1. Introduction
The dihydropyridine calcium channel antagonist amlodipine is one of the most commonly prescribed drugs for treatment of hypertension and angina [
1,
2,
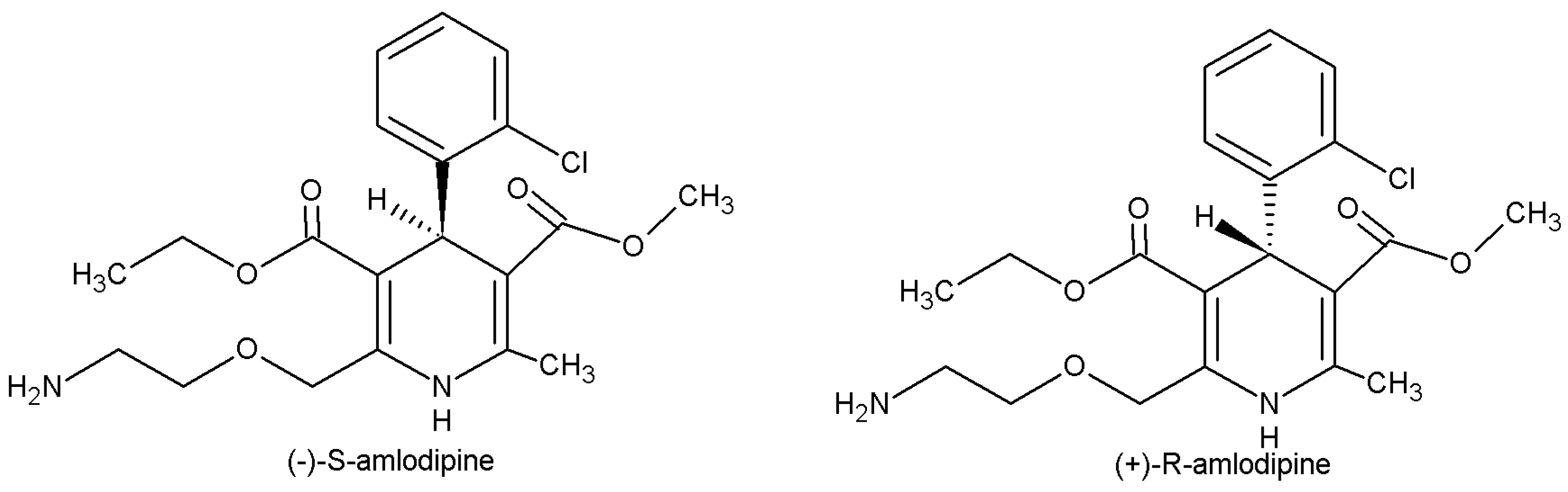
3]. It is commercially available as a racemic 1:1 mixture of
R- and
S-enantiomers (
Figure 1). However,
S-amlodipine is known to be 1000 times more pharmacologically potent than
R-amlodipine and has therefore been marketed in enantiopure form under the name levamlodipine in some Asian countries [
4,
5,
6]. Amlodipine is extensively (about 90%) converted to inactive metabolites via hepatic metabolism, with 60% of the metabolites being excreted in the urine together with 10% of the remaining parent compound. The drug is cleared via CYP3A-mediated dehydrogenation of its dihydropyridine moiety to a pyridine derivate, primarily yielding an inactive metabolite designated M9 [
1,
7].
Cytochrome P450 enzymes (CYPs) are important in the oxidative biotransformation of endogenous compounds and xenobiotics including drugs and environmental chemicals [
8,
9,
10]. Drug–drug interactions that affect CYP activity can cause major problems in patients who are undergoing multi-drug treatment or who consume certain foods: the resulting enzyme inhibition can increase the risk of adverse reactions or reduce the effectiveness of prodrugs. Amlodipine reportedly reduces the activities of various cytochromes P450 including CYP1A1, CYP3A4, CYP2B6 and CYP2C9 [
11,
12]. The genetic polymorphism of CYPs may also significantly affect the efficacy and safety of some drugs, giving rise to the so-called “poor”, “intermediate”, “extensive” (also referred to as “normal”), and ultrarapid metabolism pharmacogenetic phenotypes [
10]. This effect can be augmented by interactions between concomitantly taken drugs that are metabolized by the same enzymes. Many examples of genetic influences on the metabolism of clinically relevant drugs have been reported [
13]. The CYP2C family accounts for approximately 28% of the total CYP protein content in human liver microsomes (HLM) [
14], and CYP2C9 mediates the metabolism of 20% of all drugs that undergo phase I metabolism [
15]. To date, at least 60 allelic variants of CYP2C9 and 35 allelic variants of CYP2C19 (
www.pharmvar.org/) have been reported, many of which are associated with reduced substrate metabolism. Several studies have demonstrated the clinical significance of CYP2C9 and CYP2C19 polymorphisms [
10].
It is reasonable to expect that the enantiomers of amlodipine may have different inhibitory effects on CYP-mediated drug metabolism. To investigate the inhibitory potency of R- and S-amlodipine, their IC50 and Ki values against nine relevant CYPs were assessed in HLM, and their selective effects were characterized. Because CYP2C9 and CYP2C19 are highly polymorphic and their polymorphisms are relevant to the pharmacodynamics and pharmacokinetics of many drugs, additional experiments were performed to characterize the amlodipine enantiomers’ inhibitory effects on CYP2C9 and CYP2C19 polymorphisms associated with extensive (normal), intermediate, and no enzyme activity.
