Pharmabiotics as an Emerging Medication for Metabolic Syndrome and Its Related Diseases



Abstract
1. Introduction
2. Current Approach
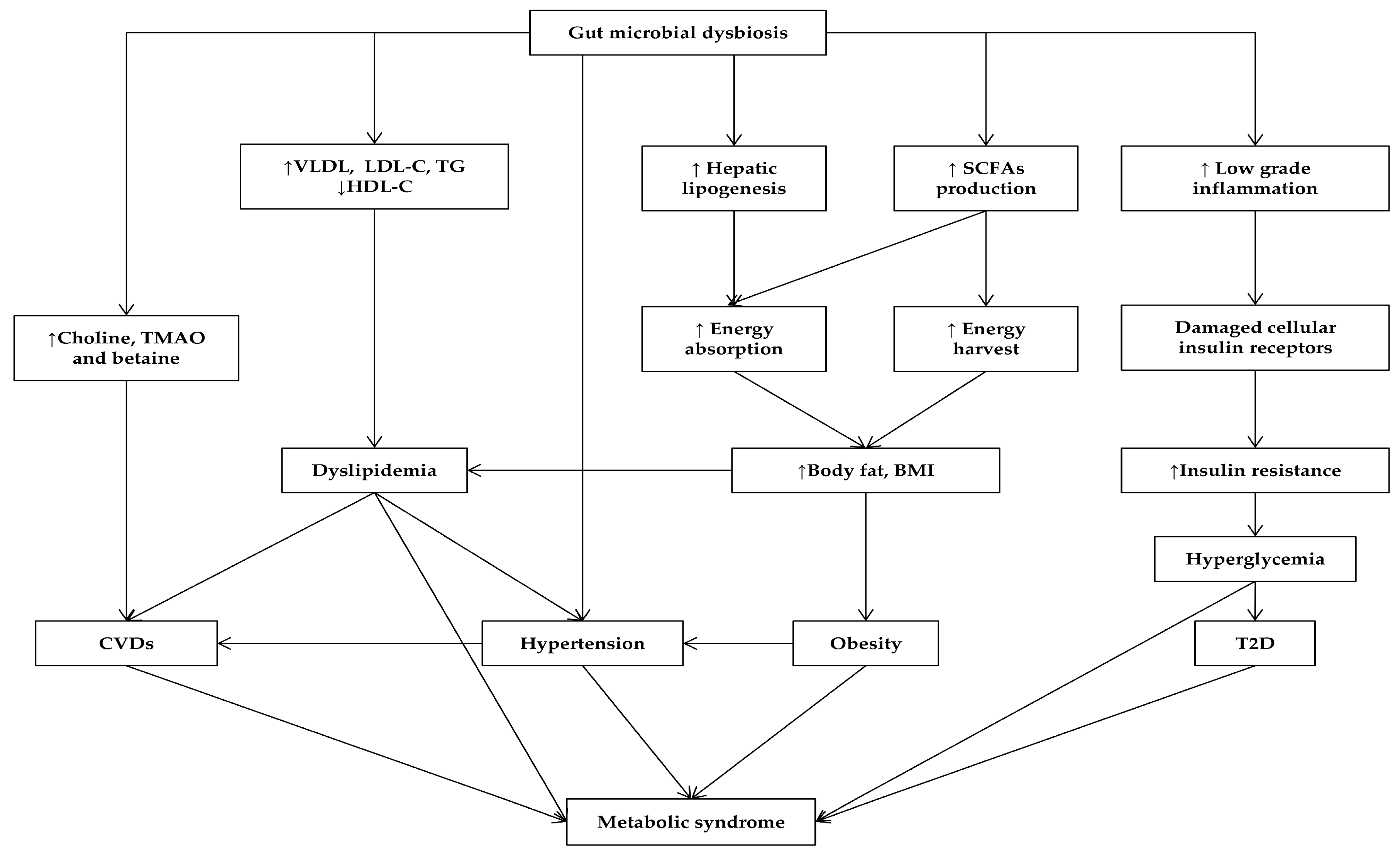
3. Gut Microbiota Affects MetS and Its Related Diseases
3.1. Gut Microbiota in Obesity
3.2. Gut Microbiota Affects Lipid Metabolism
3.3. Gut Microbiota in Diabetes
3.4. Gut Microbiota and Hypertension
3.5. Gut Microbiota and Psychiatric Problems
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alwan, A. Global Status Report on Noncommunicable Diseases 2010; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Eckel, R.H.; Alberti, K.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Samson, S.L.; Garber, A.J. Metabolic syndrome. Endocrinol. Metab. Clin. N. Am. 2014, 43, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J. A comprehensive review on metabolic syndrome. Cardiol. Res. Pract. 2014, 2014, 943162. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.; Pervanidou, P.; Kaltsas, G.; Chrousos, G. Metabolic syndrome: Definitions and controversies. BMC Med. 2011, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, I.; Tillmann, T.; Banerjee, A. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Van Vliet-Ostaptchouk, J.V.; Nuotio, M.-L.; Slagter, S.N.; Doiron, D.; Fischer, K.; Foco, L.; Gaye, A.; Gögele, M.; Heier, M.; Hiekkalinna, T. The prevalence of metabolic syndrome and metabolically healthy obesity in europe: A collaborative analysis of ten large cohort studies. BMC Endocr. Disord. 2014, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Langan, S.M.; Seminara, N.M.; Shin, D.B.; Troxel, A.B.; Kimmel, S.E.; Mehta, N.N.; Margolis, D.J.; Gelfand, J.M. Prevalence of metabolic syndrome in patients with psoriasis: A population-based study in the united kingdom. J. Investig. Dermatol. 2012, 132, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Curtin, L.R. Prevalence and trends in obesity among us adults, 1999–2008. JAMA 2010, 303, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; Naghavi, M. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and trends in diabetes among adults in the united states, 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Guariguata, L.; Whiting, D.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 137–149. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Report On Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- King, H.; Aubert, R.E.; Herman, W.H. Global burden of diabetes, 1995–2025: Prevalence, numerical estimates, and projections. Diabetes Care 1998, 21, 1414–1431. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M. Global burden of stroke and risk factors in 188 countries, during 1990–2013: A systematic analysis for the global burden of disease study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef]
- Daviglus, M.L.; Talavera, G.A.; Avilés-Santa, M.L.; Allison, M.; Cai, J.; Criqui, M.H.; Gellman, M.; Giachello, A.L.; Gouskova, N.; Kaplan, R.C. Prevalence of major cardiovascular risk factors and cardiovascular diseases among hispanic/latino individuals of diverse backgrounds in the united states. JAMA 2012, 308, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Dahlöf, B. Cardiovascular disease risk factors: Epidemiology and risk assessment. Am. J. Cardiol. 2010, 105, 3A–9A. [Google Scholar] [CrossRef] [PubMed]
- Towfighi, A.; Saver, J.L. Stroke declines from third to fourth leading cause of death in the united states. Stroke 2011, 42, 2351–2355. [Google Scholar] [CrossRef] [PubMed]
- Scholze, J.; Alegria, E.; Ferri, C.; Langham, S.; Stevens, W.; Jeffries, D.; Uhl-Hochgraeber, K. Epidemiological and economic burden of metabolic syndrome and its consequences in patients with hypertension in germany, spain and italy; a prevalence-based model. BMC Public Health 2010, 10, 529. [Google Scholar] [CrossRef] [PubMed]
