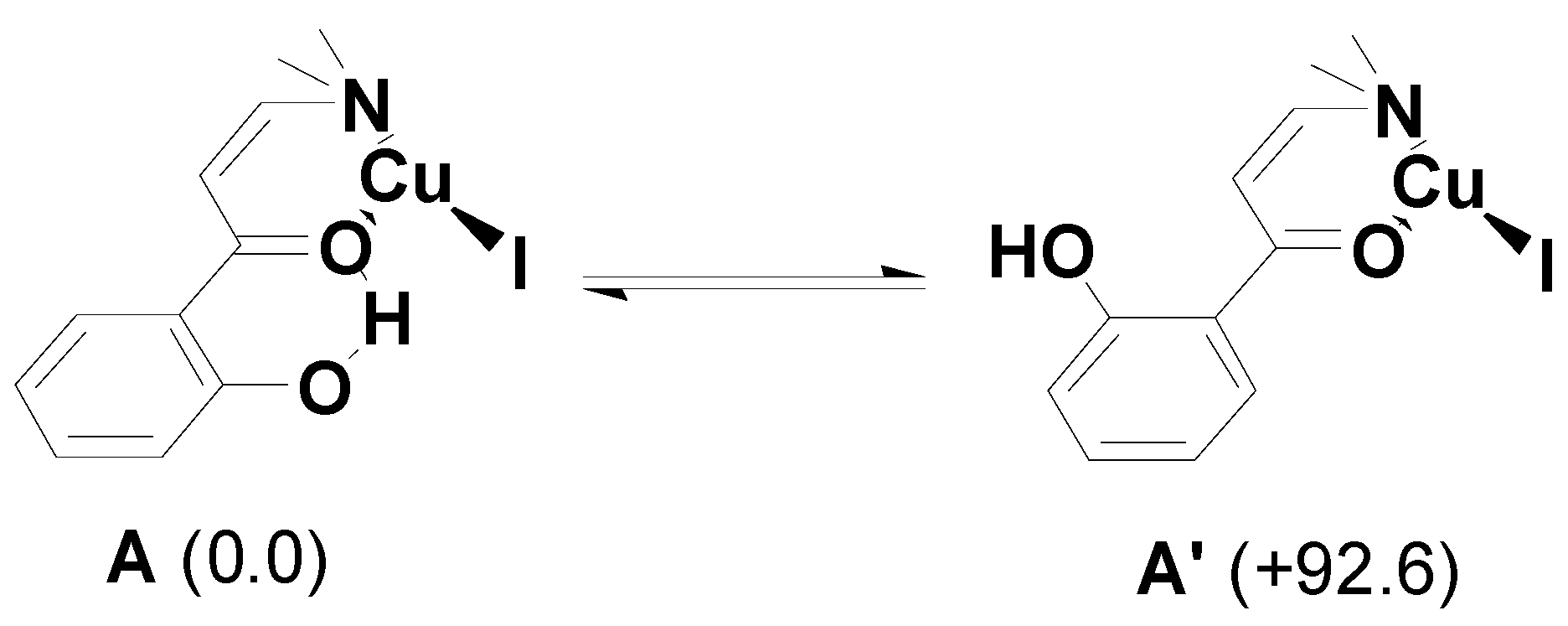
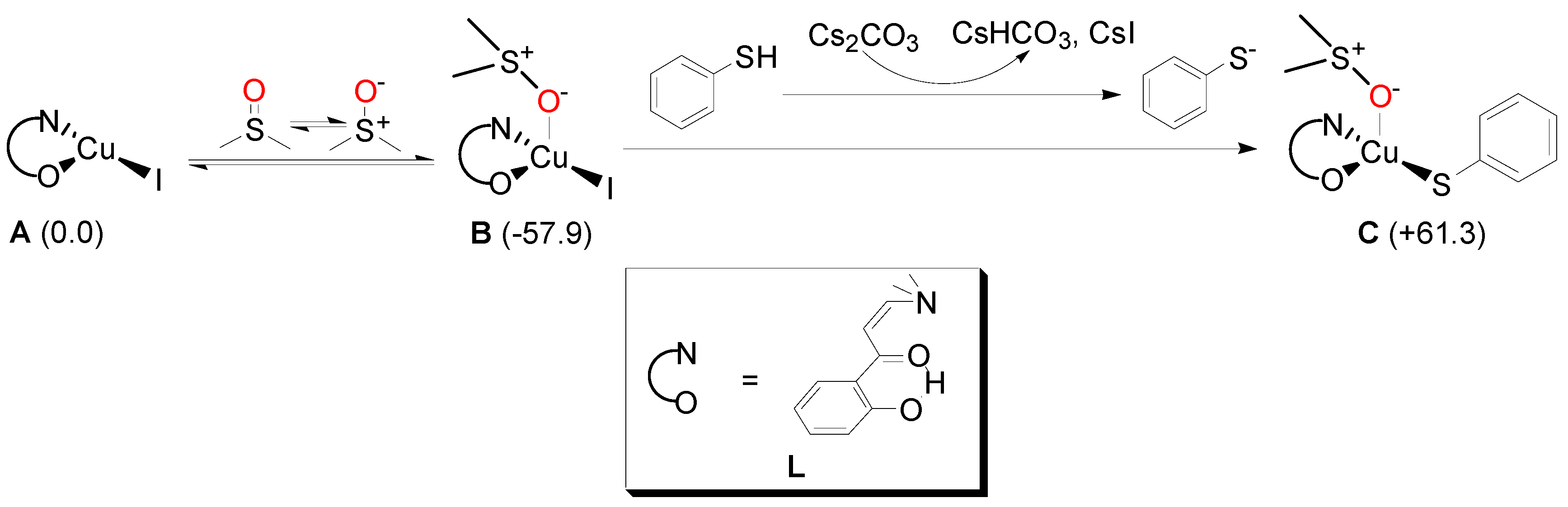
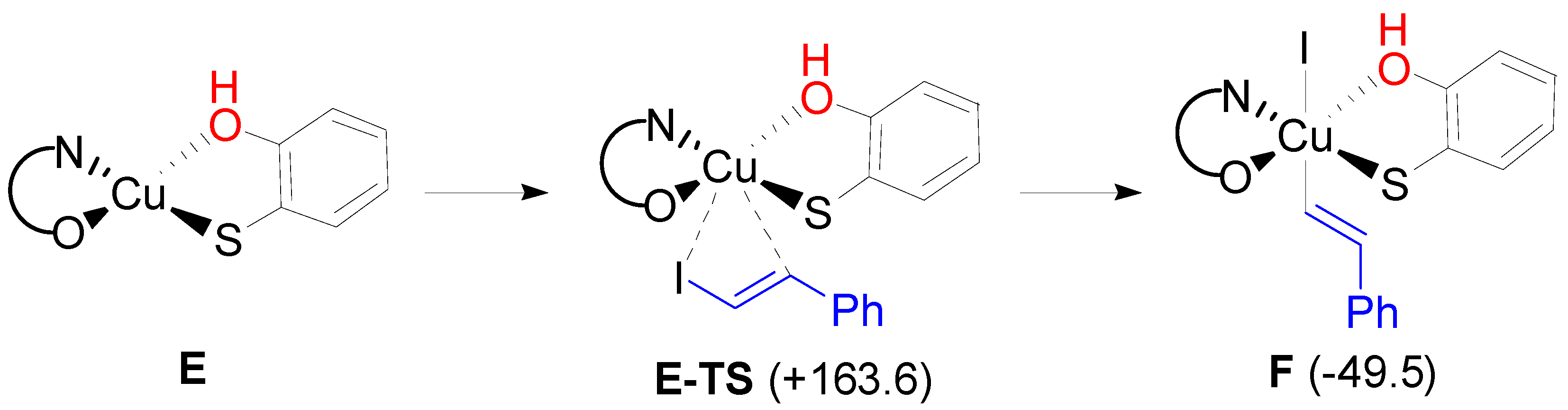
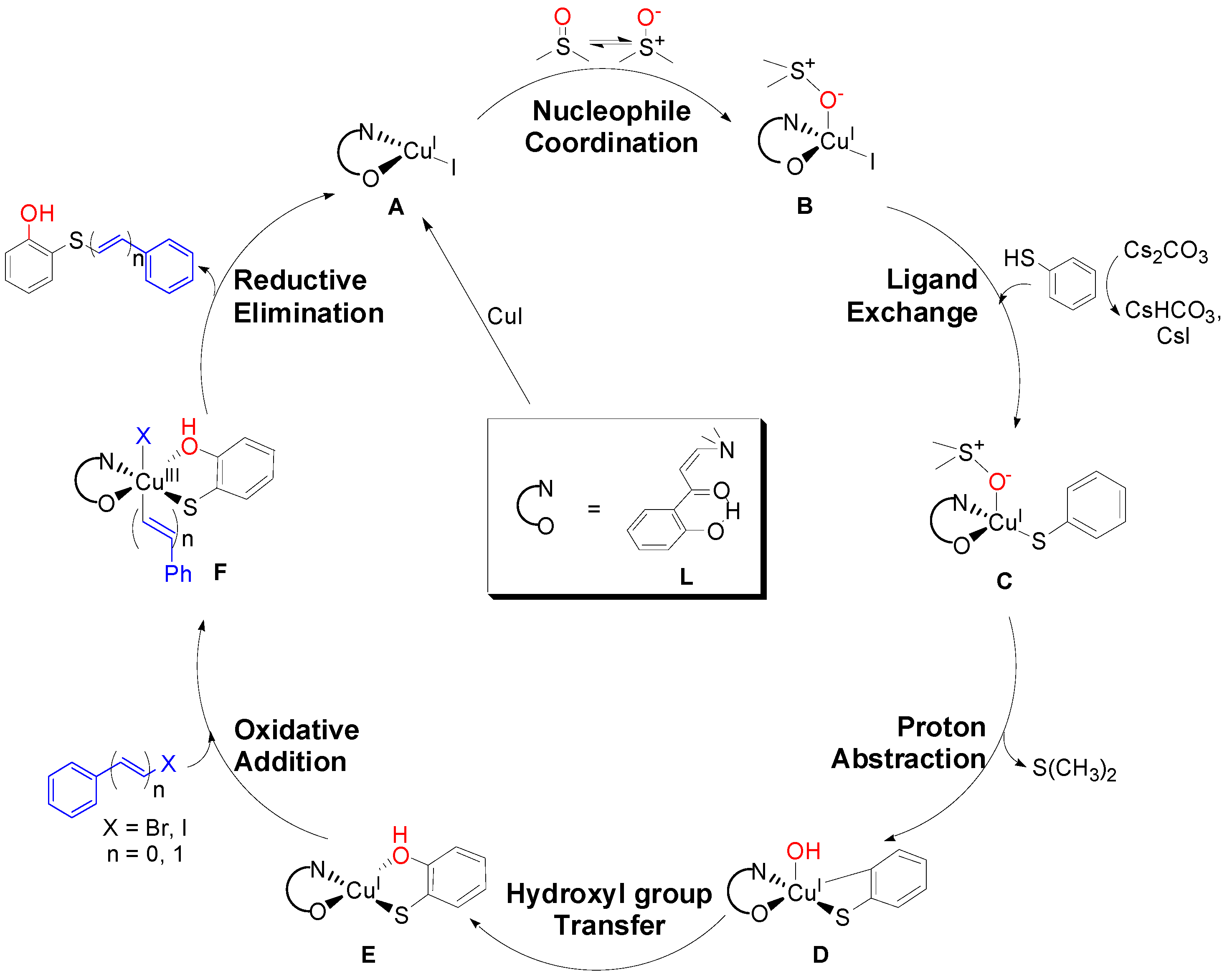
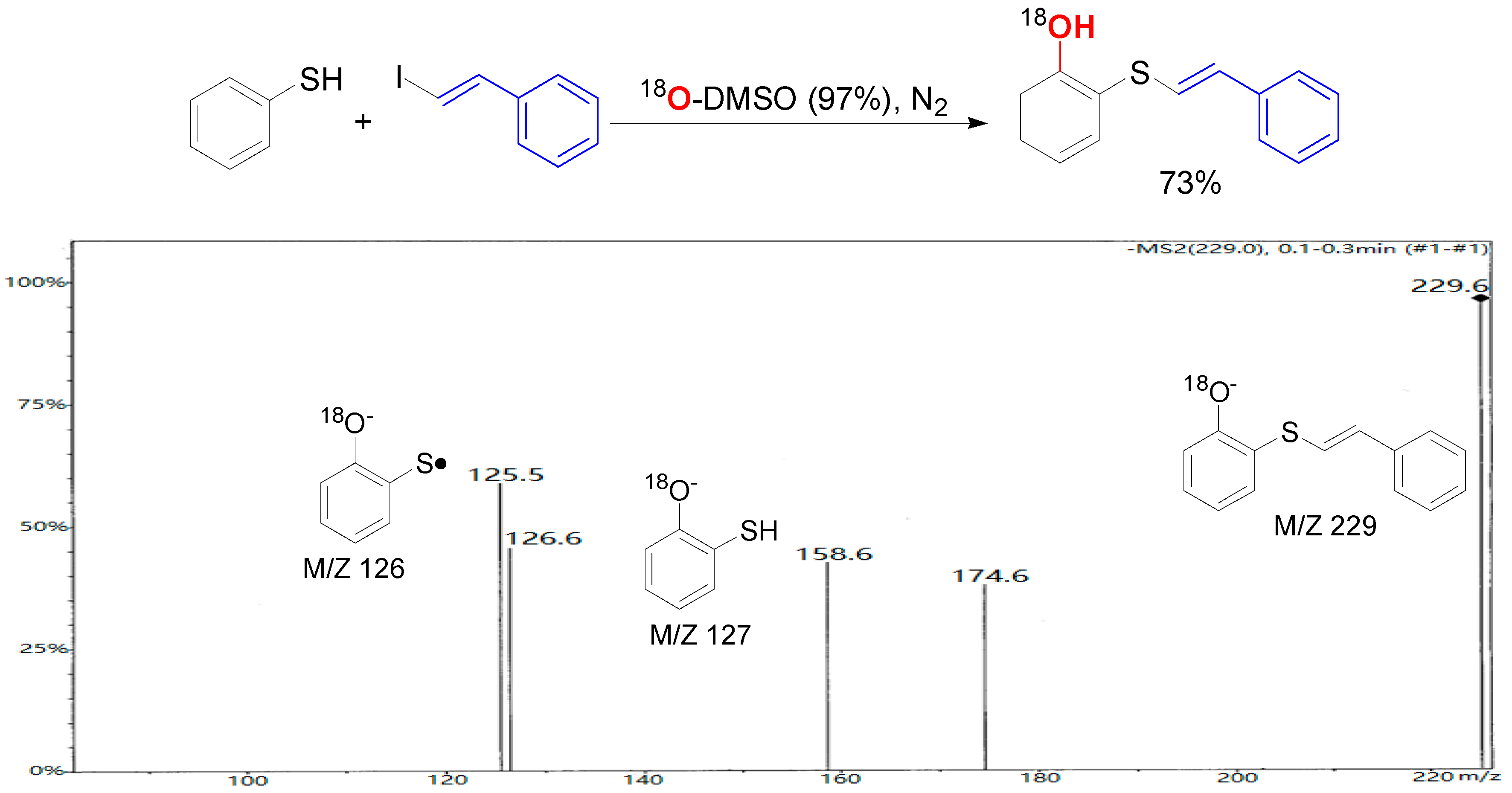
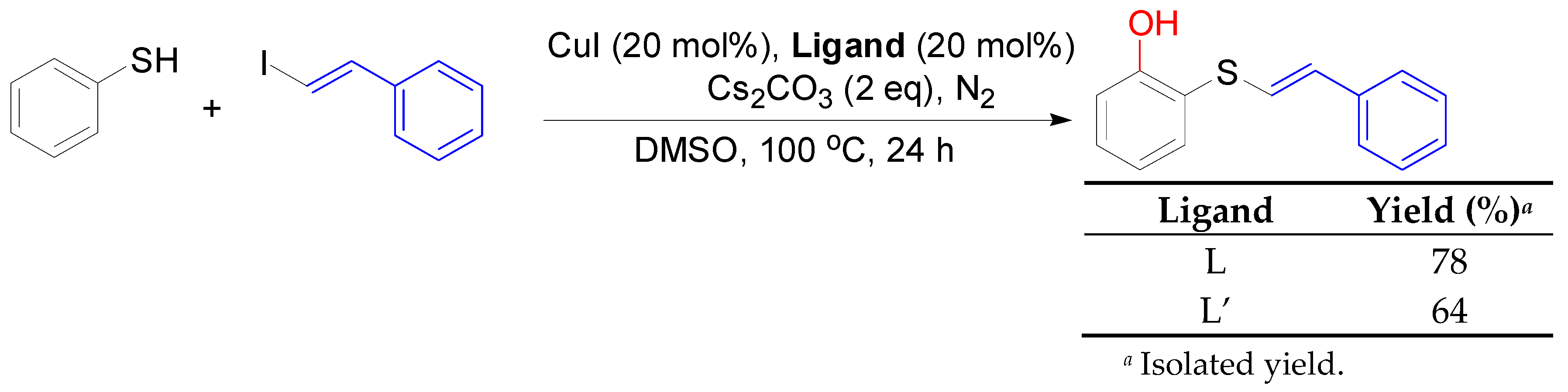
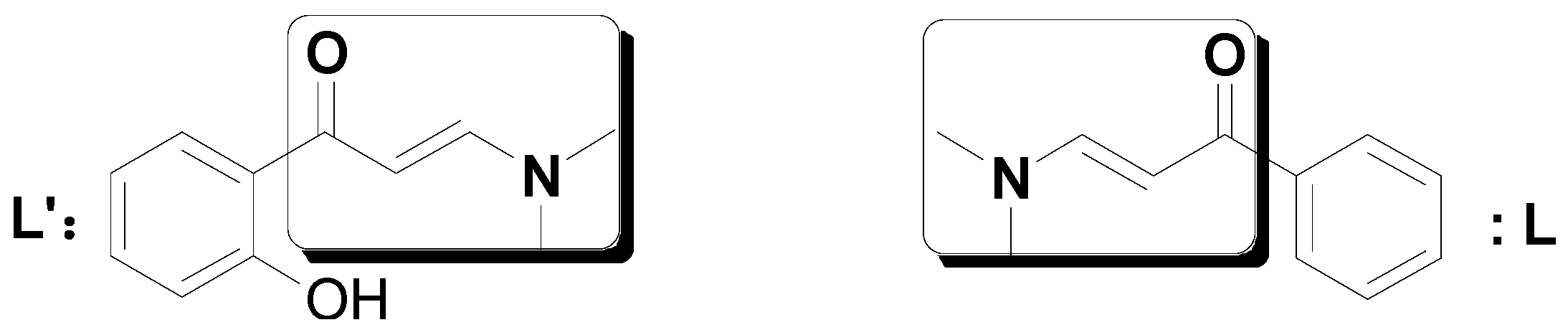
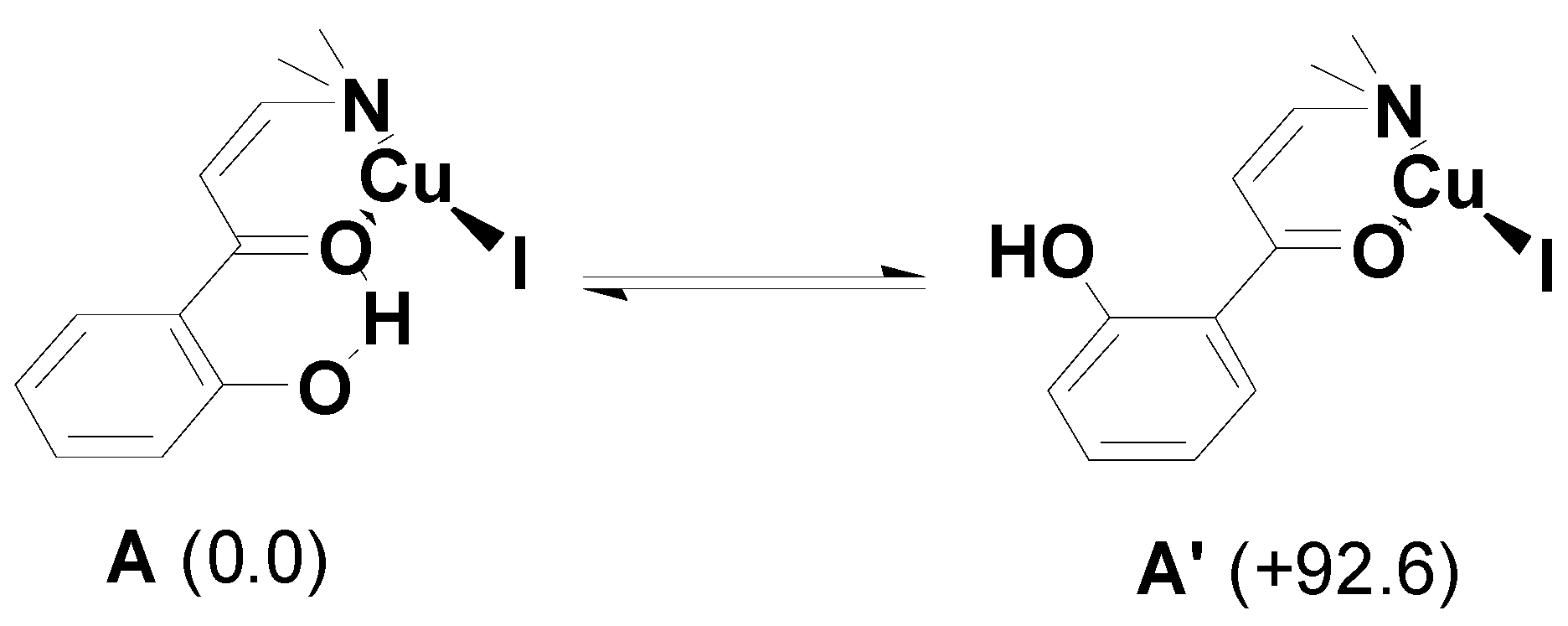
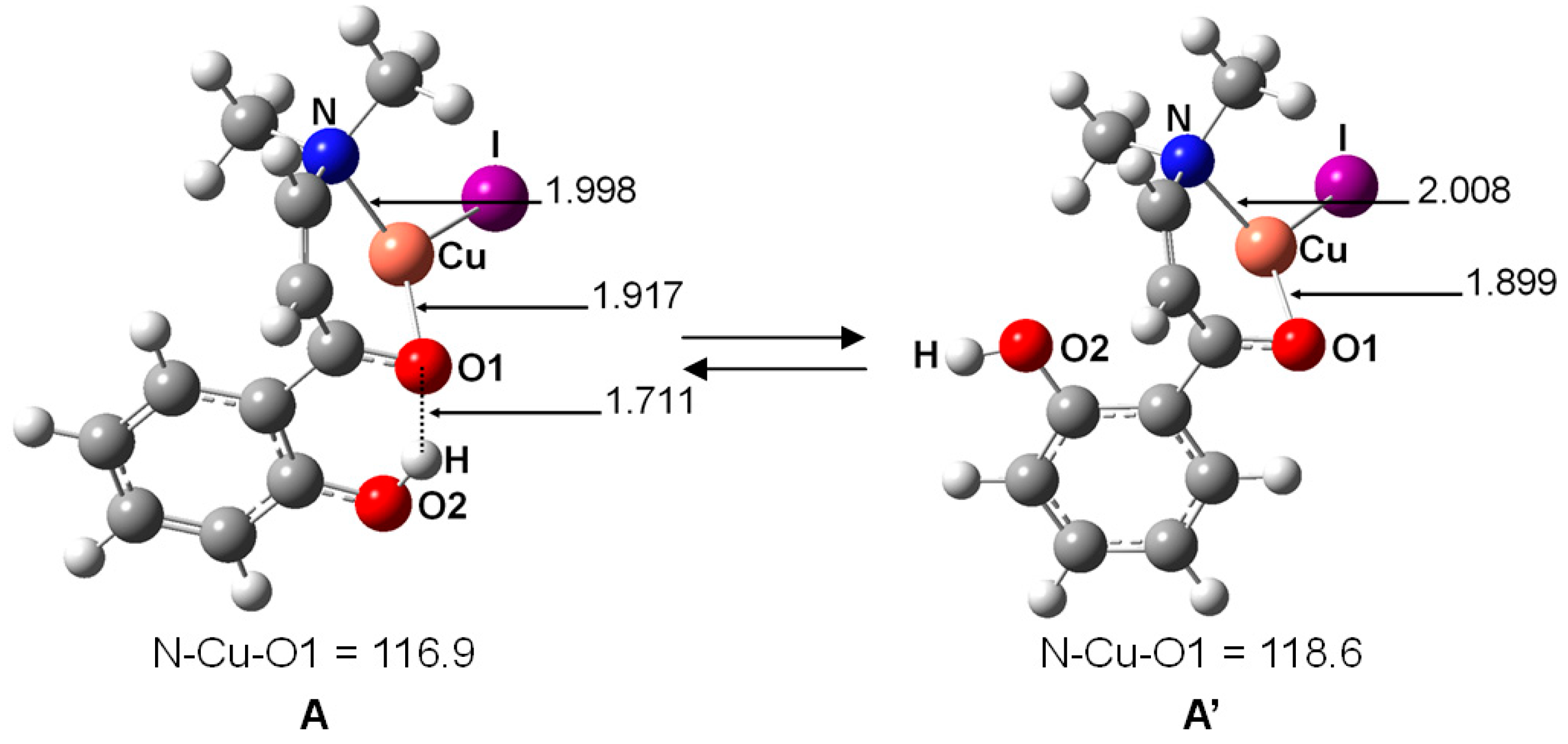
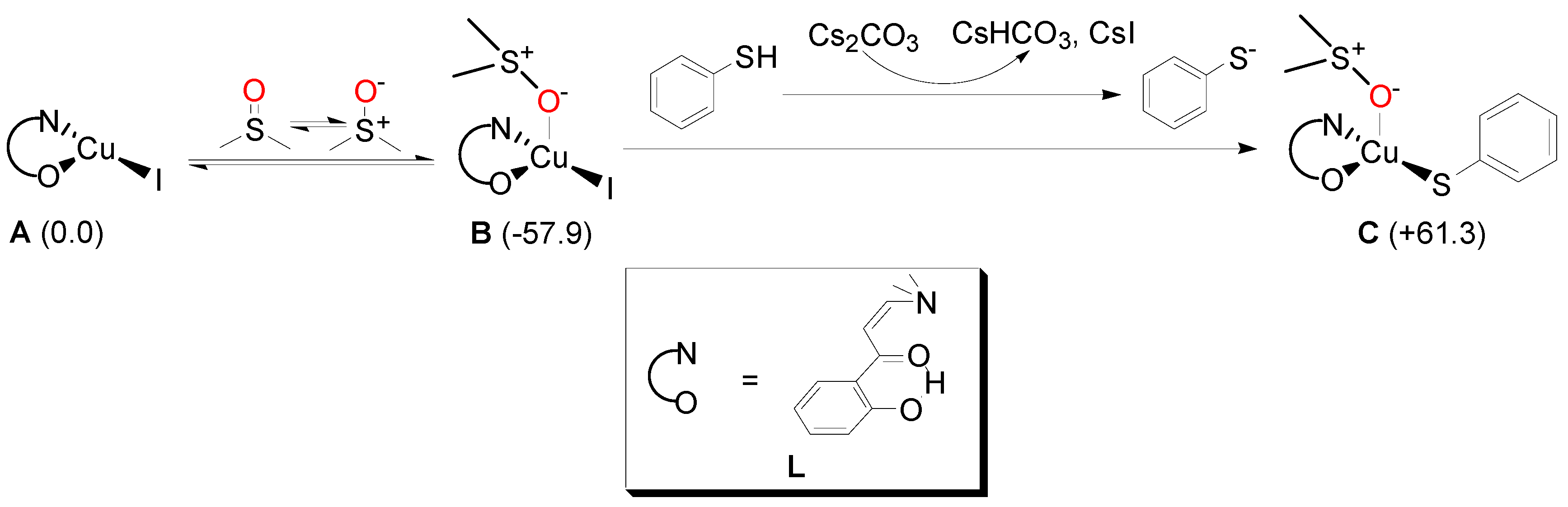
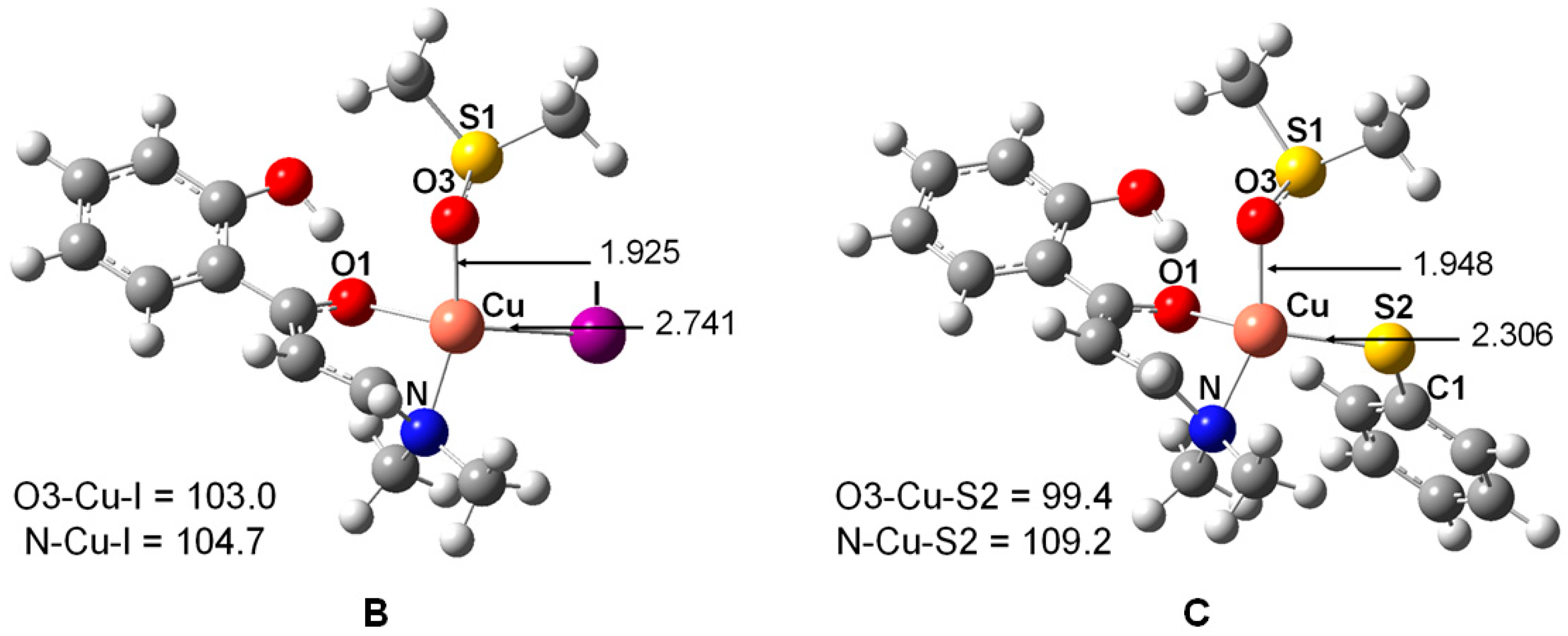
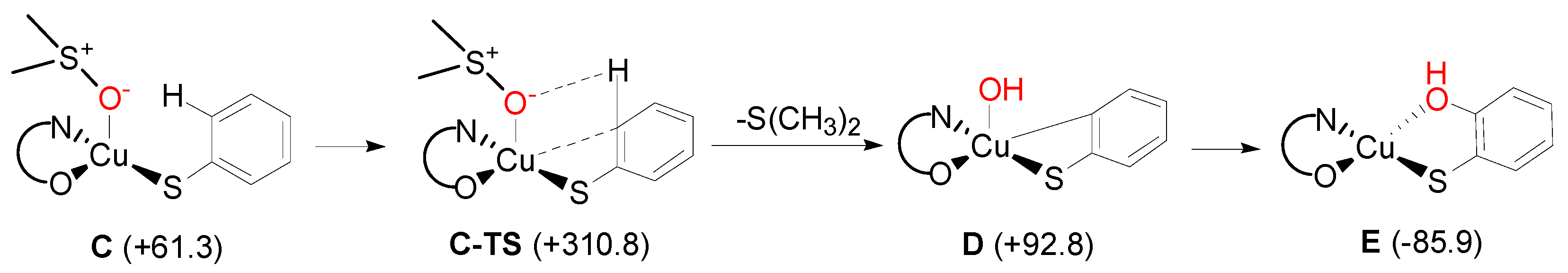
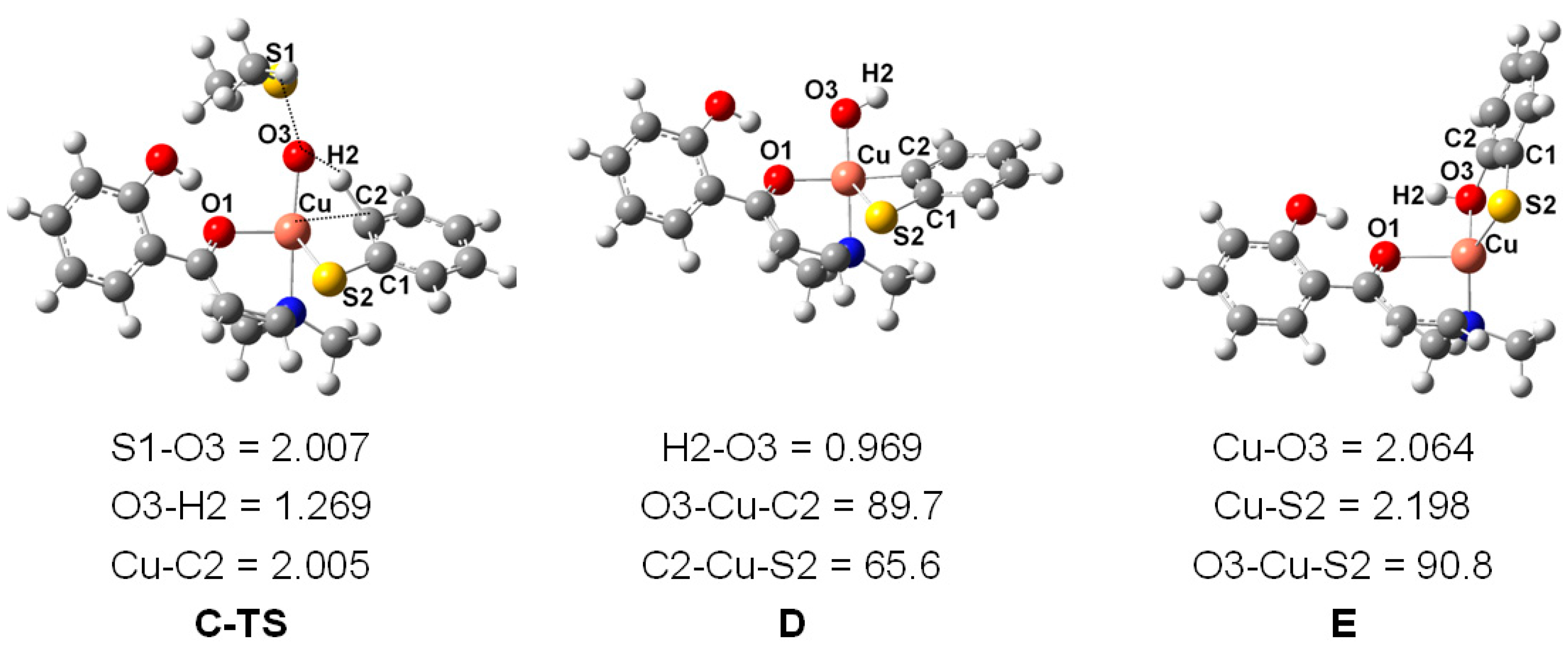
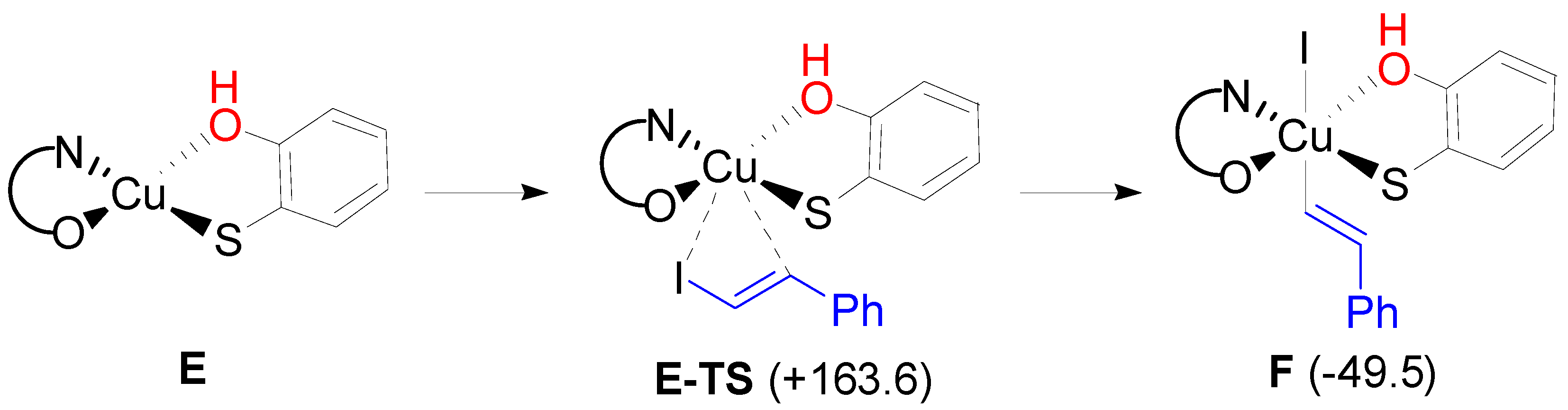
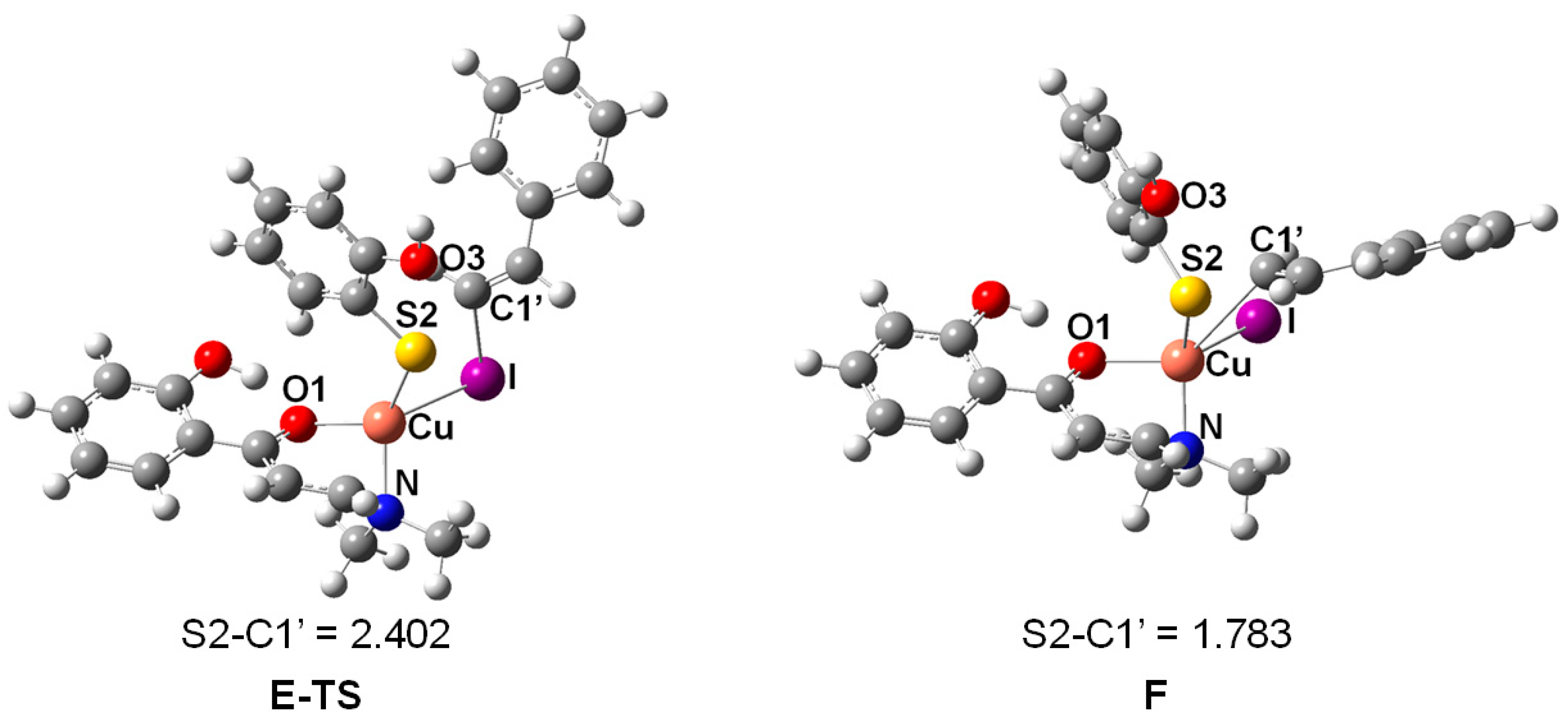
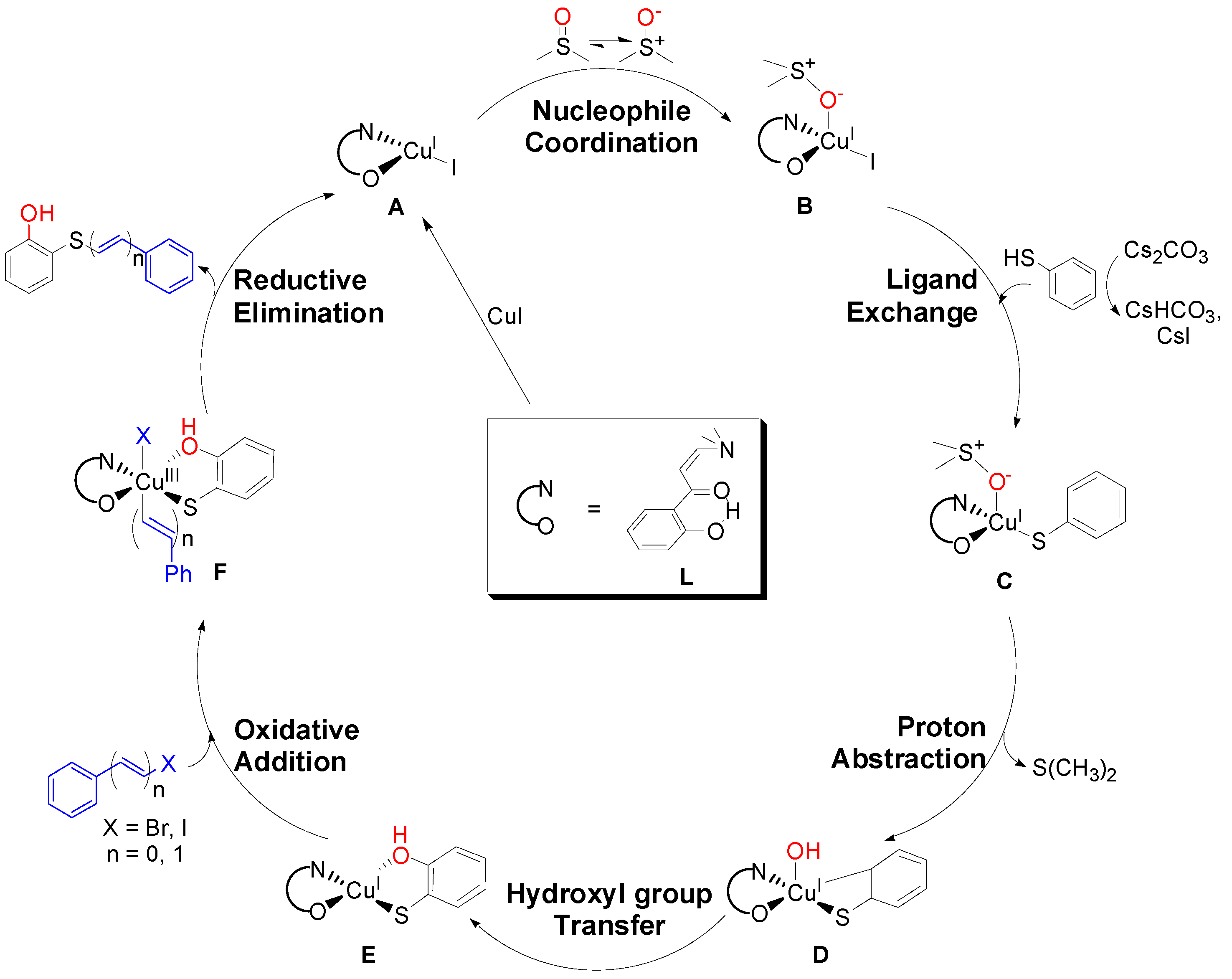
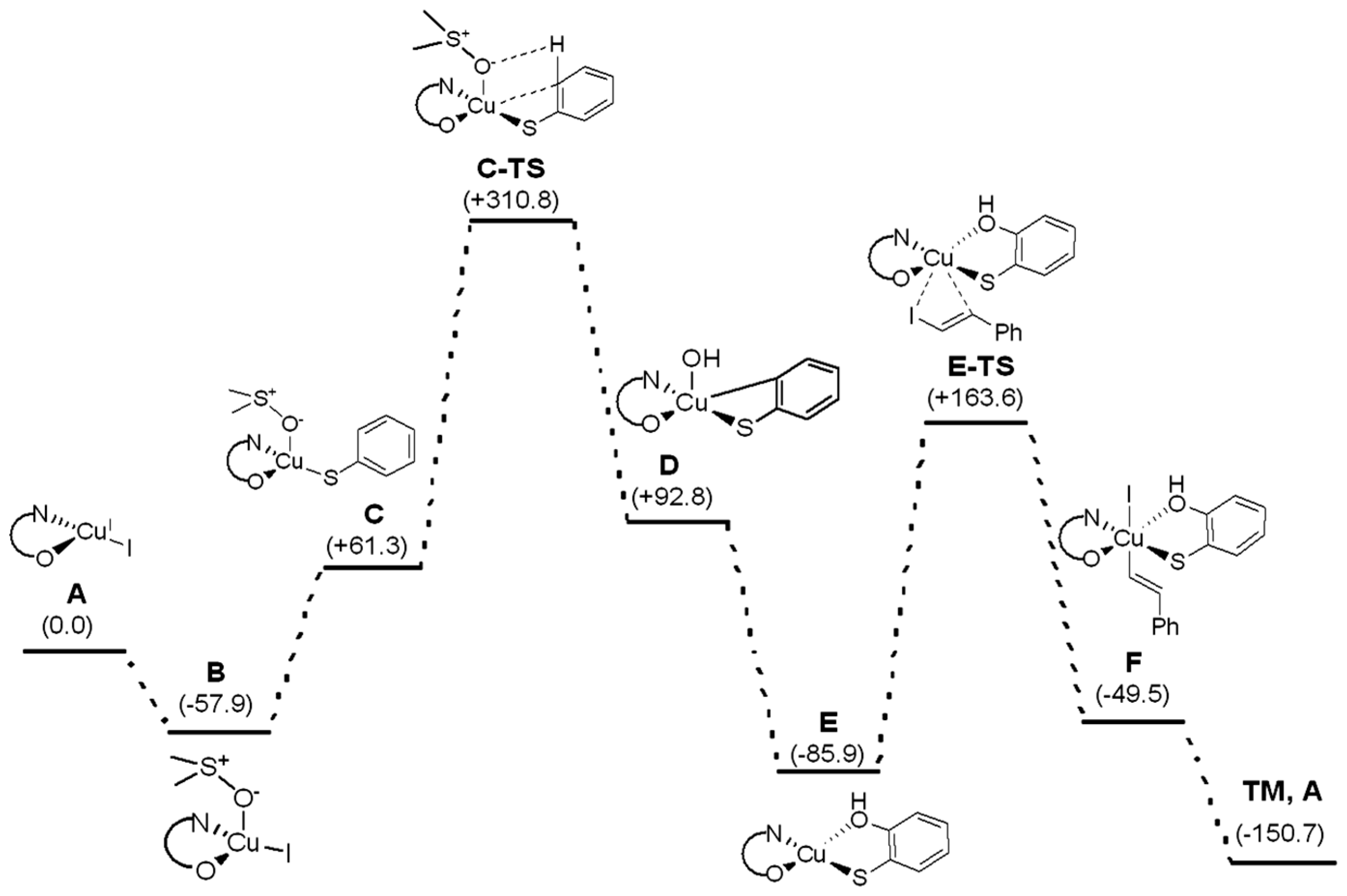
2. Results and Discussion
3. Experimental Section
3.1. General Procedure for the Preparation of Ligand L
3.2. General Procedure for the Preparation of 2-(Styrylthio)phenols
3.3. General Procedure for Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Godula, K.; Sames, D. CH bond functionalization in complex organic synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Activation and Functionalization of C-H Bonds; Goldberg, K.I.; Alan, S.G. (Eds.) American Chemical Society: Washington, DC, USA, 2004. [Google Scholar]