3. Discussion
Amlodipine is clinically available as a racemic mixture of
R- and
S-enantiomers, which are known to have different pharmacokinetic and pharmacodynamic properties. Compared to the
R-enantiomer, the
S-enantiomer exhibits approximately 1000-fold greater pharmacological activity, is eliminated more slowly, and has a longer t
1/2 [
17]. In addition, no racemization occurs in vivo when enantiopure AML is administered to humans [
18]. In 2014, amlodipine was the sixth most commonly prescribed drug in the United States [
19]. As a typical CYP3A substrate, it is known to influence the metabolism of several coadministered CYP3A substrates. A typical example is its effects on statins, with which it is commonly coprescribed in patients with hypertension and hypercholesterolemia: amlodipine can raise the level of simvastatin and increase the risk of myopathy. Consequently, the US Food and Drug administration has issued a recommended dose limitation for this drug combination [
1,
20]. The inhibition of CYP3A by amlodipine is also responsible for a diminished pharmacodynamic response to clopidogrel [
21,
22]. Also there was reported a significant in vivo interaction between amlodipine and tacrolimus resulting in rapid increase in tacrolimus blood concentrations [
23]. The CYP3A4 is predominant hepatic form but also CYP3A5 contributes significantly to total liver CYP3A.Their substrate specificity overlaps and therefore it is referred here to these two enzymes together as CYP3A [