- De Onis, M.; Blössner, M.; Borghi, E. Global prevalence and trends of overweight and obesity among preschool children. Am. J. Clin. Nutr. 2010, 92, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Friend, A.; Craig, L.; Turner, S. The prevalence of metabolic syndrome in children: A systematic review of the literature. Metab. Syndr. Relat. Disord. 2013, 11, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lim, H. The Global Childhood Obesity Epidemic And The Association between Socio-Economic Status And Childhood Obesity; Taylor & Francis: Boca Raton, FL, USA, 2012. [Google Scholar]
- Poyrazoglu, S.; Bas, F.; Darendeliler, F. Metabolic syndrome in young people. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Hills, A.P.; Mokhtar, N.; Brownie, S.; Byrne, N.M. Childhood obesity in Asia: The value of accurate body composition methodology. Asia Pac. J. Clin. Nutr. 2014, 23, 339–343. [Google Scholar] [PubMed]
- Mitchell, A.J.; Vancampfort, D.; Sweers, K.; van Winkel, R.; Yu, W.; De Hert, M. Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders—A systematic review and meta-analysis. Schizophr. Bull. 2011, 39, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Vancampfort, D.; Vansteelandt, K.; Correll, C.U.; Mitchell, A.J.; De Herdt, A.; Sienaert, P.; Probst, M.; De Hert, M. Metabolic syndrome and metabolic abnormalities in bipolar disorder: A meta-analysis of prevalence rates and moderators. Am. J. Psychiatry 2013, 170, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Drug therapy of the metabolic syndrome: Minimizing the emerging crisis in polypharmacy. Nat. Rev. Drug Discov. 2006, 5, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Ioannides-Demos, L.L.; Piccenna, L.; McNeil, J.J. Pharmacotherapies for obesity: Past, current, and future therapies. J. Obes. 2010, 2011, 179674. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.G.; Park, C.-Y. Anti-obesity drugs: A review about their effects and safety. Diabetes Metab. J. 2012, 36, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Marvasti, T.B.; Adeli, K. Pharmacological management of metabolic syndrome and its lipid complications. DARU J. Pharma. Sci. 2010, 18, 146–154. [Google Scholar]
- Beckett, R.D.; Schepers, S.M.; Gordon, S.K. Risk of new-onset diabetes associated with statin use. SAGE Open Med. 2015, 3, 2050312115605518. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.K.; Quon, M.J.; Han, S.H.; Lee, Y.; Kim, S.J.; Shin, E.K. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J. Am. Coll. Cardiol. 2010, 55, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimof, S.; Mirmiran, P. Nutritional approaches for prevantion and treatment of metabolic syndrome in adults. J. Paramed. Sci. 2013, 4. [Google Scholar] [CrossRef]
- Durstine, J.L.; Gordon, B.; Wang, Z.; Luo, X. Chronic disease and the link to physical activity. J. Sport Health Sci. 2013, 2, 3–11. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Torgerson, J.S.; Hauptman, J.; Boldrin, M.N.; Sjostrom, L. Xenical in the prevention of diabetes in obese subjects (xendos) study: A randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 2004, 27, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Douglas, I.J.; Langham, J.; Bhaskaran, K.; Brauer, R.; Smeeth, L. Orlistat and the risk of acute liver injury: Self controlled case series study in uk clinical practice research datalink. BMJ (Clin. Res. Ed.) 2013, 346, f1936. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.B.; Barros, L.T.; Turatti, A.; Martinello, F.; Modiano, P.; Ribeiro-Silva, A.; Vespucio, M.V.; Uyemura, S.A. The anti-obesity agent orlistat is associated to increase in colonic preneoplastic markers in rats treated with a chemical carcinogen. Cancer Lett. 2006, 240, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Katsuki, S.; Takahashi, Y.; Ohi, M.; Nojiri, S.; Sakamaki, S.; Kato, J.; Kogawa, K.; Miyake, H.; Niitsu, Y. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 1998, 339, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Franson, K.; Rossner, S. Fat intake and food choices during weight reduction with diet, behavioural modification and a lipase inhibitor. J. Intern. Med. 2000, 247, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, W.J.; Grottick, A.J.; Menzaghi, F.; Reyes-Saldana, H.; Espitia, S.; Yuskin, D.; Whelan, K.; Martin, M.; Morgan, M.; Chen, W.; et al. Lorcaserin, a novel selective human 5-hydroxytryptamine2c agonist: In vitro and in vivo pharmacological characterization. J. Pharmacol. Exp. Ther. 2008, 325, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.A.; Fletcher, P.J. Therapeutic potential of 5-ht2c receptor agonists for addictive disorders. ACS Chem. Neurosci. 