- Schwarz, H. Chemistry with methane: Concepts rather than recipes. Angew. Chem. Int. Ed. 2011, 50, 10096–10115. [Google Scholar] [CrossRef] [PubMed]
- Roithová, J.; Schröder, D. Selective activation of alkanes by gas-phase metal ions. Chem. Rev. 2010, 110, 1170–1211. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H. How and why do cluster size, charge state, and ligands affect the course of metal-mediated gas-phase activation of methane? Isr. J. Chem. 2014, 54, 1413–1431. [Google Scholar] [CrossRef]
- Desai, L.V.; Stowers, K.J.; Sanford, M.S. Insights into directing group ability in palladium-catalyzed C−H bond functionalization. J. Am. Chem. Soc. 2008, 130, 13285–13293. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C−H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. Selectivity enhancement in functionalization of C–H bonds: A review. Org. Biomol. Chem. 2010, 8, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, D.; Fagnou, K. Overview of the mechanistic work on the concerted metallation-deprotonation pathway. Chem. Lett. 2010, 39, 1118–1126. [Google Scholar] [CrossRef]
- Winstein, S.; Traylor, T.G. Mechanisms of reaction of organomercurials. II. Electrophilic substitution on saturated carbon. Acetolysis of dialkylmercury compounds. J. Am. Chem. Soc. 1955, 77, 3747–3752. [Google Scholar] [CrossRef]
- Sen, A. Catalytic functionalization of carbon−hydrogen and carbon−carbon bonds in protic media. Acc. Chem. Res. 1998, 31, 550. [Google Scholar] [CrossRef]
- Jones, W.D. Conquering the carbon-hydrogen bond. Science 2000, 287, 1942. [Google Scholar] [CrossRef]
- Ritleng, V.; Sirlin, C.; Pfeffer, M. Ru-, Rh-, and Pd-catalyzed C−C bond formation involving C−H activation and addition on unsaturated substrates: Reactions and mechanistic aspects. Chem. Rev. 2002, 102, 1731. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Privalov, T. A theoretical study of the essential role of DMSO as a solvent/ligand in the Pd (OAc) 2/DMSO catalyst system for aerobic oxidation. Organometallics 2005, 24, 6019. [Google Scholar] [CrossRef]
- Parshina, L.N.; Grishchenko, L.A.; Larina, L.I.; Novikova, L.N.; Trofimov, B.A. Cross-coupling of propargylated arabinogalactan with 2-bromothiophene. Carbohyd. Polym. 2016, 150, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Corwin, A.H.; Naylor, M.A., Jr. Aromatic substitution. The cleavage of diphenylmercury1. J. Am. Chem. Soc. 1947, 69, 1004–1009. [Google Scholar] [CrossRef]
