Advanced Prodrug Strategies in Nucleoside and Non-Nucleoside Antiviral Agents: A Review of the Recent Five Years



{kind=link}
{kind=link}
{kind=link}
{kind=link}
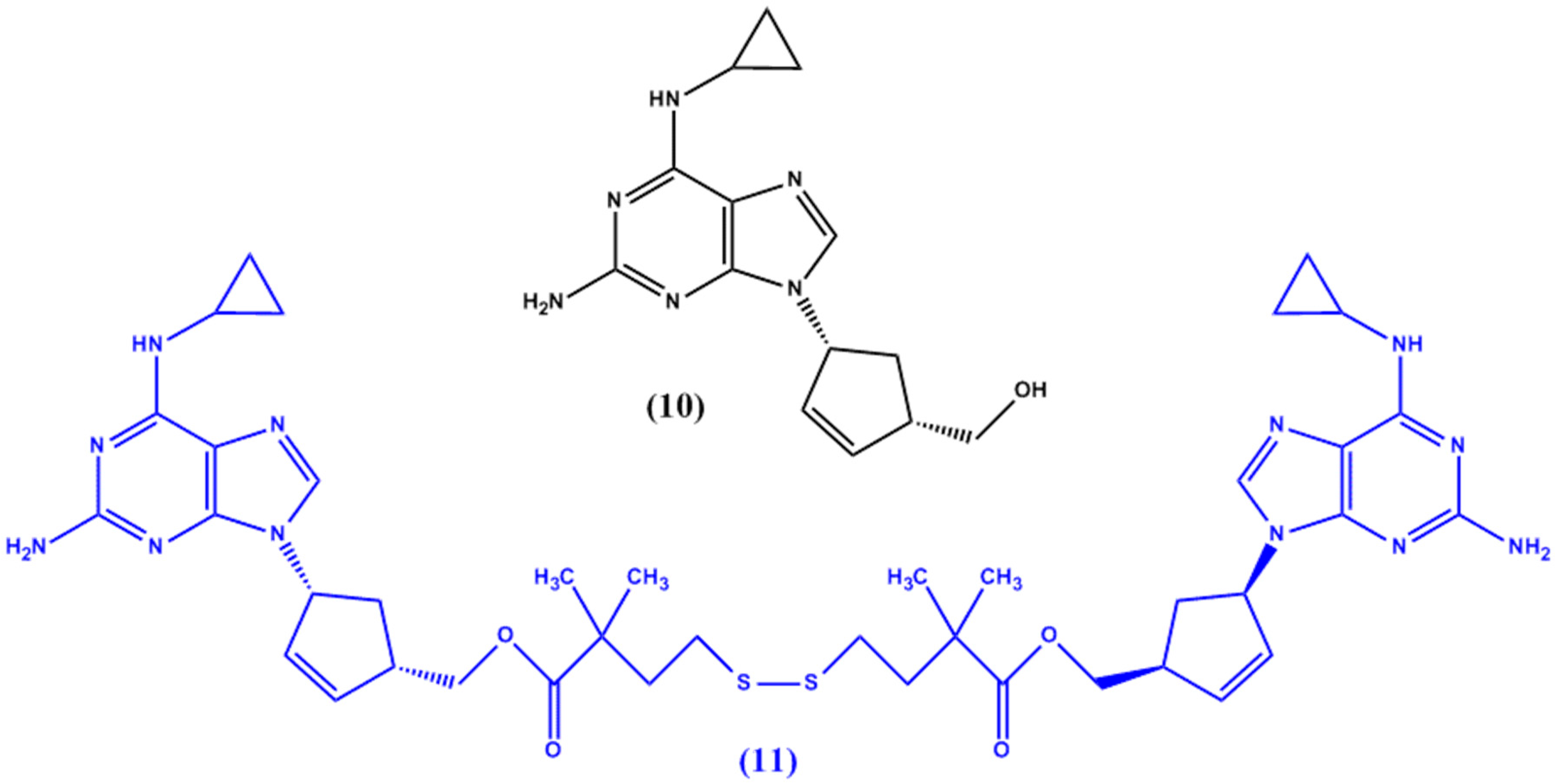{kind=link}
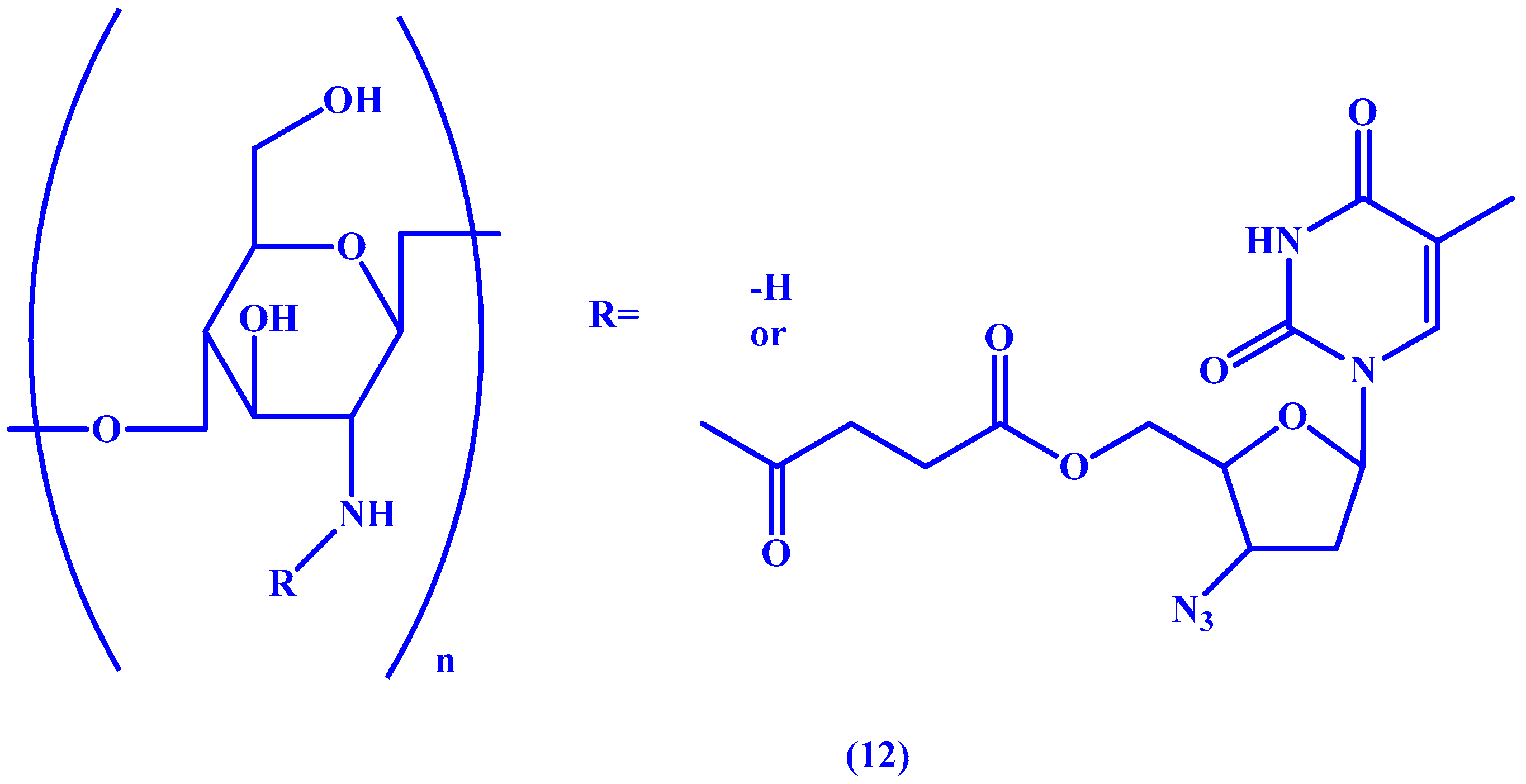{kind=link}
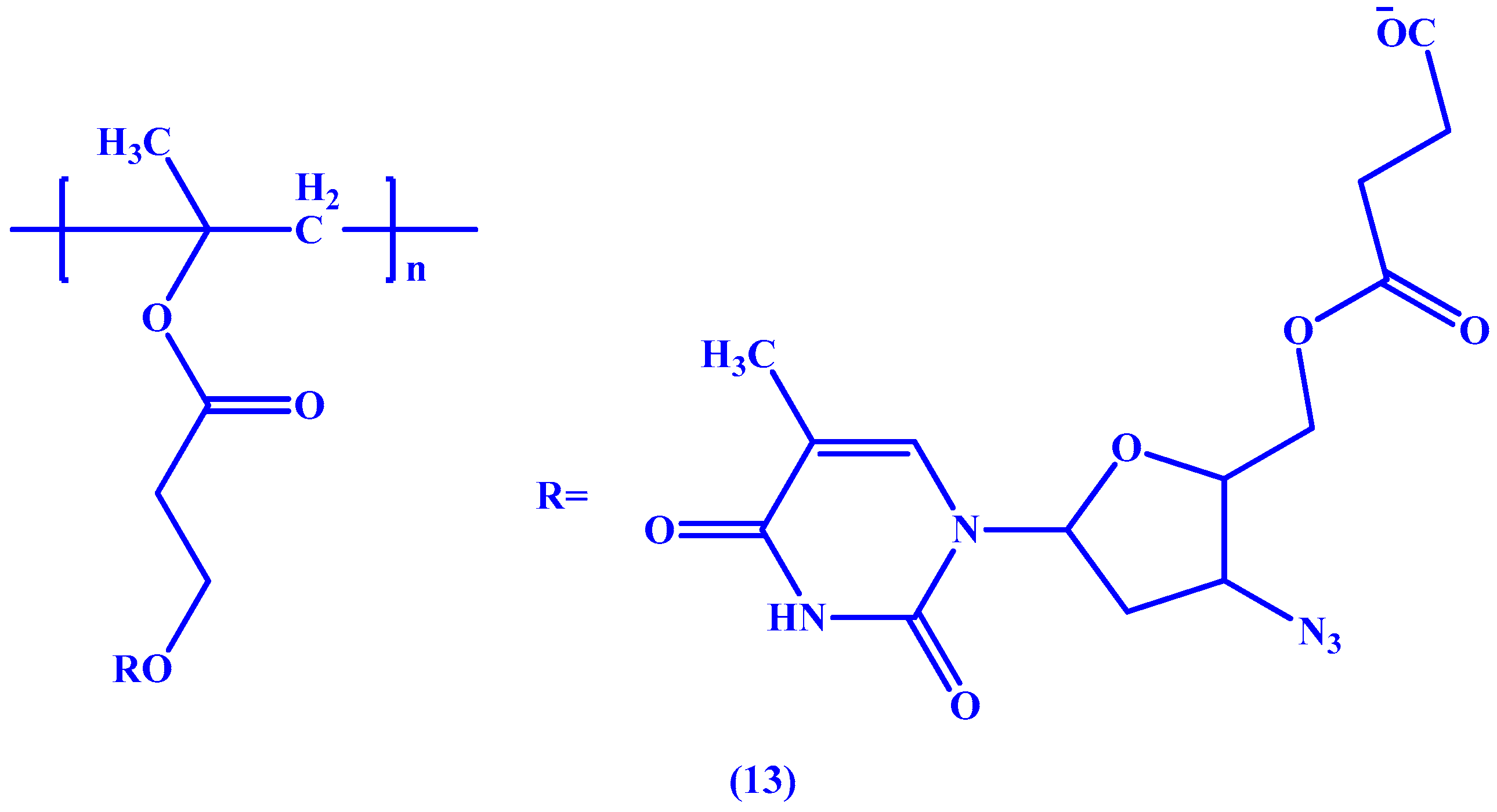{kind=link}
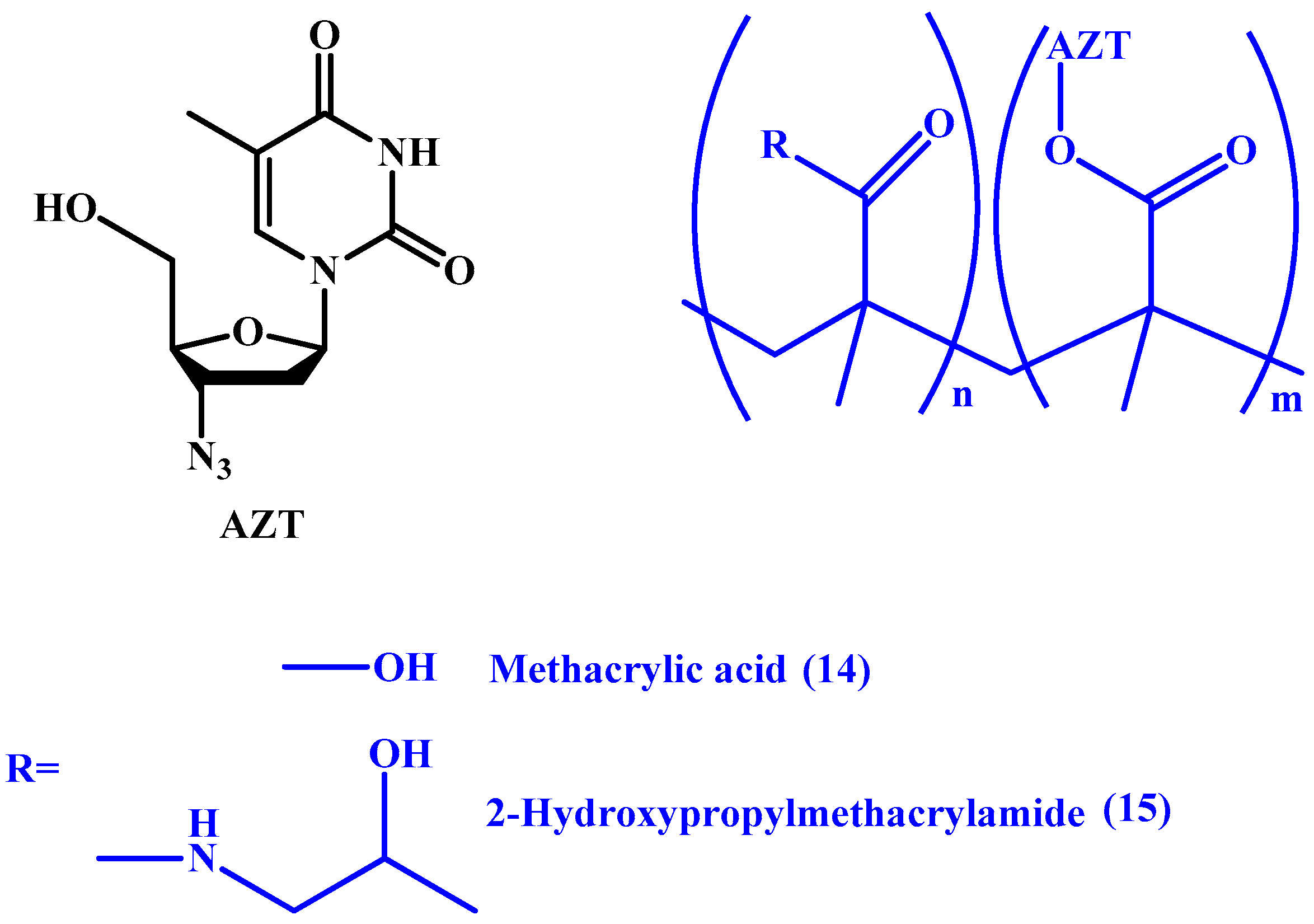{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Ester Prodrugs
3. Targeted Transporters Approach
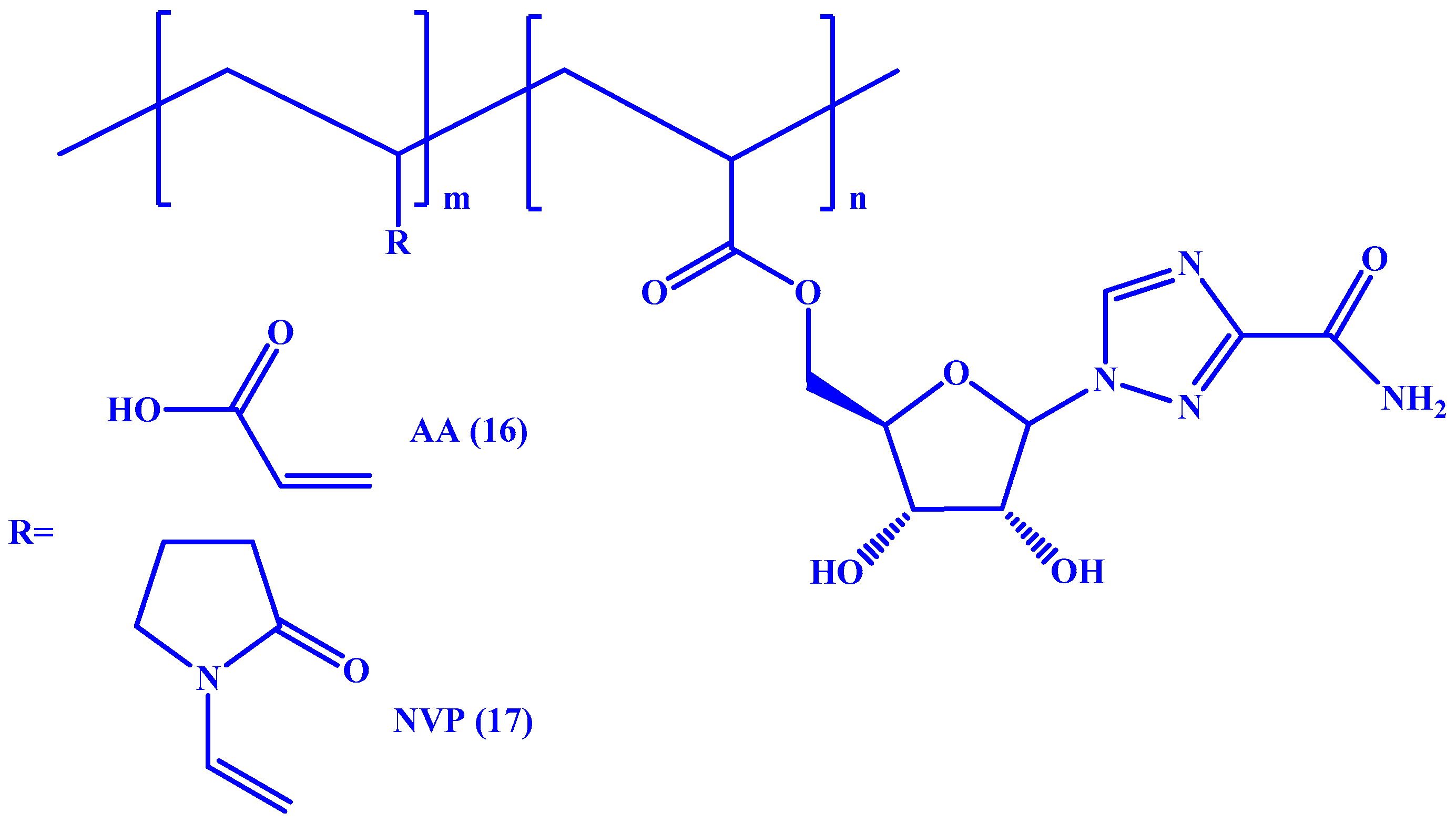
4. Macromolecular Prodrugs
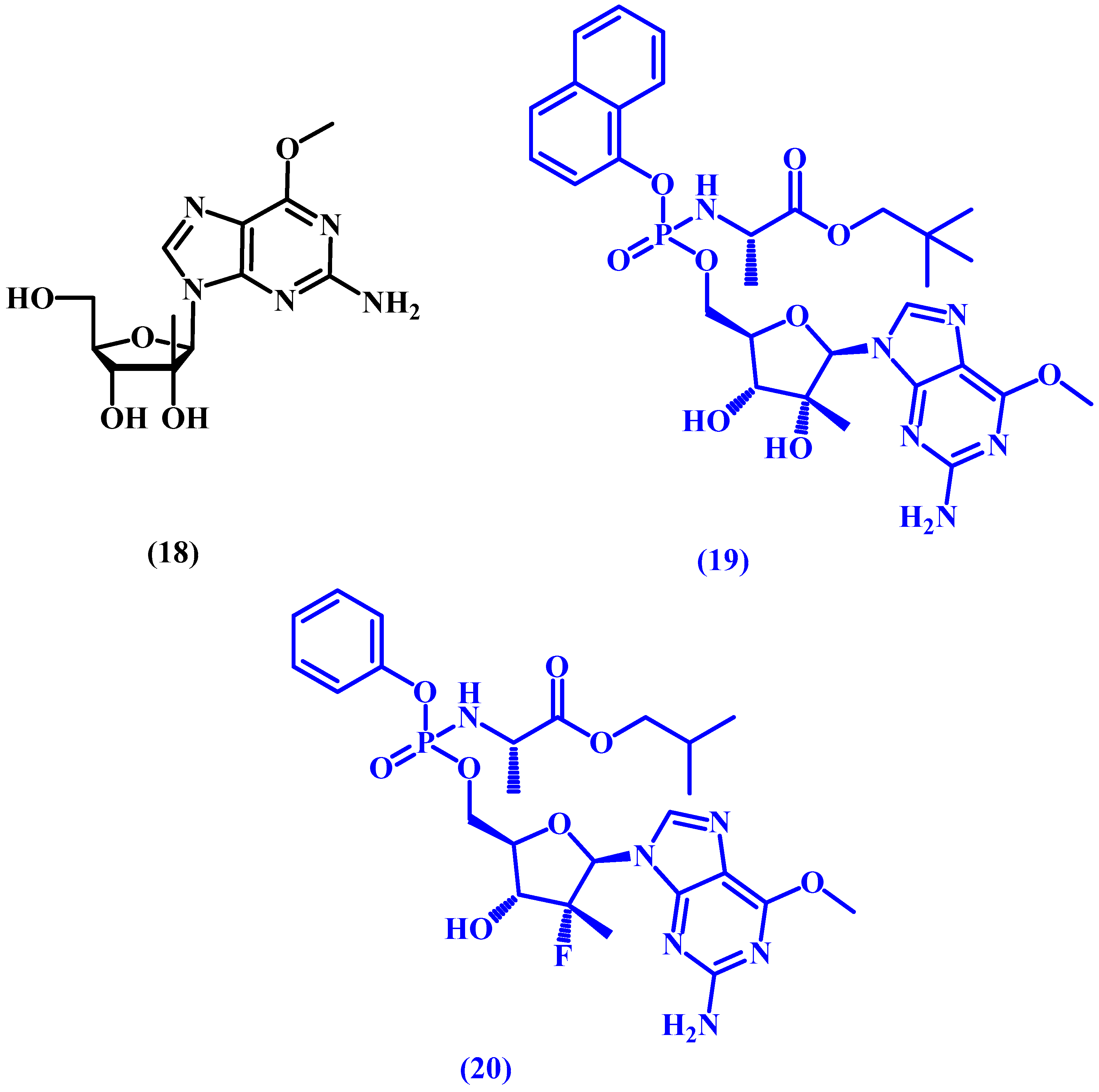
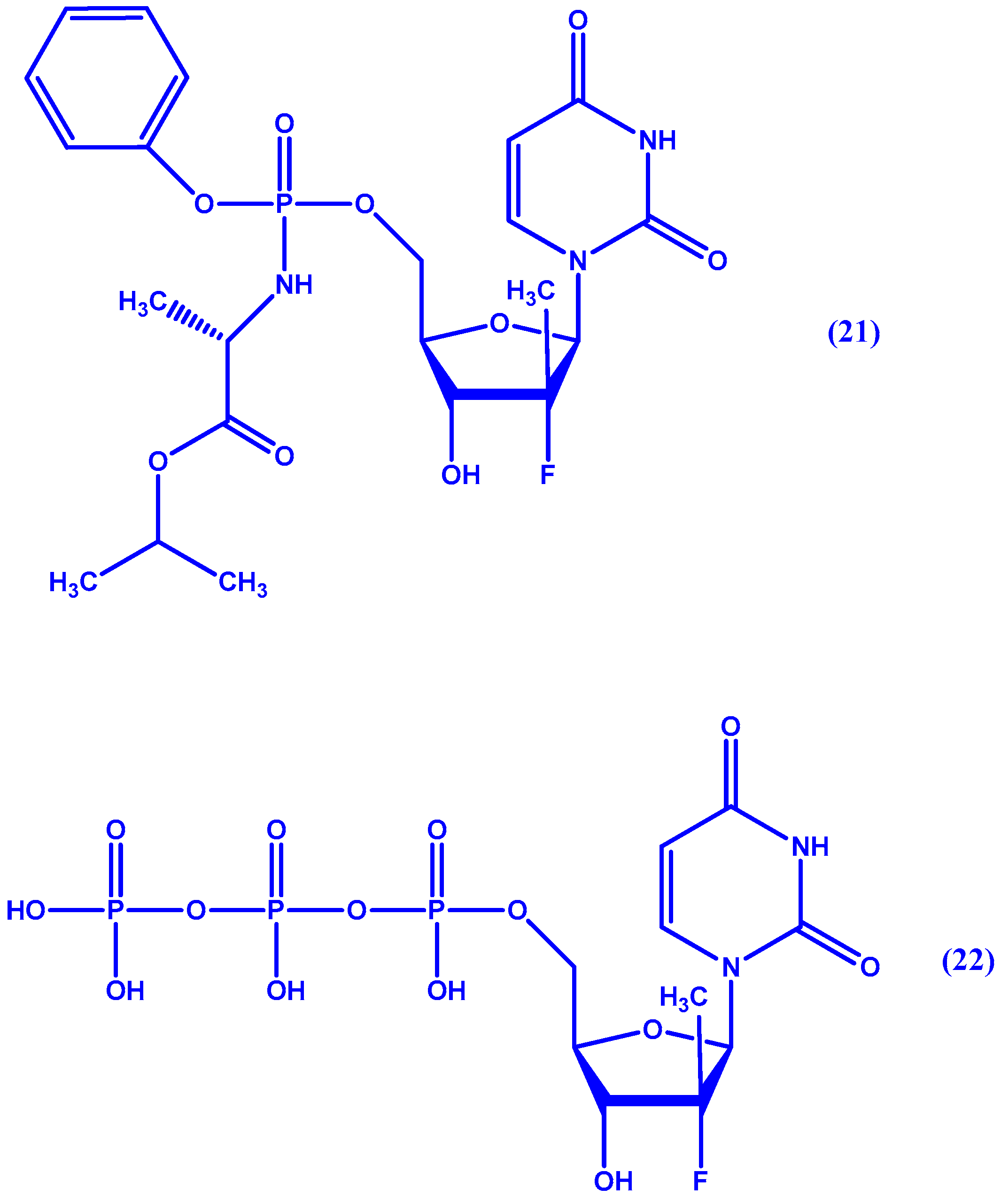
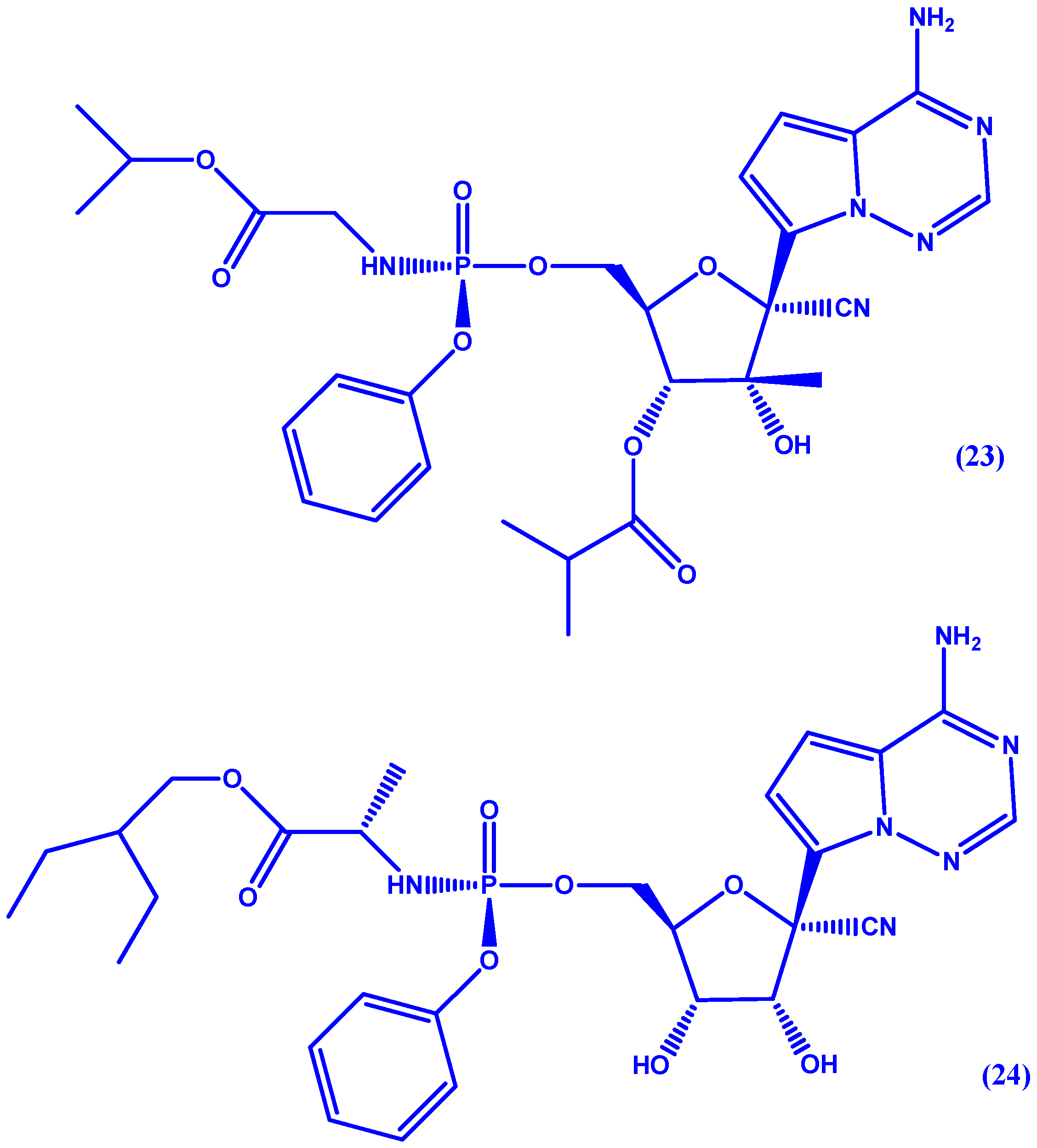
5. ProTides and Nucleoside Analogues
6. Non-Nucleoside Antiviral Agents
7. Nanoparticles
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. People Living with HIV/AIDS. Available online: http://www.who.int/gho/hiv/en/ (accessed on 11 May 2017).
- World Health Organization. Hepatitis B Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs204/en/ (accessed on 11 May 2017).
- Karaman, R. Prodrugs Design—A New Era; Nova Science Publishers: New York, NY, USA, 2014; p. 278. [Google Scholar]
- Amly, W.; Karaman, R. Recent updates in utilizing prodrugs in drug delivery (2013–2015). Expert Opin. Drug Deliv. 2016, 13, 571–591. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Zimmermann, E.M.; Ben-Shabat, S. Modern prodrug design for targeted oral drug delivery. Molecules 2014, 19, 16489–16505. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Shun-Shin, M.; Gill, P.; Perera, R.; Harnden, A. Neuraminidase inhibitors for preventing and treating influenza in children (published trials only). Cochrane Database Syst. Rev. 2012, CD002744. [Google Scholar] [CrossRef]
- Andrei, G.; Snoeck, R. Emerging drugs for varicella-zoster virus infections. Expert Opin. Emerg. Drugs 2011, 16, 507–535. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The clinical potential of the acyclic (and cyclic) nucleoside phosphonates: The magic of the phosphonate bond. Biochem. Pharmacol. 2011, 82, 99–109. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Highlights in antiviral drug research: Antivirals at the horizon. Med. Res. Rev. 2013, 33, 1215–1248. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J.; Chang, W.; Furman, P.A.; Mosley, R.T.; Ross, B.S. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B rna-dependent RNA-polymerase. J. Med. Chem. 2012, 55, 2481–2531. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; McKenna, C.E. Prodrug approaches to improving the oral absorption of antiviral nucleotide analogues. Expert Opin. Drug Deliv. 2009, 6, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Pertusat, F.; Serpi, M.; McGuigan, C. Medicinal chemistry of nucleoside phosphonate prodrugs for antiviral therapy. Antivir. Chem. Chemother. 