2015, 6, 1071–1088. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.M.; Smith, J.M.; Tsai, J.H.; Schultz, J.A.; Gilson, C.A.; Estrada, S.A.; Chen, R.R.; Park, D.M.; Prieto, E.B.; Gallardo, C.S.; et al. Discovery and sar of new benzazepines as potent and selective 5-ht(2c) receptor agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2005, 15, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Serotonin 5-ht2c receptors as a target for the treatment of depressive and anxious states: Focus on novel therapeutic strategies. Therapie 2005, 60, 441–460. [Google Scholar] [CrossRef] [PubMed]
- Lorcaserin. In obesity: Unacceptable risks. Prescrire Int. 2014, 23, 117–120. [Google Scholar]
- Lipska, K.J.; Bailey, C.J.; Inzucchi, S.E. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care 2011, 34, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Klepser, T.B.; Kelly, M.W. Metformin hydrochloride: An antihyperglycemic agent. Am. J. Health Syst. Pharm. 1997, 54, 893–903. [Google Scholar] [PubMed]
- Norris, S.L.; Carson, S.; Thakurta, S.; Chan, B.K.S. Drug class reviews. In Drug Class Review: Thiazolidinediones: Final Report Update 1; Oregon Health & Science University Oregon Health & Science University: Portland, OR, USA, 2008. [Google Scholar]
- Byrne, C.D.; Wild, S.H. Review: Thiazolidinediones increase risk for heart failure in type 2 diabetes. Evid. Based Med. 2008, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Seino, S. Cell signalling in insulin secretion: The molecular targets of atp, camp and sulfonylurea. Diabetologia 2012, 55, 2096–2108. [Google Scholar] [CrossRef] [PubMed]
- Blickle, J.F. Meglitinide analogues: A review of clinical data focused on recent trials. Diabetes Metab. 2006, 32, 113–120. [Google Scholar] [CrossRef]
- Singh, S.; Chang, H.Y.; Richards, T.M.; Weiner, J.P.; Clark, J.M.; Segal, J.B. Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: A population-based matched case-control study. JAMA Intern. Med. 2013, 173, 534–539. [Google Scholar]
- Bunck, M.C.; Diamant, M.; Corner, A.; Eliasson, B.; Malloy, J.L.; Shaginian, R.M.; Deng, W.; Kendall, D.M.; Taskinen, M.R.; Smith, U.; et al. One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: A randomized, controlled trial. Diabetes Care 2009, 32, 762–768. [Google Scholar] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (glp-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Ali, E.S.; Hua, J.; Wilson, C.H.; Tallis, G.A.; Zhou, F.H.; Rychkov, G.Y.; Barritt, G.J. The glucagon-like peptide-1 analogue exendin-4 reverses impaired intracellular ca(2+) signalling in steatotic hepatocytes. Biochim. Biophys. Acta 2016, 1863, 2135–2146. [Google Scholar] [PubMed]
- Salvo, F.; Moore, N.; Arnaud, M.; Robinson, P.; Raschi, E.; De Ponti, F.; Begaud, B.; Pariente, A. Addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia: Systematic review and meta-analysis. BMJ (Clin. Res. Ed.) 2016, 353, i2231. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.A.; Bergman, A.; Liu, F.; Stevens, C.; Wang, A.Q.; Zeng, W.; Chen, L.; Snyder, K.; Hilliard, D.; Tanen, M.; et al. Pharmacokinetics and pharmacodynamic effects of the oral dpp-4 inhibitor sitagliptin in middle-aged obese subjects. J. Clin. Pharmacol. 2006, 46, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Alsaad, A.A.; Dhannoon, S.M.; Pantin, S.A.; Porter, I.E. Rare allergic reaction of the kidney: Sitagliptin-induced acute tubulointerstitial nephritis. BMJ Case Rep. 2016, 2016, bcr2016216297. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Krum, H.; Abraham, W.T.; Dickstein, K.; Kober, L.V.; Desai, A.S.; Solomon, S.D.; Greenlaw, N.; Ali, M.A.; Chiang, Y.; et al. Aliskiren, enalapril, or aliskiren and enalapril in heart failure. N. Engl. J. Med. 2016, 374, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.P.; Fish, B.L.; Moulder, J.E. Clinically relevant doses of enalapril mitigate multiple organ radiation injury. Radiat. Res. 2016, 185, 313–318. [Google Scholar] [PubMed]