24].
When dealing with racemic mixtures of compounds that inhibit CYP-mediated metabolism in vitro, it is important to consider the inhibitory potency of each enantiomer individually because several cases of stereoselectivity in CYP inhibition have been reported. For instance,
R-omeprazole inhibited the 4-hydroxylation of diclofenac by CYP2C9 16 times more strongly than did
S-omeprazole [
25], the K
i value for (+)-ketoconazole towards CYP3A (6β-hydroxylation of testosterone) is five times lower than that for (−)-ketoconazole [
26], the K
i value for
R-tamsulosin towards CYP3A (6β-hydroxylation of testosterone) is five times lower than that for
S-tamsulosin [
27]. We have studied the inhibition of CYP3A enzymes extensively because these enzymes are responsible for most known drug biotransformation reactions, including the oxidation of amlodipine (which is mediated almost exclusively by CYP3A). Evidence for stereoselective inhibition of CYPs by some other dihydropyridine calcium channel blockers has been presented recently. For example, (+)-benidipine and (+)-felodipine were reported to strongly inhibit CYP3A and CYP2C19, respectively [
28]. This work demonstrates that both
R- and
S-amlodipine are effective reversible and time-dependent inhibitors of CYP3A enzyme activities in human liver microsomes.
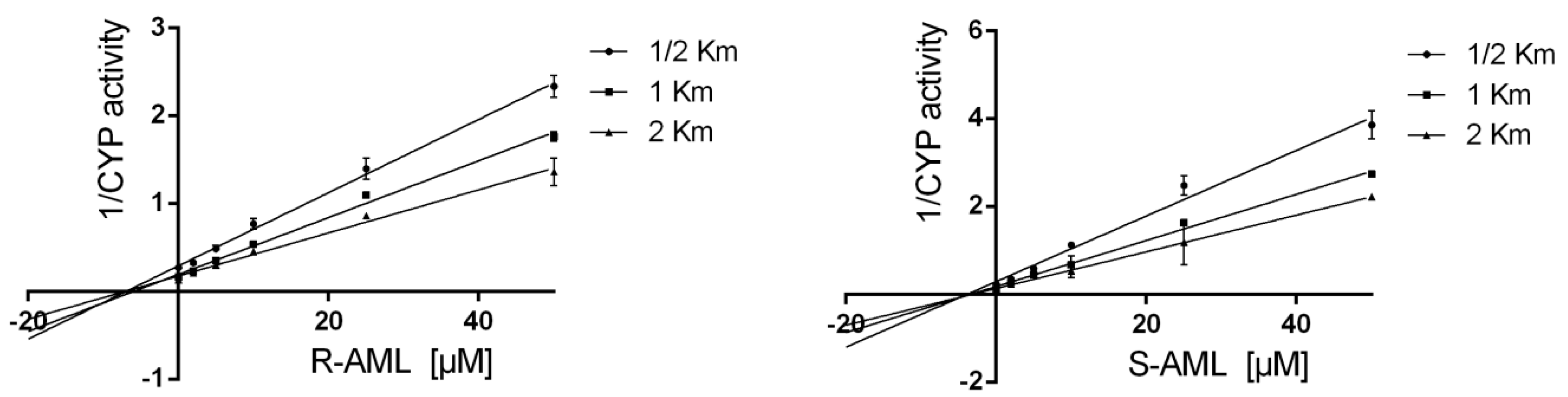
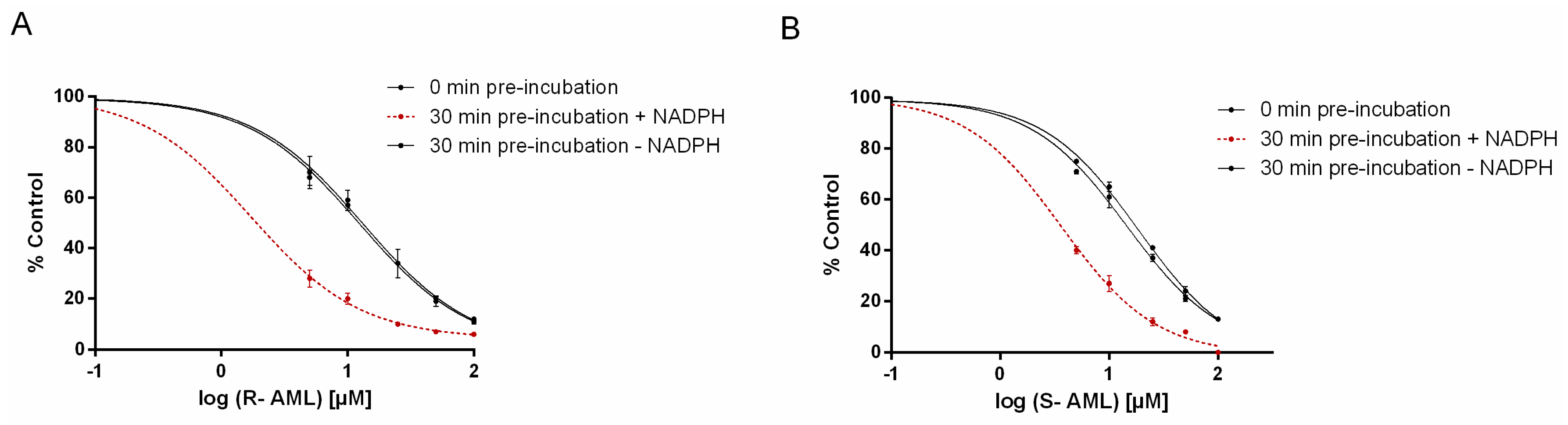
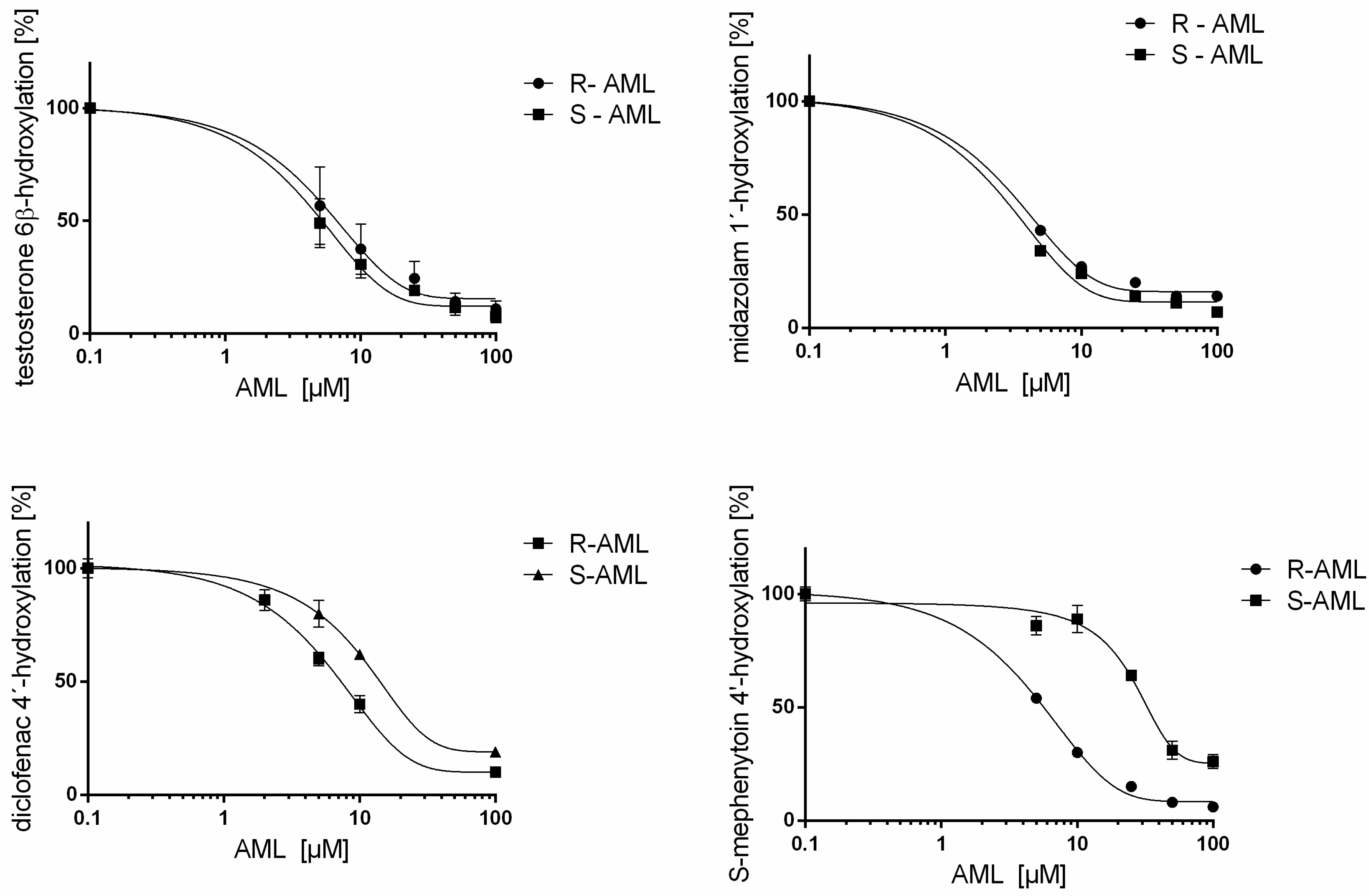
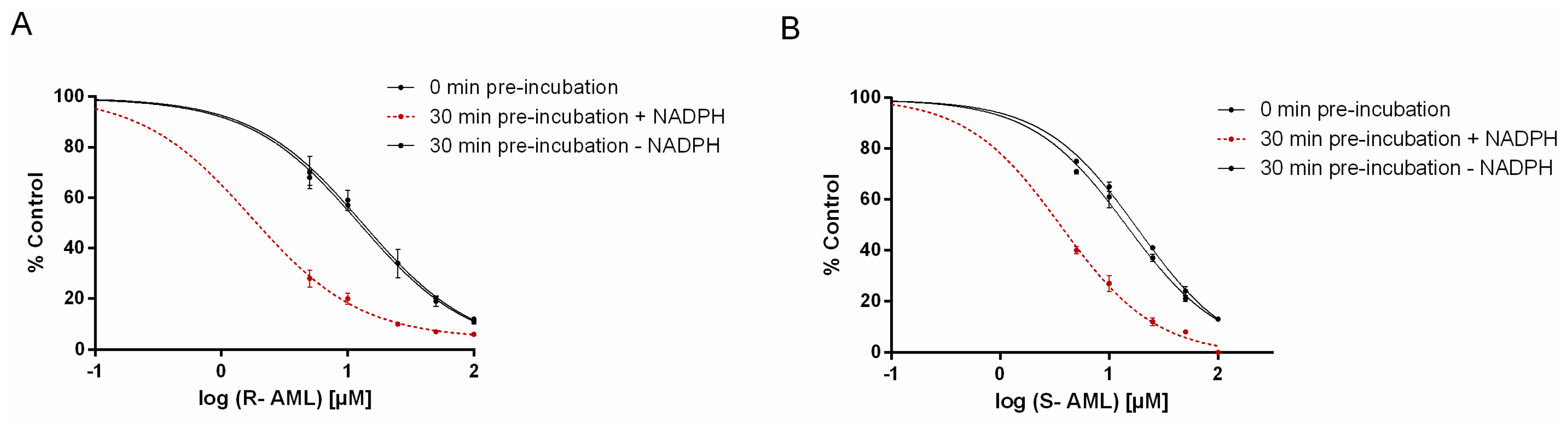
S-AML is the stronger inhibitor if one only considers competitive inhibition (clearly indicated in shorter incubation times). However, time-dependent inhibition experiments showed
R-AML to be the stronger inhibitor. Experiments with extended preincubation periods indicated that both mechanisms of inhibition become relevant in such cases (see
Table 4). Amlodipine enantiomers act as competitive inhibitors and also exhibit the time-dependent inhibition. Time-dependent inhibition encompasses all phenomena that cause enzyme activity to decline as the incubation time increases. It can result from several different processes, but two are considered especially significant. One is “mechanism-based inhibition”, which typically involves the formation of reactive electrophiles that react with the CYP to form covalent adducts of the heme or apoprotein, inactivating the enzyme. The second involves the formation of tightly bound complexes between the P450 and specific metabolites (this process is also known as “quasi-irreversible” inhibition) [
29,
30]. Increased inhibitory potency (up to 8-fold) towards CYP3A activity when preincubated in the presence of NADPH suggest that AML may be converted, at least in part, to reactive intermediates or products that contribute to the overall inhibition of CYP3A activity. Time-dependent inhibition by racemic amlodipine was previously predicted by Jones et al. by computational approaches [