2012, 22, 181–203. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and ursodeoxycholic acid conjugation: Design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.I.; Uchil, V.R.; Okello, M.; Mishra, S.; Ma, X.-H.; Nishonov, M.; Shu, Q.; Chi, G.; Nair, V. Discovery of a potent HIV integrase inhibitor that leads to a prodrug with significant anti-hiv activity. ACS Med. Chem. Lett. 2011, 2, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Shirasaka, Y.; Kuraoka, E.; Kikuchi, A.; Iguchi, M.; Suzuki, H.; Shibasaki, S.; Kurosawa, T.; Tamai, I. Intestinal absorption mechanism of tebipenem pivoxil, a novel oral carbapenem: Involvement of human oatp family in apical membrane transport. Mol. Pharm. 2010, 7, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I. Oral drug delivery utilizing intestinal oatp transporters. Adv. Drug Deliv. Rev. 2012, 64, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Kolhatkar, V.; Polli, J.E. Structural requirements of bile acid transporters: C-3 and C-7 modifications of steroidal hydroxyl groups. Eur. J. Pharm. Sci. 2012, 46, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Polli, J.E. Synthesis and in vitro evaluation of potential sustained release prodrugs via targeting asbt. Int. J. Pharm. 2010, 396, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Ambler, C.M.; Ullah, M.; Rotter, C.J.; Sun, H.; Litchfield, J.; S Fenner, K.; El-Kattan, A.F. Targeting intestinal transporters for optimizing oral drug absorption. Curr. Drug Metab. 2010, 11, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Diop-Bove, N.; Visentin, M.; Goldman, I.D. Mechanisms of membrane transport of folates into cells and across epithelia. Annu. Rev. Nutr. 2011, 31, 177–201. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Smith, D.E. Significance of peptide transporter 1 in the intestinal permeability of valacyclovir in wild-type and pept1 knockout mice. Drug Metab. Dispos. 2013, 41, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Cass, L.M.; Efthymiopoulos, C.; Bye, A. Pharmacokinetics of zanamivir after intravenous, oral, inhaled or intranasal administration to healthy volunteers. Clin. Pharmacokinet. 1999, 36, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Varghese Gupta, S.; Gupta, D.; Sun, J.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Enhancing the intestinal membrane permeability of zanamivir: A carrier mediated prodrug approach. Mol. Pharm. 2011, 8, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Varghese Gupta, S.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Increasing oral absorption of polar neuraminidase inhibitors: A prodrug transporter approach applied to oseltamivir analogue. Mol. Pharm. 2013, 10, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Varatharajan, L.; Thomas, S.A. The transport of anti-hiv drugs across blood-cns interfaces: Summary of current knowledge and recommendations for further research. Antivir. Res. 2009, 82, A99–A109. [Google Scholar] [CrossRef] [PubMed]