- Wang, W.; McKinnie, S.M.; Farhan, M.; Paul, M.; McDonald, T.; McLean, B.; Llorens-Cortes, C.; Hazra, S.; Murray, A.G.; Vederas, J.C.; et al. Angiotensin-converting enzyme 2 metabolizes and partially inactivates pyr-apelin-13 and apelin-17: Physiological effects in the cardiovascular system. Hypertension 2016, 68, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.H.; Hall, A.S. Angiotensin receptor blockers may increase risk of myocardial infarction: Unraveling the arb-mi paradox. Circulation 2006, 114, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.I. How to explain the differences between renin angiotensin system modulators. Am. J. Hypertens. 2005, 18, 134s–141s. [Google Scholar] [CrossRef] [PubMed]
- Reudelhuber, T.L. The continuing saga of the at2 receptor: A case of the good, the bad, and the innocuous. Hypertension 2005, 46, 1261–1262. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A. 11c-Labeled Telmisartan, an Angiotensin II Type 1 Receptor Antagonist. In Molecular Imaging and Contrast Agent Database (Micad); National Center for Biotechnology Information (US): Bethesda, MD, USA, 2004. [Google Scholar]
- Prasa, D.; Hoffmann-Walbeck, P.; Barth, S.; Stedtler, U.; Ceschi, A.; Farber, E.; Genser, D.; Seidel, C.; Deters, M. Angiotensin ii antagonists—An assessment of their acute toxicity. Clin. Toxicol. (Phila) 2013, 51, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.A.; Opatrny, L.; Brophy, J.M.; Suissa, S. Drug drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. Can. Med. Assoc. J. 2007, 177, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Santucci, L.; Wallace, J.L.; Sardina, M.; Romano, M.; del Soldato, P.; Morelli, A. Interaction of a selective cyclooxygenase-2 inhibitor with aspirin and no-releasing aspirin in the human gastric mucosa. Proc. Natl. Acad. Sci. USA 2003, 100, 10937–10941. [Google Scholar] [CrossRef] [PubMed]
- Maessen-Visch, M.B.; de Roos, K.P. Dutch venous ulcer guideline update. Phlebology 2014, 29, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr. Opin. Pharmacol. 2013, 13, 632–640. [Google Scholar] [CrossRef] [PubMed]
- McCrindle, B.W.; Ose, L.; Marais, A.D. Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: A multicenter, randomized, placebo-controlled trial. J. Pediatr. 2003, 143, 74–80. [Google Scholar] [CrossRef]
- Nissen, S.E.; Nicholls, S.J.; Sipahi, I.; Libby, P.; Raichlen, J.S.; Ballantyne, C.M.; Davignon, J.; Erbel, R.; Fruchart, J.C.; Tardif, J.C.; et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: The asteroid trial. JAMA 2006, 295, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Sever, P.S.; Dahlof, B.; Poulter, N.R.; Wedel, H.; Beevers, G.; Caulfield, M.; Collins, R.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T.; et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the anglo-scandinavian cardiac outcomes trial--lipid lowering arm (ascot-lla): A multicentre randomised controlled trial. Drugs 2004, 64 (Suppl. 2), 43–60. [Google Scholar]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the collaborative atorvastatin diabetes study (cards): Multicentre randomised placebo-controlled trial. Lancet (Lond. Engl.) 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Kostapanos, M.S.; Liamis, G.L.; Milionis, H.J.; Elisaf, M.S. Do statins beneficially or adversely affect glucose homeostasis? Curr. Vasc. Pharmacol. 2010, 8, 612–631. [Google Scholar] [CrossRef] [PubMed]
- Ghirlanda, G.; Oradei, A.; Manto, A.; Lippa, S.; Uccioli, L.; Caputo, S.; Greco, A.V.; Littarru, G.P. Evidence of plasma coq10-lowering effect by hmg-coa reductase inhibitors: A double-blind, placebo-controlled study. J. Clin. Pharmacol. 1993, 33, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Gehlbach, P.; Li, T.; Hatef, E. Statins for age-related macular degeneration. Cochrane Database Syst. Rev. 2016, Cd006927. [Google Scholar]