31].
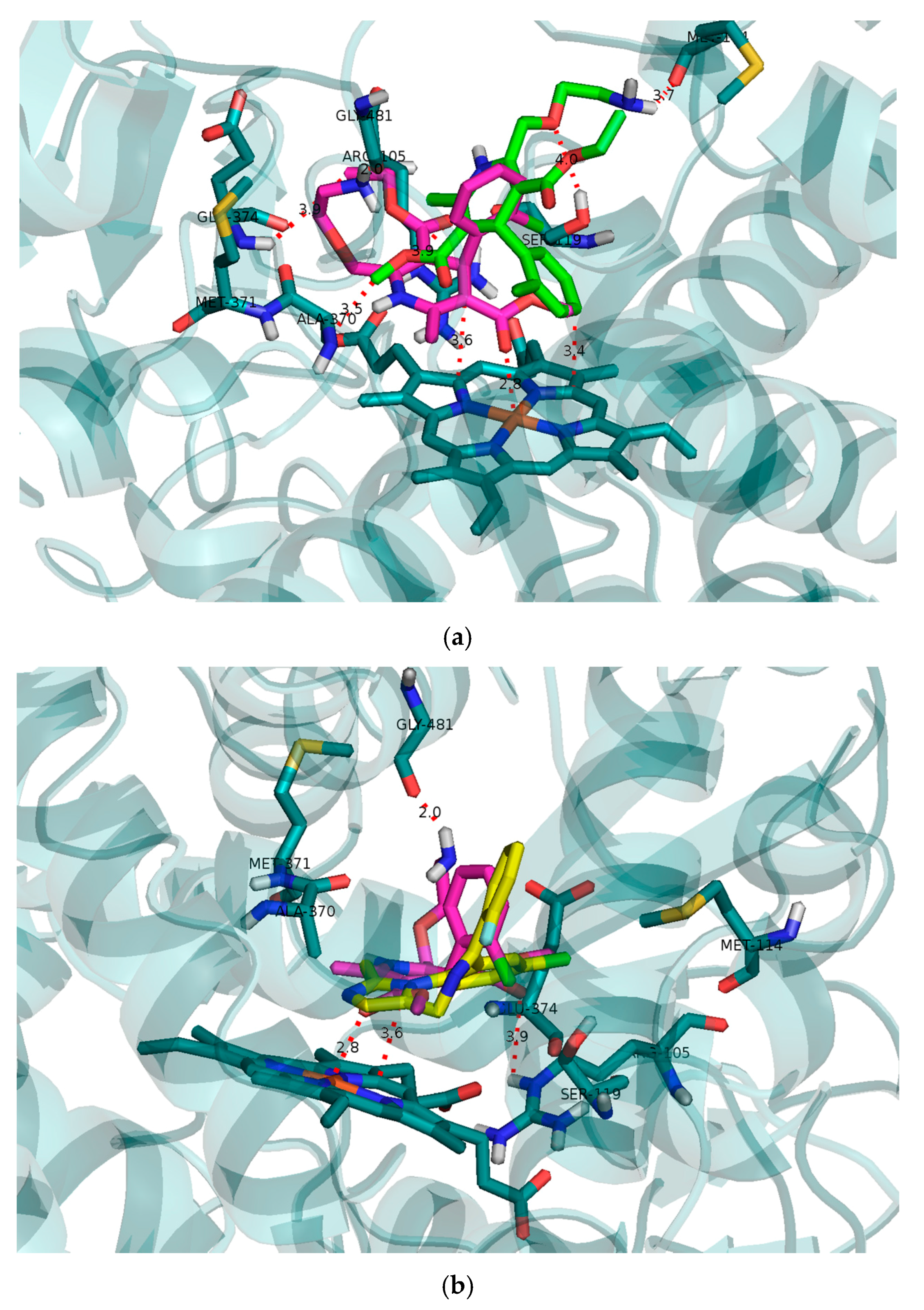
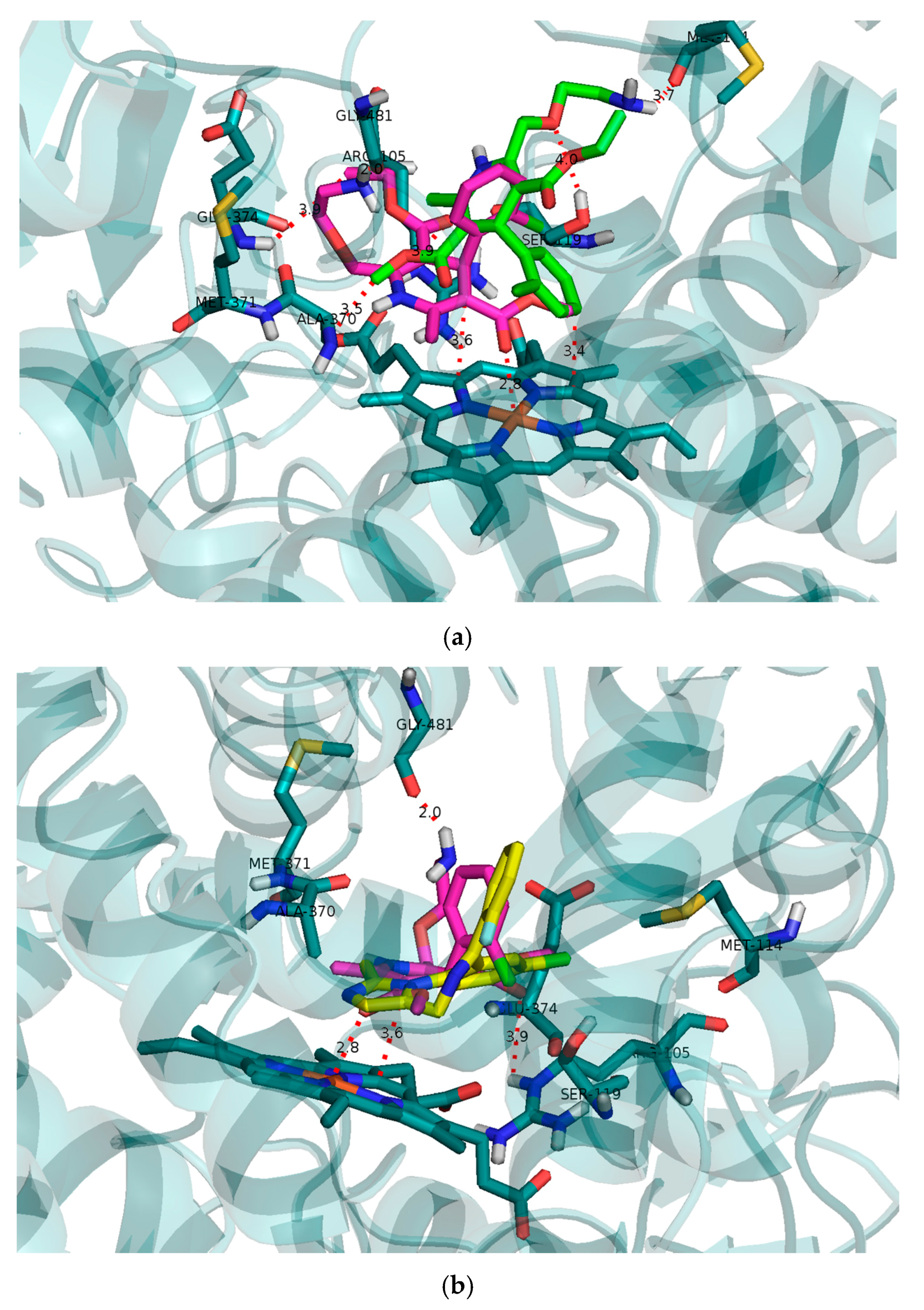
Molecular modelling of the initial binding poses for
R- and
S-AML in CYP3A4 was performed to compare their predicted capabilities as reversible competitive inhibitors to the results of the in vitro experiments. CYP3A4 has a relatively large and flexible active site cavity that can accommodate multiple substrate molecules to achieve optimal activity [
32,
33,
34]. The modelling results confirmed the expected difference between the interactions of
R- and
S-AML with the CYP3A4 cavity (
Figure 4a). Both testosterone and midazolam adopt binding positions that place their sites of oxidation by CYP3A4 in close proximity to the heme located at the bottom of the cavity.
S-AML occupies a position closer to the heme than does
R-AML, and is therefore more effective at preventing the approach of other substrate molecules. Together with the potential formation of π–π stacking interactions between the heme and the dihydropyridine ring of
S-AML, this may explain
S-AML’s greater inhibitory activity. The binding free energy of −7.6 kcal/mol for
S-AML also reflects its higher affinity towards the active site compared to the
R-isomer (−6.7 kcal/mol).