- Namanja, H.A.; Emmert, D.; Davis, D.A.; Campos, C.; Miller, D.S.; Hrycyna, C.A.; Chmielewski, J. Toward eradicating HIV reservoirs in the brain: Inhibiting P-glycoprotein at the blood-brain barrier with prodrug abacavir dimers. J. Am. Chem. Soc. 2012, 134, 2976–2980. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, J.; Hrycyna, C. Research spotlight: Tools for eradicating hiv in the brain: Prodrug dimeric inhibitors of P-gp. Ther. Deliv. 2012, 3, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, J.; Canal, F.; Lucas, R.; Vicent, M.J. Polymer-drug conjugates for novel molecular targets. Nanomedicine 2010, 5, 915–935. [Google Scholar] [CrossRef] [PubMed]
- Alconcel, S.N.; Baas, A.S.; Maynard, H.D. Fda-approved poly (ethylene glycol)–protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar] [CrossRef]
- Najjar, A.; Rajabi, N.; Karaman, R. Recent approaches to platinum(IV) prodrugs: A variety of strategies for enhanced delivery and efficacy. Curr. Pharm. Des. 2017, 23, 2366–2376. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Mura, S.; Brambilla, D.; Mackiewicz, N.; Couvreur, P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, S.; Huang, Y.; Chen, X.; Jing, X. Biodegradable block copolymer-doxorubicin conjugates via different linkages: Preparation, characterization, and in vitro evaluation. Biomacromolecules 2010, 11, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Murphy, C.J.; Zhang, B.; Shen, Y.; Van Kirk, E.A.; Murdoch, W.J.; Radosz, M. Curcumin polymers as anticancer conjugates. Biomaterials 2010, 31, 7139–7149. [Google Scholar] [CrossRef] [PubMed]
- Pirrone, V.; Wigdahl, B.; Krebs, F.C. The rise and fall of polyanionic inhibitors of the human immunodeficiency virus type 1. Antivir. Res. 2011, 90, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Haldar, J.; De Cienfuegos, L.Á.; Tumpey, T.M.; Gubareva, L.V.; Chen, J.; Klibanov, A.M. Bifunctional polymeric inhibitors of human influenza a viruses. Pharm. Res. 2010, 27, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Makimura, Y.; Itoh, M.; Yamamoto, T.; Mine, T.; Mitani, S.; Simizu, I.; Ashida, H.; Yamamoto, K. One-step synthesis of efficient binding-inhibitor for influenza virus through multiple addition of sialyloligosaccharides on chitosan. Carbohydr. Polym. 2010, 81, 330–334. [Google Scholar] [CrossRef]
- Nazemi, A.; Haeryfar, S.M.; Gillies, E.R. Multifunctional dendritic sialopolymersomes as potential antiviral agents: Their lectin binding and drug release properties. Langmuir 2013, 29, 6420–6428. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.A.; Kryger, M.B.; Wohl, B.M.; Ruiz-Sanchis, P.; Zuwala, K.; Tolstrup, M.; Zelikin, A.N. Macromolecular (pro) drugs in antiviral research. Polym. Chem. 2014, 5, 6407–6425. [Google Scholar] [CrossRef]