- Guymer, R.H.; Baird, P.N.; Varsamidis, M.; Busija, L.; Dimitrov, P.N.; Aung, K.Z.; Makeyeva, G.A.; Richardson, A.J.; Lim, L.; Robman, L.D. Proof of concept, randomized, placebo-controlled study of the effect of simvastatin on the course of age-related macular degeneration. PLoS ONE 2013, 8, e83759. [Google Scholar] [CrossRef] [PubMed]
- Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. Slco1b1 variants and statin-induced myopathy—A genomewide study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [PubMed]
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The clinical pharmacogenetics implementation consortium guideline for slco1b1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.H.; Davidson, M.H.; Stein, E.A.; Bays, H.E.; McKenney, J.M.; Miller, E.; Cain, V.A.; Blasetto, J.W. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (stellar* trial). Am. J. Cardiol. 2003, 92, 152–160. [Google Scholar] [CrossRef]
- McTaggart, F.; Jones, P. Effects of statins on high-density lipoproteins: A potential contribution to cardiovascular benefit. Cardiovasc. Drugs Ther. 2008, 22, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L.; Deeks, E.D. Alirocumab: A review in hypercholesterolemia. Am. J. Cardiovasc. Drugs 2016, 16, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, T.; Sasaki, J.; Ishibashi, S.; Birou, S.; Daida, H.; Dohi, S.; Egusa, G.; Hiro, T.; Hirobe, K.; Iida, M.; et al. Diabetes mellitus. Executive summary of the japan atherosclerosis society (jas) guidelines for the diagnosis and prevention of atherosclerotic cardiovascular diseases in japan-2012 version. J. Atheroscler. Thromb. 2014, 21, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Chanson, N.; Bossi, P.; Schneider, L.; Bourry, E.; Izzedine, H. Rhabdomyolysis after ezetimibe/simvastatin therapy in an hiv-infected patient. NDT Plus 2008, 1, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Merten, D.F.; Grossman, H. Intestinal obstruction associated with cholestyramine therapy. AJR Am. J. Roentgenol. 1980, 134, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A.; Armani, A.; McKenney, J.M.; Guyton, J.R. Safety considerations with gastrointestinally active lipid-lowering drugs. Am. J. Cardiol. 2007, 99, 47c–55c. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, Y. Role of bile acid sequestrants in the treatment of type 2 diabetes. Diabetes Care 2011, 34 (Suppl. 2), S244–S250. [Google Scholar] [CrossRef] [PubMed]
- Beigel, F.; Teich, N.; Howaldt, S.; Lammert, F.; Maul, J.; Breiteneicher, S.; Rust, C.; Goke, B.; Brand, S.; Ochsenkuhn, T. Colesevelam for the treatment of bile acid malabsorption-associated diarrhea in patients with crohn’s disease: A randomized, double-blind, placebo-controlled study. J. Crohns Colitis 2014, 8, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.A.; Rosenstock, J.; Wang, A.C.; Truitt, K.E.; Jones, M.R. Colesevelam hcl improves glycemic control and reduces ldl cholesterol in patients with inadequately controlled type 2 diabetes on sulfonylurea-based therapy. Diabetes Care 2008, 31, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Rodney, G.; Uhlendorf, P.; Maxwell, R.E. The hypolipidaemic effect of gemfibrozil (ci-719) in laboratory animals. Proc. R. Soc. Med. 1976, 69 (Suppl. 2), 6–10. [Google Scholar]
- Fitzgerald, J.E.; Sanyer, J.L.; Schardein, J.L.; Lake, R.S.; McGuire, E.J.; de la Iglesia, F.A. Carcinogen bioassay and mutagenicity studies with the hypolipidemic agent gemfibrozil. J. Natl. Cancer Inst. 1981, 67, 1105–1116. [Google Scholar] [PubMed]