Because
S-AML is the pharmacologically active component of racemic amlodipine, the use of enantiopure
S-AML could impose fewer burdens on patients and reduce the risk of drug–drug interactions. The absence of
R-AML may be also beneficial in terms of cytochrome P450 inhibition. This suggestion was strongly confirmed by the finding that
R-AML was a stronger inhibitor of both CYP2C9 and CYP2C19, having IC
50 values against these enzymes that were two- and twelve-fold lower than those for
S-AML, respectively (see
Table 1). The differences between
R- and
S-AML in terms of their inhibition of CYP3A were minor and probably clinically insignificant, but treatment with enantiopure
S-AML would halve the required amlodipine dosage required for effective treatment. If one also considers that
R-AML causes venodilation and is responsible for side effects associated with racemic amlodipine, the use of the pure
S-enantiomer becomes even more appealing [
5]. Enantiopure
S-AML is already used as a drug in India, China and some other Asian countries. Levamlodipine has a better safety profile than the racemate because it is administered at half the dosage and offers better tolerability with a reduced incidence of peripheral edema while retaining the antihypertensive effectiveness of conventional amlodipine [
6].
Approximately 40% of human cytochrome P450 drug metabolism is carried out by polymorphic enzymes [
35]. Extensive and intermediate metabolizers are more susceptible to drug interactions resulting from CYP inhibition than poor metabolizers. As discussed above, the pharmacokinetics of amlodipine in humans are stereoselective. In our inhibition studies, both enantiomers exhibited inhibition potential towards two polymorphic cytochromes P450: CYP2C9 and CYP2C19. We were interested in the possible differences between poor, intermediate and extensive metabolizer phenotypes of these two enzymes in terms of their stereoselective inhibition by
R- and
S-AML, and therefore investigated their responses to this drug using genotyped human liver microsomes (see
Table 5). Kim et al. have previously evaluated the effects of different CYP3A5 genotypes on the disposition of the amlodipine enantiomers in humans, and found them to be minor [
36]. This also seems to be true for CYP2C inhibition because we observed no significant differences in inhibition in extensive or intermediate metabolizers. The measured IC
50 values indicate that the stereoselectivity of CYP2C inhibition was allele-independent:
R-AML was the stronger inhibitor of CYP2C9*1/*1, CYP2C9*1/*2, CYP2C19*1/*1, and CYP2C19*1/*2. Naturally, the presence of loss-of-function variants in patient genome (CYP2C9 low and CYP2C19*2/*2) will be reflected in increase of plasma concentrations of the parent drug if the drug is a substrate of the CYP in question, which may lead to drug toxicity in some cases. These loss-of-function variants of CYP2C9 and CYP2C19 cannot hence be inhibited.
Based on the above observations and the typical plasma concentrations (5 to 50 nM) of AML [
17,
37], its enantiomers can be regarded as weak to moderate inhibitors of CYP2C9 and CYP2C19 that exhibit stereoselective patterns only under special conditions (drug accumulation, overdosing, etc.). On the other hand, the inhibition of CYP3A by both enantiomers may affect CYP3A-mediated drug interactions in vivo. Our data are consistent with the results of a previous study that reported IC
50 values for racemic amlodipine against CYP3A (5 µM), CYP2C9 (14 µM) and CYP2D6 (88 µM) [
12]. However, this study provides new evidence on the mechanism of CYP3A inhibition, of reversible and of time-dependent CYP3A inhibition by amlodipine enantiomers. This work highlights the importance of accounting for stereochemistry when evaluating drug–drug interactions. We tested the influence of amlodipine enantiomers on the inhibition of important drug-metabolizing cytochromes P450 and showed that the enantiomers should be treated as pharmacologically independent entities. Our results showed for the first time that both amlodipine enantiomers are reversible (specifically, competitive) and time-dependent inhibitors of CYP3A activity, and inhibitors of CYP2C9 and CYP2C19. Differences in the inhibition potency of the AML enantiomers towards different metabolism phenotypes of CYP2C9 and CYP2C19 were also investigated, but found to be seemingly irrelevant. However, the individual AML enantiomers exhibited stereoselective inhibition of CYP3A and CYP2C.
4. Materials and Methods
R-amlodipine and
S-amlodipine were purchased from Santa Cruz Biotechnology Inc. (Heidelberg, Germany). Ethoxyresorufin, 7-ethoxy-4-(trifluoromethyl)coumarin and 4′-hydroxydiclofenac were purchased from Fluka (Buchs, Switzerland). Coumarin, testosterone, diclofenac, bufuralol, chlorzoxazone, resorufin, 7-hydroxycoumarin, 7-hydroxy-4-(trifluoromethyl)coumarin, 1′-hydroxymidazolam and 1′-hydroxybufuralol were obtained from Sigma-Aldrich (Prague, Czech Republic). Midazolam was purchased from Abcam (Cambridge, UK), 6β-hydroxytestosterone was purchased from Ultrafine (Manchester, UK), and paclitaxel from Chemos CZ (Prague, Czech Republic). The 6-hydroxypaclitaxel and
S-mephenytoin were purchased from Santa Cruz Biotechnology (Heidelberg, Germany), and (
S)-4-hydroxy mephenytoin was bought from Toronto Research Chemicals Inc. (Toronto, Canada). Human liver microsomes and genotyped human liver microsomes (
Table 5) were obtained from XenoTech (Lenexa, KS, USA). The activities of the CYP1A, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A enzymes can be accessed from the XenoTech web site (
www.xenotechllc.com).
4.1. Enzyme Assays
The activities of the individual CYP forms are listed in
Table 6 and were determined using established protocols (
Table 6) and references therein [
38]. The formation of metabolites from specific substrates was monitored using a Prominence HPLC system (Shimadzu, Kyoto, Japan) equipped with a LiChroCART 250-4 LiChrospher 100 RP-18 column or a Chromolith
® HighResolution RP-18 endcapped column (Merck, Darmstadt, Germany) and UV or fluorescence detection as specified in the relevant publications. Preliminary experiments were performed to determine the Michaelis constant (K
m,) and limiting velocity (V
max) of individual CYP forms. The incubation conditions used in each experiment were specific for the CYP form under consideration. In all cases, the conditions were chosen to be within the linear range for the reaction’s V
max in terms of time of incubation, substrate concentration (which was set equal to the enzyme’s K
m), and the quantity of HLM in the reaction mixture (see
Table 6). The reaction conditions used in the inhibition studies were identical to those used for the determination of individual CYP activities. The reaction mixtures were buffered with 100 mM K/PO
4 (pH 7.4) or 50 mM K/PO
4 (pH 7.4), and contained a NADPH-generating system consisting of isocitrate dehydrogenase, NADP+, isocitric acid, and MgSO
4. Parallel methanol controls were performed for each experiment; in all such cases, the content of methanol in the reaction mixture was kept below 0.1% to exclude the possibility of enzyme inhibition at higher organic solvent (e.g., [
39]).