- De Smedt, S.C.; Demeester, J.; Hennink, W.E. Cationic polymer based gene delivery systems. Pharm. Res. 2000, 17, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Troev, K.D.; Mitova, V.A.; Ivanov, I.G. On the design of polymeric 5′-O-ester prodrugs of 3′-azido-2′,3′-dideoxythymidine (AZT). Tetrahedron Lett. 2010, 51, 6123–6125. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, H.; Chen, L.; Hu, L.; Li, Z. Preparation, characterization and in vitro release of chitosan-stavudine conjugate nano-prodrug. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2013, 28, 617–621. [Google Scholar]
- Smith, A.A.; Wohl, B.M.; Kryger, M.B.; Hedemann, N.; Guerrero-Sanchez, C.; Postma, A.; Zelikin, A.N. Macromolecular prodrugs of ribavirin: Concerted efforts of the carrier and the drug. Adv. Healthc. Mater. 2014, 3, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Wohl, B.M.; Smith, A.A.; Kryger, M.B.; Zelikin, A.N. Narrow therapeutic window of ribavirin as an inhibitor of nitric oxide synthesis is broadened by macromolecular prodrugs. Biomacromolecules 2013, 14, 3916–3926. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.A.; Zuwala, K.; Kryger, M.B.; Wohl, B.M.; Guerrero-Sanchez, C.; Tolstrup, M.; Postma, A.; Zelikin, A.N. Macromolecular prodrugs of ribavirin: Towards a treatment for co-infection with HIV and HCV. Chem. Sci. 2015, 6, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sanchis, P.; Wohl, B.M.; Smith, A.A.; Zuwala, K.; Melchjorsen, J.; Tolstrup, M.; Zelikin, A.N. Highly active macromolecular prodrugs inhibit expression of the hepatitis C virus genome in the host cells. Adv. Healthc. Mater. 2015, 4, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Brookes, S.; Biessels, P.; Ng, N.F.; Woods, C.; Bell, D.N.; Adamson, G. Synthesis and characterization of a hemoglobin-ribavirin conjugate for targeted drug delivery. Bioconj. Chem. 2006, 17, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.A.; Adamson, G.; Phillips, M.J.; Scrocchi, L.A.; Fung, L.; Biessels, P.; Ng, N.F.; Ghanekar, A.; Rowe, A.; Ma, M.X. Targeted delivery of ribavirin improves outcome of murine viral fulminant hepatitis via enhanced anti-viral activity. Hepatology 2006, 43, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Kato, D.; Era, S.; Watanabe, I.; Arihara, M.; Sugiura, N.; Kimata, K.; Suzuki, Y.; Morita, K.; Hidari, K.I.; Suzuki, T. Antiviral activity of chondroitin sulphate E targeting dengue virus envelope protein. Antivir. Res. 2010, 88, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Gong, T.; Sun, X.; Tang, J.Z.; Zhang, Z. Chitosan oligomers as drug carriers for renal delivery of zidovudine. Carbohydr. Polym. 2012, 87, 2284–2290. [Google Scholar] [CrossRef]