- Yang, L.P.; Keating, G.M. Fenofibric acid: In combination therapy in the treatment of mixed dyslipidemia. Am. J. Cardiovasc. Drugs 2009, 9, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Simo, R.; Mitchell, P. Fenofibrate—A potential systemic treatment for diabetic retinopathy? Am. J. Ophthalmol. 2012, 154, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Keene, D.; Price, C.; Shun-Shin, M.J.; Francis, D.P. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and cetp inhibitors: Meta-analysis of randomised controlled trials including 117,411 patients. BMJ (Clin. Res. Ed.) 2014, 349, g4379. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Sharma, A.; Krishnamoorthy, P.; Garg, J.; Virmani, D.; Sharma, T.; Stefanini, G.; Kostis, J.B.; Mukherjee, D.; Sikorskaya, E. Role of niacin in current clinical practice: A systematic review. Am. J. Med. 2017, 130, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Katzmarzyk, P.T.; Leon, A.S.; Wilmore, J.H.; Skinner, J.S.; Rao, D.C.; Rankinen, T.; Bouchard, C. Targeting the metabolic syndrome with exercise: Evidence from the heritage family study. Med. Sci. Sports Exerc. 2003, 35, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Feldeisen, S.E.; Tucker, K.L. Nutritional strategies in the prevention and treatment of metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Lakka, T.A.; Laaksonen, D.E. Physical activity in prevention and treatment of the metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.B.; Gadde, K.M.; Garvey, W.T.; Peterson, C.A.; Schwiers, M.L.; Najarian, T.; Tam, P.Y.; Troupin, B.; Day, W.W. Controlled-release phentermine/topiramate in severely obese adults: A randomized controlled trial (equip). Obesity 2012, 20, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Meyer, M.; Trinkley, K.E. Phentermine/topiramate for the treatment of obesity. Ann. Pharmacother. 2013, 47, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Khera, R.; Murad, M.H.; Chandar, A.K.; Dulai, P.S.; Wang, Z.; Prokop, L.J.; Loomba, R.; Camilleri, M.; Singh, S. Association of pharmacological treatments for obesity with weight loss and adverse events: A systematic review and meta-analysis. JAMA 2016, 315, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- James, W.P.T.; Caterson, I.D.; Coutinho, W.; Finer, N.; Van Gaal, L.F.; Maggioni, A.P.; Torp-Pedersen, C.; Sharma, A.M.; Shepherd, G.M.; Rode, R.A.; et al. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N. Engl. J. Med. 2010, 363, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Berstein, L.M. Metformin in obesity, cancer and aging: Addressing controversies. Aging (Albany N. Y.) 2012, 4, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiome shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Lei, L.; Duan, Y.; Zhang, K.-Q.; Yang, J. Culture-independent methods for studying environmental microorganisms: Methods, application, and perspective. Appl. Microbiol. Biotechnol. 2012, 93, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R.E. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.M.P. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel. Dis. 2016, 22, 1137. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Wostmann, B.S.; Larkin, C.; Moriarty, A.; Bruckner-Kardoss, E. Dietary intake, energy metabolism, and excretory losses of adult male germfree wistar rats. Lab. Anim. Sci. 1983, 33, 46–50. [Google Scholar] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Hold, G.L.; Barcenilla, A.; Stewart, C.S.; Flint, H.J. Roseburia intestinalis sp. Nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int. J. Syst. Evol. Microbiol. 2002, 52, 1615–1620. [Google Scholar] [PubMed]
- Drissi, F.; Merhej, V.; Angelakis, E.; El Kaoutari, A.; Carriere, F.; Henrissat, B.; Raoult, D. Comparative genomics analysis of lactobacillus species associated with weight gain or weight protection. Nutr. Diabetes 2014, 4, e109. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Greiner, T.; Bäckhed, F. Effects of the gut microbiota on obesity and glucose homeostasis. Trends Endocrinol. Metab. 2011, 22, 117–123. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Chassaing, B.; Zhang, L.; San Yeoh, B.; Xiao, X.; Kumar, M.; Baker, M.T.; Cai, J.; Walker, R.; Borkowski, K. Microbiota-dependent hepatic lipogenesis mediated by stearoyl coa desaturase 1 (scd1) promotes metabolic syndrome in tlr5-deficient mice. Cell Metab. 