- Brown, H.C.; Nelson, K.L. An interpretation of meta orientation in the alkylation of toluene. The relative reactivity and isomer distribution in the chloromethylation and mercuration of benzene and toluene1, 2. J. Am. Chem. Soc. 1953, 75, 6292–6299. [Google Scholar] [CrossRef]
- Brown, H.C.; McGary, C.W. The rates of mercuration of the monoalkyl-and the polymethylbenzenes. calculation of relative reactivities for the mercuration reaction1, 2. J. Am. Chem. Soc. 1955, 77, 2310–2312. [Google Scholar] [CrossRef]
- Kresge, A.; Brennan, J. Perchloric Acid catalyzed aromatic mercuration in acetic acid solution. II. The Substitution Process. J. Org. Chem. 1967, 32, 752–755. [Google Scholar]
- Fernandez-Rodriguez, M.A.; Shen, Q.; Hartwig, J.F. A general and long-lived catalyst for the palladium-catalyzed coupling of aryl halides with thiols. J. Am. Chem. Soc. 2006, 128, 2180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Xu, S.; Pan, C.; Sui, X.; Gu, Z. Catalytically asymmetric Pd/Norbornene catalysis: Enantioselective synthesis of (+)-Rhazinal, (+)-Rhazinilam, and (+)-Kopsiyunnanine C1–3. Org. Lett. 2016, 18, 3782–3785. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.S.; Wan, J.P.; Mao, H.; Pan, Y.J. Facile synthesis of 2-(phenylthio)phenols by copper(I)-catalyzed tandem transformation of C-S coupling/C-H functionalization. J. Am. Chem. Soc. 2010, 132, 15531. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.S.; Yue, L.; Pan, Y.J. Regioselective copper (I)-catalyzed C–H hydroxylation/C–S coupling: Expedient construction of 2-(styrylthio) phenols. Tetrahedron 2012, 68, 5046. [Google Scholar] [CrossRef]
- Strieter, E.R.; Bhayana, B.; Buchwald, S.L. Mechanistic studies on the copper-catalyzed N-arylation of amides. J. Am. Chem. Soc. 2009, 131, 78. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y. The first phosphine oxide ligand precursors for transition metal catalyzed cross-coupling reactions: C−C, C−N, and C−S bond formation on unactivated aryl chlorides. Angew. Chem. Int. Ed. 2001, 40, 1513. [Google Scholar] [CrossRef]
- Peng, X.; Ma, C.; Tung, C.H.; Xu, Z. Cu-catalyzed three-component coupling of aryne, alkyne, and benzenesulfonothioate: Modular synthesis of o-alkynyl arylsulfides. Org. Lett. 2016, 18, 4154–4157. [Google Scholar] [CrossRef] [PubMed]