- Neeraj, A.; Chandrasekar, M.; Sara, U.; Rohini, A. Poly (hema-zidovudine) conjugate: A macromolecular pro-drug for improvement in the biopharmaceutical properties of the drug. Drug Deliv. 2011, 18, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Zuwala, K.; Smith, A.A.; Postma, A.; Guerrero-Sanchez, C.; Ruiz-Sanchis, P.; Melchjorsen, J.; Tolstrup, M.; Zelikin, A.N. Polymers fight hiv: Potent (pro) drugs identified through parallel automated synthesis. Adv. Healthc. Mater. 2015, 4, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, B.E.; Knight, V. Biochemistry and clinical applications of ribavirin. Antimicrob. Agents Chemother. 1986, 30, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Kryger, M.B.L.; Wohl, B.M.; Smith, A.A.A.; Zelikin, A.N. Macromolecular prodrugs of ribavirin combat side effects and toxicity with no loss of activity of the drug. Chem. Commun. 2013, 49, 2643–2645. [Google Scholar] [CrossRef] [PubMed]
- Kryger, M.B.; Smith, A.A.; Wohl, B.M.; Zelikin, A.N. Macromolecular prodrugs for controlled delivery of ribavirin. Macromol. Biosci. 2014, 14, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Tyssen, D.; Henderson, S.A.; Johnson, A.; Sterjovski, J.; Moore, K.; La, J.; Zanin, M.; Sonza, S.; Karellas, P.; Giannis, M.P. Structure activity relationship of dendrimer microbicides with dual action antiviral activity. PLoS ONE 2010, 5, e12309. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Rodriguez, J.; Barber, P.; das Neves, J.; Sarmento, B.; Nair, M. The potential of HIV-1 nanotherapeutics: From in vitro studies to clinical trials. Nanomedicine (Lond. Engl.) 2015, 10, 3597–3609. [Google Scholar] [CrossRef] [PubMed]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Coppola, N.; Buonomo, A.R.; Zappulo, E.; Borgia, G. Investigational nucleoside and nucleotide polymerase inhibitors and their use in treating hepatitis C virus. Expert Opin. Investig. Drugs 2014, 23, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Madela, K.; Aljarah, M.; Gilles, A.; Brancale, A.; Zonta, N.; Chamberlain, S.; Vernachio, J.; Hutchins, J.; Hall, A. Design, synthesis and evaluation of a novel double pro-drug: Inx-08189. A new clinical candidate for hepatitis C virus. Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Bao, D.; Chun, B.-K.; Naduthambi, D.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Zhang, H.-R.; Bansal, S.; et al. Discovery of PSI-353661, a novel purine nucleotide prodrug for the treatment of hcv infection. ACS Med. Chem. Lett. 2011, 2, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Herbst, D.A., Jr.; Reddy, K.R. Sofosbuvir, a nucleotide polymerase inhibitor, for the treatment of chronic hepatitis c virus infection. Expert Opin. Investig. Drugs 2013, 22, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H.M.M.; Bao, D.; Chang, W.; Espiritu, C.; Bansal, S.; Lam, A.M.; et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Xing, W.; Chan, K.; Niedziela-Majka, A.; Brendza, K.M.; Kirschberg, T.; Kato, D.; Link, J.O.; Cheng, G.; Liu, X.; et al. Direct binding of ledipasvir to hcv ns5a: Mechanism of resistance to an hcv antiviral agent. PLoS ONE 2015, 10, e0122844. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Taylor, J.G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.; Kirschberg, T.; Sun, J.; Squires, N.; et al. Discovery of ledipasvir (GS-5885): A potent, once-daily oral NS5A inhibitor for the treatment of hepatitis c virus infection. J. Med. Chem. 2014, 57, 2033–2046. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Marcellin, P.; Afdhal, N.; Kowdley, K.V.; Zeuzem, S.; Hunt, S.L. Treatment with ledipasvir and sofosbuvir improves patient-reported outcomes: Results from the ion-1, -2, and -3 clinical trials. Hepatology 2015, 61, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.; Zhang, L.; Xu, J.; Lee, R.; Butler, T.; Metobo, S.; Aktoudianakis, V.; Lew, W.; Ye, H.; Clarke, M.; et al. Discovery of the first c-nucleoside hcv polymerase inhibitor (GS-6620) with demonstrated antiviral response in hcv infected patients. J. Med. Chem. 2014, 57, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Hui, H.C.; Doerffler, E.; Clarke, M.O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine c-nucleoside (GS-5734) for the treatment of ebola and emerging viruses. J. Med. Chem. 2017, 60, 1648–1661. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.A.; He, G.-X.; Eisenberg, E.; Cihlar, T.; Swaminathan, S.; Mulato, A.; Cundy, K.C. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 2005, 49, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Zolopa, A.; Squires, K.; Ruane, P.; Coakley, D.; Kearney, B.; Zhong, L.; Wulfsohn, M.; Miller, M.D.; Lee, W.A. Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the hiv reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J. Antimicrob. Chemother. 2014, 69, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, K.; Fung, S.K.; Nguyen, T.T.; Cheng, W.; Sicard, E.; Ryder, S.D.; Flaherty, J.F.; Lawson, E.; Zhao, S.; Subramanian, G.M.; et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis b infection. J. Hepatol. 2015, 62, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Ruane, P.J.; DeJesus, E.; Berger, D.; Markowitz, M.; Bredeek, U.F.; Callebaut, C.; Zhong, L.; Ramanathan, S.; Rhee, M.S.; Fordyce, M.W.; et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1-positive adults. J. Acquir. Immune Defic. Syndr. 2013, 63, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E.; Daar, E.S.; Raffi, F.; Brinson, C.; Ruane, P.; DeJesus, E.; Johnson, M.; Clumeck, N.; Osiyemi, O.; Ward, D.; et al. Efficacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate given as fixed-dose combinations containing emtricitabine as backbones for treatment of HIV-1 infection in virologically suppressed adults: A randomised, double-blind, active-controlled phase 3 trial. Lancet HIV 2016, 3, e158–e165. [Google Scholar] [PubMed]