2015, 22, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, V.R.; Hezaveh, R.; Reigstad, C.S.; Gopalacharyulu, P.; Yetukuri, L.; Islam, S.; Felin, J.; Perkins, R.; Borén, J.; Orešič, M. The gut microbiota modulates host energy and lipid metabolism in mice. J. Lipid. Res. 2010, 51, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Inturri, R.; Lazzara, F.; Di Rosa, M.; Malaguarnera, L. Impact of gut microbiota on diabetes mellitus. Diabetes Metab. 2016, 42, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Festi, D.; Schiumerini, R.; Eusebi, L.H.; Marasco, G.; Taddia, M.; Colecchia, A. Gut microbiota and metabolic syndrome. World J. Gastroenterol. 2014, 20, 16079–16094. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Eisenbarth, G.S. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet 2001, 358, 221–229. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in european women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H. Microbiota associated with type 2 diabetes and its related complications. Food Sci. Hum. Wellness 2013, 2, 167–172. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Cani, P.D. Gut microbiota and the pathogenesis of insulin resistance. Curr. Diabetes Rep. 2011, 11, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J. Gut dysbiosis is linked to hypertensionnovelty and significance. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Fetissov, S.O. Role of the gut microbiota in host appetite control: Bacterial growth to animal feeding behaviour. Nat. Rev. Endocrinol. 2017, 13, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; D’Souza, R.; Hong, S.-T. The role of gut microbiota in the gut-brain axis: Current challenges and perspectives. Protein Cell 2013, 4, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Vancampfort, D.; Correll, C.U.; Wampers, M.; Sienaert, P.; Mitchell, A.; De Herdt, A.; Probst, M.; Scheewe, T.W.; De Hert, M. Metabolic syndrome and metabolic abnormalities in patients with major depressive disorder: A meta-analysis of prevalences and moderating variables. Psychol. Med. 2014, 44, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A., III. Metabolic changes associated with antipsychotic use. Prim. Care Companion J. Clin. Psychiatry 2004, 6, 8. [Google Scholar] [PubMed]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Kiely, B.; Cryan, J.; Dinan, T. Effects of the probiotic bifidobacterium infantis in the maternal separation model of depression. Neuroscience 2010, 170, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of lactobacillus strain regulates emotional behavior and central gaba receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Groups | Drug Classes | Name, Functions | Possible Side Effects | References |
|---|---|---|---|---|
| Obesity treatment | Lipase inhibitors | Orlistat Prevent fats absorption | Steatorrhea, fecal incontinence, frequent or urgent bowel movements, liver injury, acute kidney injury, colon carcinogenesis | [39,40,41,42,43] |
| Serotonin agonists | Lorcaserin Regulate appetite, mood and endocrine secretion | Upper respiratory tract infection, depression, anxiety, hallucinogenic, cardiac valvulopathy, suicidal ideation, cancer | [44,45,46,47,48] | |
| Hyperglycemia treatment | Insulin-sensitizing agents | Metformin or Thiazolidinediones Decrease glucose production and increase the insulin sensitivity | Diarrhea, nausea, abdominal pain, hypoglycemia, high blood lactic acid level, edema, weight gain, heart failure, bone fractures, certain types of cancer | [49,50,51,52] |
| Insulin secretagogues | Sulfonylureas or Meglitinides Increase fusion of insulin granulae, insulin secretion, release from the beta cells in the pancreas | Weight gain, hypoglycemia gastrointestinal upset, headache, hypersensitivity reactions, adenomas of the thyroid and liver, cardiovascular mortality | [53,54] | |
| The glucagon-like peptide (GLP) agonists | Exenatide Suppress pancreatic release of glucagon in response to eating, slow down gastric emptying, decrease the rate, and decrease liver fat content | Gastroesophageal reflux disease, belching, diarrhea, heartburn, indigestion, nausea, vomiting, dizziness, headache, pancreatitis, thyroid cancer | [55,56,57,58] | |