- Munakata, M.; Kuroda-Sowa, T.; Maekawa, M.; Hirota, A.; Kitagawa, S. Building of 2D sheet of tetrakis (methylthio) tetrathiafulvalenes coordinating to copper (I) halides with zigzag and helical frames and the 3D network through the S. cntdot.. cntdot.. cntdot. S contacts. Inorg. Chem. 1995, 34, 2705. [Google Scholar] [CrossRef]
- Peariso, K.; Huffman, D.L.; Penner-Hahn, J.E.; O’Halloran, T.V. The PcoC copper resistance protein coordinates Cu(I) via novel S-methionine interactions. J. Am. Chem. Soc. 2003, 125, 342. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, B.; Liu, J.; Li, H.L.; Na, R. Brønsted acid or lewis acid catalyzed [3+3] cycloaddition of azomethine imines with N-benzyl azomethine ylide: A facile access to bicyclic N-heterocycles. Synlett 2016, 27, 282–286. [Google Scholar] [CrossRef]
- Cotton, F.A.; Dikarev, E.V.; Petrukhina, M.A.; Striba, S.-E. Studies of dirhodium (II) tetra (trifluoroacetate). 5. Remarkable examples of the ambidentate character of dimethyl sulfoxide. Inorg. Chem. 2000, 39, 1748. [Google Scholar] [CrossRef] [PubMed]
- Thornley, W.A.; Bitterwolf, T.E. Photolysis of Isoelectronic ruthenium nitrosyl and diazonium complexes in frozen PVC matrices: Retention of dinitrogen on ruthenium following photochemical phenyl radical loss. Eur. J. Inorg. Chem. 2016, 2016, 464–468. [Google Scholar] [CrossRef]