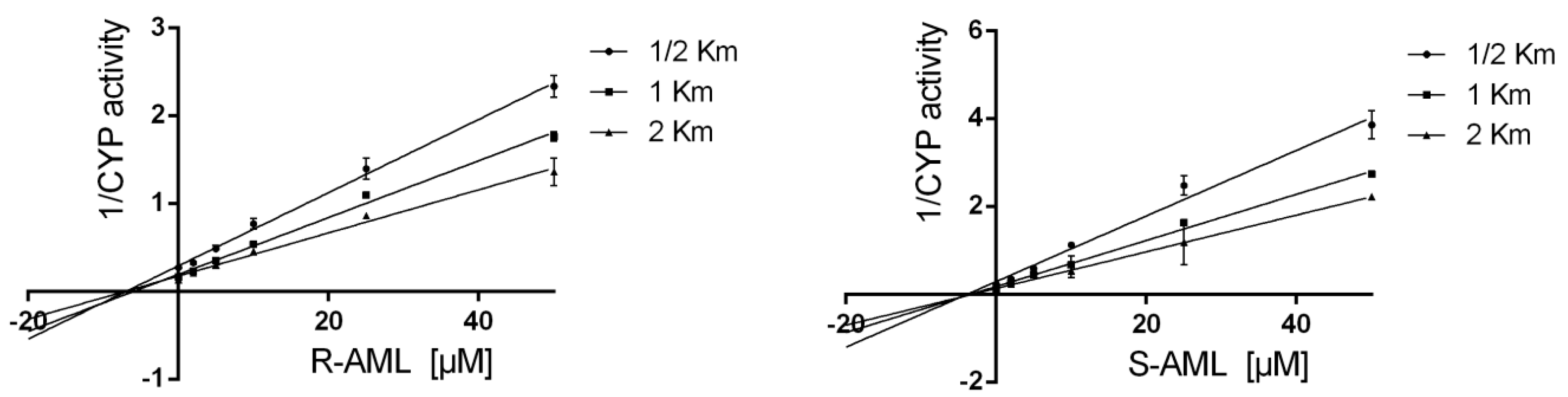
The assays were performed using AML concentrations between 0 and 100 µM. Experiments were performed with the individual enantiomers alongside drug-free controls. Each incubation was performed in triplicate at 37 °C, and two independent measurements were performed for each replicate. Apparent Ki values were determined by performing additional measurements using substrate concentrations corresponding to 1/2 Km, Km, and 2 Km in cases where inhibition was observed (i.e., where the tested drug achieved an IC50 < 10 μM).
Inhibition of individual CYP activities was evaluated by plotting the remaining activity against the inhibitor concentration using GraphPad Prism (La Jolla, CA, USA). IC
50 values were obtained by analyzing plots of the logarithm of the inhibitor concentration against the percentage of activity remaining after inhibition using the Sigma Plot 12 scientific graphing software (version 13.3.0, SPSS, Chicago, IL, USA). Apparent K
i values were determined by nonlinear regression analysis with GraphPad Prism, and are shown in
Table 4. Prism was used to fit the experimental data to the equations presented by Copeland et al. [
40]. The enzyme inhibition model (uncompetitive, mixed-model, competitive, or noncompetitive) applied to each data set was selected on the basis of visual inspection of Lineweaver–Burk, Dixon, and Scatchard plots, together with evaluations of model-derived parameters,
R2 values, and absolute sum of squares values obtained from GraphPad Prism plots.
Stereoselective differences in the inhibitory effects of individual amlodipine enantiomers were analyzed using the Statistica 12 software package (12.1 StatSoft, Prague, Czech Republic). The Shapiro–Wilks test was used as a test of normality. The t-test was used for parametric data and the Mann–Whitney test for nonparametric data.