| DPP-4 inhibitors | Sitagliptin Increase insulin secretion and suppress glucagon release by the alpha cells of the pancreas | Nausea, common cold-like symptoms, photosensitivity, hypoglycemia | [59,60,61] | |
| Hypertensive treatment | Angiotensin converting enzyme (ACE) inhibitors | Enalapril Decrease blood pressure | Increase serum creatinine, dizziness, low blood pressure, dry cough, airway compressive angioedema | [62,63,64] |
| Angiotensin receptor blockers (ARBs) | Azilsartan or Telmisartan Decrease blood pressure | Dizziness, headache, hyperkalemia, hypotension, rash, diarrhea, abnormal liver function, muscle cramp, back pain, insomnia, renal impairment, pharyngitis, myocardial infarction, tachycardia, brachycardia. | [65,66,67,68,69] | |
| Preventive cardiovascular treatment | Antiplatelet | Aspirin Suppress prostaglandins and thromboxanes production, platelets function | Gastrointestinal bleeding, gastric mucosal erosion, temporary tinnitus, Reye’s syndrome, swelling, headache, kidney injury, cerebral microbleeds, ischemic stroke, intracerebral hemorrhage, Alzheimer’s disease | [70,71,72,73,74] |
| Dyslipidemia treatment | Statins | Atorvastatin, Simvastatin or Rosuvastatin Decrease cholesterol production, increase levels of HDL-C and prevent the events associated with CVDs | T2D, diarrhea, dyspepsia, myalgia, nausea, memory loss, forgetfulness, eczema, muscle cramps, rhabdomyolysis, heartburn, depression, chest pain, jaundice, extreme tiredness, loss of appetite, flu-like symptoms, unusual bleeding or bruising, worse glycemic control, cholestatic hepatitis, hepatic cirrhosis | [75,76,77,78,79,80,81,82,83,84,85,86] |
| PCSK9 inhibitors | Alirocumab or Evolocumab Decrease LDL-C level 60–70%, prevent early death from cardiovascular disease | Nose irritation, flu-like symptoms, urinary tract infection, diarrhea, bronchitis, muscle pain, soreness, spasms | [87] | |
| Cholesterol absorption inhibitors | Ezetimibe Decrease LDL-C by decreasing cholesterol absorption in the small intestine | Headache, diarrhea, myalgia, hypersensitivity reactions, myopathy, myalgia, rhabdomyolysis | [88,89,90] | |
| Bile acid sequestrants | Cholestyramine, Colestipol or Colesevelam Decrease blood LDL-C, increase HDL-C | Increased TG, transaminase, headache, flatulence, vomiting, diarrhea, dyspepsia, abdominal pain, nausea, myalgia, intestinal obstruction, liver injury, kidney injury | [91,92,93,94,95] | |
| Fibrates | Gemfibrozil or Fenofibrate Decrease elevated LDL-C, total cholesterol, TG, apo B, increase HDL-C | Headache, back pain, nausea, diarrhea, upper respiratory tract infection, gastrointestinal distress, musculoskeletal pain, gallstone, hypokalemia, cancer. | [96,97,98,99] | |
| Nicotinic acid | Niacin immediate release (Niacor) or Niacin extended release (Niaspan) Decrease LDL-C, increase HDL-C in the blood, decrease TG levels by 15–25% | Flushing of the face and neck along with warmth, headache, burning, sweating, chills, dizziness, stomach upset, heartburn, vomiting, diarrhea, indigestion, nausea, liver failure and hyperglycemia | [100,101] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.B.; Jin, Y.Y.; Chung, H.-J.; Hong, S.-T. Pharmabiotics as an Emerging Medication for Metabolic Syndrome and Its Related Diseases. Molecules 2017, 22, 1795. https://doi.org/10.3390/molecules22101795
Nguyen TTB, Jin YY, Chung H-J, Hong S-T. Pharmabiotics as an Emerging Medication for Metabolic Syndrome and Its Related Diseases. Molecules. 2017; 22(10):1795. https://doi.org/10.3390/molecules22101795
Chicago/Turabian StyleNguyen, Thi Thanh Binh, Yan Yan Jin, Hea-Jong Chung, and Seong-Tschool Hong. 2017. "Pharmabiotics as an Emerging Medication for Metabolic Syndrome and Its Related Diseases" Molecules 22, no. 10: 1795. https://doi.org/10.3390/molecules22101795
APA StyleNguyen, T. T. B., Jin, Y. Y., Chung, H.-J., & Hong, S.-T. (2017). Pharmabiotics as an Emerging Medication for Metabolic Syndrome and Its Related Diseases. Molecules, 22(10), 1795. https://doi.org/10.3390/molecules22101795