- Epstein, W.W.; Sweat, F.W. Dimethyl sulfoxide oxidations. Chem. Rev. 1967, 67, 247. [Google Scholar] [CrossRef]
- Barnes, I.; Hjorth, J.; Mihalopoulos, N. Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere. Chem. Rev. 2006, 106, 940. [Google Scholar] [CrossRef] [PubMed]
- Cella, J.A.; Bacon, S.W. Preparation of dialkyl carbonates via the phase-transfer-catalyzed alkylation of alkali metal carbonate and bicarbonate salts. J. Org. Chem. 1984, 49, 1122. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Buchwald, S.L. Scope and limitations of the Pd/BINAP-catalyzed amination of aryl bromides. J. Org. Chem. 2000, 65, 1144. [Google Scholar] [CrossRef] [PubMed]
- Reichart, B.; de la Cruz, G.G.; Zangger, K.; Kappe, C.O.; Glasnov, T. Copper/nafion-catalyzed hydroarylation process involving ketenimine intermediates: A novel and synthetic approach to 4-sulfonamidoquinoline-2-ones and derivatives thereof. Adv. Syn. Catal. 2016, 358, 50–55. [Google Scholar] [CrossRef]
- Olah, G.A.; Reddy, V.P.; Prakash, G.K.S. Long-lived cyclopropylcarbinyl cations. Chem. Rev. 1992, 92, 69. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 03, Revision 05; Gaussian Inc.: Pittsburgh, PA, USA, 2003.