- Painter, G.R.; Almond, M.R.; Trost, L.C.; Lampert, B.M.; Neyts, J.; De Clercq, E.; Korba, B.E.; Aldern, K.A.; Beadle, J.R.; Hostetler, K.Y. Evaluation of hexadecyloxypropyl-9-R-[2-(phosphonomethoxy)propyl]-adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob. Agents Chemother. 2007, 51, 3505–3509. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhuang, X.; Qian, K.; Sun, L.; Wang, X.; Li, H.; Lee, K.; Xie, L. Prodrug design, synthesis and pharmacokinetic evaluation of (3′R,4′R)-3-hydroxymethyl-4-methyl-3′,4′-di-O-(S)-camphanoyl-(+)-cis-khellactone. Acta Pharm. Sin. B 2012, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Yu, D.; Wild, C.; Allaway, G.; Turpin, J.; Smith, P.C.; Lee, K.-H. Anti-aids agents. 52. Synthesis and anti-hiv activity of hydroxymethyl (3′R,4′R)-3′,4′-di-O-(s)-camphanoyl-(+)-cis-khellactone derivatives. J. Med. Chem. 2004, 47, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yuan, X.; Yu, D.; Lee, K.H.; Chen, C.H. Mechanism of action and resistant profile of anti-HIV-1 coumarin derivatives. Virology 2005, 332, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Li, Y.; Smith, P.C.; Swenberg, J.A.; Martin, D.E.; Morris-Natschke, S.L.; Lee, K.H. Anti-aids agents 65: Investigation of the in vitro oxidative metabolism of 3′,4′-di-O-(−)-camphanoyl-(+)-cis-khellactone derivatives as potent anti-hiv agents. Drug Metab. Dispos. Biol. Fate Chem. 2005, 33, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Regueiro-Ren, A.; Xue, Q.M.; Swidorski, J.J.; Gong, Y.F.; Mathew, M.; Parker, D.D.; Yang, Z.; Eggers, B.; D'Arienzo, C.; Sun, Y.; et al. Inhibitors of human immunodeficiency virus type 1 (HIV-1) attachment. 12. Structure-activity relationships associated with 4-fluoro-6-azaindole derivatives leading to the identification of 1-(4-benzoylpiperazin-1-yl)-2-(4-fluoro-7-[1,2,3]triazol-1-yl-1H-pyrrolo[2,3-c]py ridin-3-yl)ethane-1,2-dione (BMS-585248). J. Med. Chem. 2013, 56, 1656–1669. [Google Scholar] [PubMed]
- Lalezari, J.P.; Latiff, G.H.; Brinson, C.; Echevarría, J.; Treviño-Pérez, S.; Bogner, J.R.; Thompson, M.; Fourie, J.; Sussmann Pena, O.A.; Mendo Urbina, F.C.; et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of ai438011, a phase 2b, randomised controlled trial. Lancet HIV 2015, 2, e427–e437. [Google Scholar] [CrossRef]