4.2. The CYP3A Time-Dependent Inhibition
The single point assay was done as follows:
R- or
S-AML (25 μM) were preincubated with or without NADPH and HLM at a concentration of CYP 10-fold higher than required in the assay (125.6 pmol, to minimize potential reversible inhibition). After preincubation, an aliquot of the preincubation mixture was diluted 10-fold with buffer containing midazolam (at a final concentration corresponding to the K
m) and incubated at 37 °C [
41]. Subsequently IC
50 values were determined under three different conditions: with 0-min of preincubation, with a 30-min preincubation in the absence of NADPH, and with a 30-min preincubation in the presence of NADPH to determined IC
50 shift. The preincubation mixtures also contained HLM and the appropriate concentration of an AML enantiomer. Midazolam was added to the reaction mixture after the preincubation period, and the samples were then processed according to the standard inhibition protocol [
42]. To find out the efficiency of inactivation, the K
I and K
inact values were assessed. The K
inact is the maximal rate of enzyme inactivation at a saturating concentration of the inhibitor, while the K
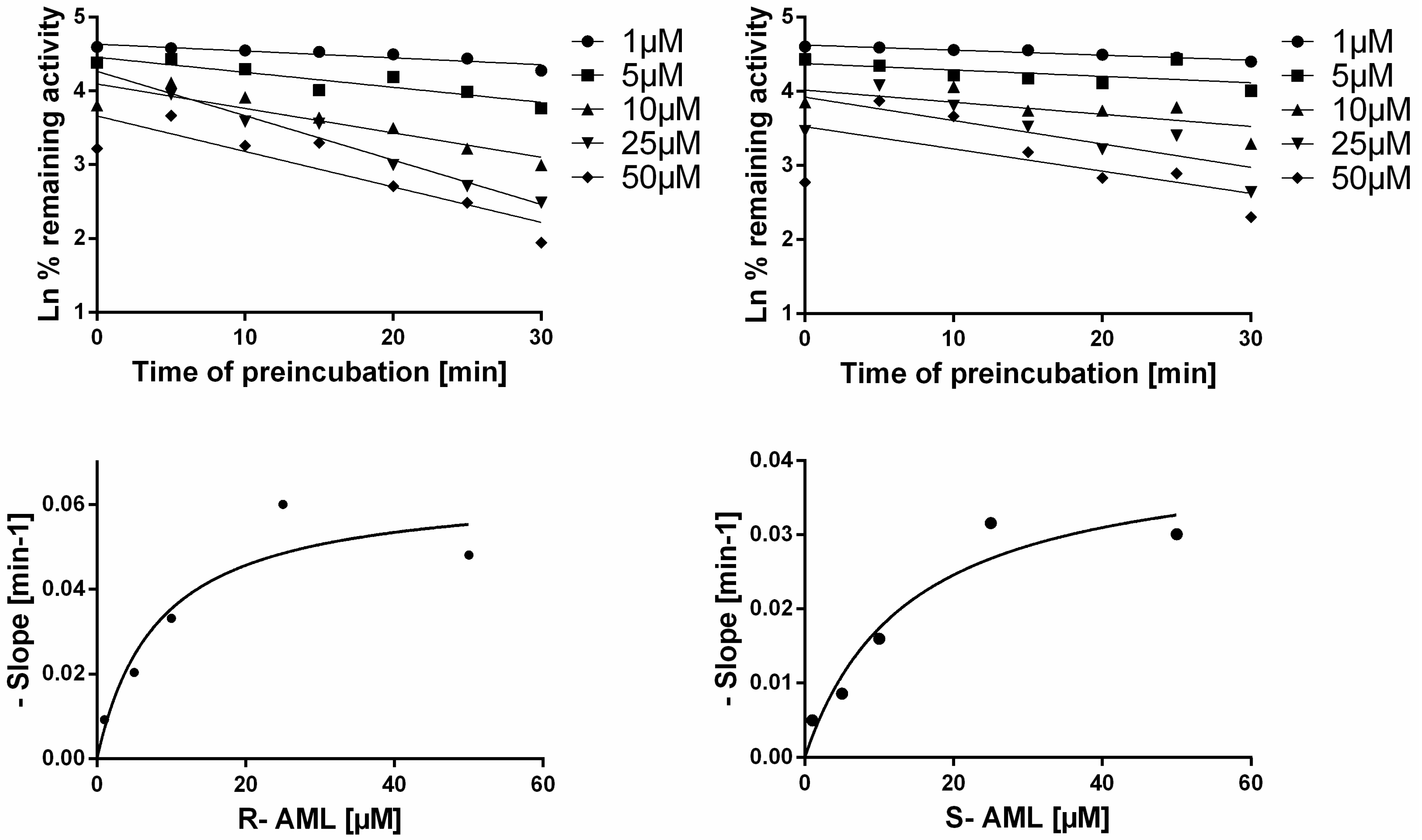
I is the inhibitor concentration that gives half the maximal rate of inactivation. The appropriate AML enantiomer at a concentration in the range of 0–100 μM was preincubated with HLM and NADPH for 0, 5, 10, 15, 20, 25, or 30 min, then an aliquot of the preincubation mixture was diluted with buffer containing midazolam (at a concentration equal to 5 times the K
m) and NADPH. Experiments were also performed with methanol as a vehicle control. Each incubation was performed in duplicate at 37 °C, and two independent measurements were performed for enzyme inactivation at each enantiomer concentration. The natural logarithm of the corrected % remaining activity was then plotted against the preincubation time for each concentration of the inhibitor, and linear regression was performed using GraphPad Prism. The negative slopes obtained in this way (representing the observed initial rates of enzyme inactivation) were plotted against the inhibitor concentration, and the data were fitted by nonlinear regression (using GraphPad Prism) using the following equation:
where
λ represents the rate constant for inactivation at each inhibitor concentration [
I] [
41,
43].
4.3. Computational Docking to the CYP3A4 Active Site
Docking studies were carried out using the Molecular Operating Environment (MOE) software package (version 2015.1001; Chemical Computing Group Inc., Montreal, PQ, Canada) [
40,
41,
43,
44]. The CYP3A4 target structure was prepared from Protein Data Bank (PDB) structure 2V0M by removing the ligand and nonbonding water molecules and then subjecting the resulting structure to an energy minimization was performed (Gradient: 0.001 RMS kcal/mol/A2). Preliminary modeling results (data not shown) suggest that 2VOM geometry is the most favorable geometry to accommodate multiple ligands. The MOE site finder tool was used to identify the active site, the ligand structures were constructed using MOE’s incorporated ligand builder, hydrogens were added, partial charges assigned using the AMBER 94 forcefield, and energy was minimized to relieve strain within the protein structure. The compounds were then docked using the Alpha PMI placement algorithm (Samples per conformation: 30; No. of poses: 250). Rescoring was performed using the Affinity dG scoring function, then pose refinement was done using the induced fit method (Gradient: 0.001 kcal/mol; No. of iterations: 500, Cutoff distance 6 Å), and a second rescoring was performed using the GBVI/WSA dG scoring function. The Root-Mean-Square Deviation between the most highly scored crystallized inhibitor pose and the redocked 2V0M ligand was 1.05 Å.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}