Sample Availability: Samples of the compounds is not available from authors. |

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, R.; Cai, R.; Zhou, S.; Zhou, Z.; Li, B.; Xu, D. Mechanistic Study of Copper-Catalyzed C-H Hydroxylation/C-S Coupling by ESI-HR MS and DFT Calculations. Molecules 2017, 22, 1912. https://doi.org/10.3390/molecules22111912
Xu R, Cai R, Zhou S, Zhou Z, Li B, Xu D. Mechanistic Study of Copper-Catalyzed C-H Hydroxylation/C-S Coupling by ESI-HR MS and DFT Calculations. Molecules. 2017; 22(11):1912. https://doi.org/10.3390/molecules22111912
Chicago/Turabian StyleXu, Runsheng, Rongrong Cai, Sixian Zhou, Zhuoda Zhou, Beibei Li, and Dihui Xu. 2017. "Mechanistic Study of Copper-Catalyzed C-H Hydroxylation/C-S Coupling by ESI-HR MS and DFT Calculations" Molecules 22, no. 11: 1912. https://doi.org/10.3390/molecules22111912
APA StyleXu, R., Cai, R., Zhou, S., Zhou, Z., Li, B., & Xu, D. (2017). Mechanistic Study of Copper-Catalyzed C-H Hydroxylation/C-S Coupling by ESI-HR MS and DFT Calculations. Molecules, 22(11), 1912. https://doi.org/10.3390/molecules22111912