- Schade, D.; Kotthaus, J.; Riebling, L.; Kotthaus, J.; Muller-Fielitz, H.; Raasch, W.; Hoffmann, A.; Schmidtke, M.; Clement, B. Zanamivir amidoxime- and N-hydroxyguanidine-based prodrug approaches to tackle poor oral bioavailability. J. Pharm. Sci. 2015, 104, 3208–3219. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, S.; Ingle, A.; Gade, A.; Rai, M.; Falanga, A.; Incoronato, N.; Russo, L.; Galdiero, S.; Galdiero, M. Antiviral activity of mycosynthesized silver nanoparticles against herpes simplex virus and human parainfluenza virus type 3. Int. J. Nanomed. 2013, 8, 4303–4314. [Google Scholar]
- Vijayakumar, S.; Ganesan, S. Gold nanoparticles as an hiv entry inhibitor. Curr. HIV Res. 2012, 10, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Vitiello, M.; Cantisani, M.; Marra, V.; Galdiero, M. Silver nanoparticles as potential antiviral agents. Molecules 2011, 16, 8894–8918. [Google Scholar] [CrossRef] [PubMed]
- Vonnemann, J.; Sieben, C.; Wolff, C.; Ludwig, K.; Bottcher, C.; Herrmann, A.; Haag, R. Virus inhibition induced by polyvalent nanoparticles of different sizes. Nanoscale 2014, 6, 2353–2360. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Gaglianone, N.; Baldassari, S.; Parodi, B.; Cafaggi, S.; Zibana, C.; Donalisio, M.; Cagno, V.; Lembo, D.; Caviglioli, G. Preparation, characterization and in vitro antiviral activity evaluation of foscarnet-chitosan nanoparticles. Colloids Surf. B Biointerfaces 2014, 118, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, R.; Donalisio, M.; Bisazza, A.; Civra, A.; Ranucci, E.; Ferruti, P.; Lembo, D. Enhanced antiviral activity of acyclovir loaded into nanoparticles. Methods Enzymol. 2012, 509, 1–19. [Google Scholar] [PubMed]
- Kumar, P.; Lakshmi, Y.S.; C, B.; Golla, K.; Kondapi, A.K. Improved safety, bioavailability and pharmacokinetics of zidovudine through lactoferrin nanoparticles during oral administration in rats. PLoS ONE 2015, 10, e0140399. [Google Scholar] [CrossRef] [PubMed]
















© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinokrot, H.; Smerat, T.; Najjar, A.; Karaman, R. Advanced Prodrug Strategies in Nucleoside and Non-Nucleoside Antiviral Agents: A Review of the Recent Five Years. Molecules 2017, 22, 1736. https://doi.org/10.3390/molecules22101736
Sinokrot H, Smerat T, Najjar A, Karaman R. Advanced Prodrug Strategies in Nucleoside and Non-Nucleoside Antiviral Agents: A Review of the Recent Five Years. Molecules. 2017; 22(10):1736. https://doi.org/10.3390/molecules22101736
Chicago/Turabian StyleSinokrot, Hanadi, Tasneem Smerat, Anas Najjar, and Rafik Karaman. 2017. "Advanced Prodrug Strategies in Nucleoside and Non-Nucleoside Antiviral Agents: A Review of the Recent Five Years" Molecules 22, no. 10: 1736. https://doi.org/10.3390/molecules22101736
APA StyleSinokrot, H., Smerat, T., Najjar, A., & Karaman, R. (2017). Advanced Prodrug Strategies in Nucleoside and Non-Nucleoside Antiviral Agents: A Review of the Recent Five Years. Molecules, 22(10), 1736. https://doi.org/10.3390/molecules22101736