ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussions
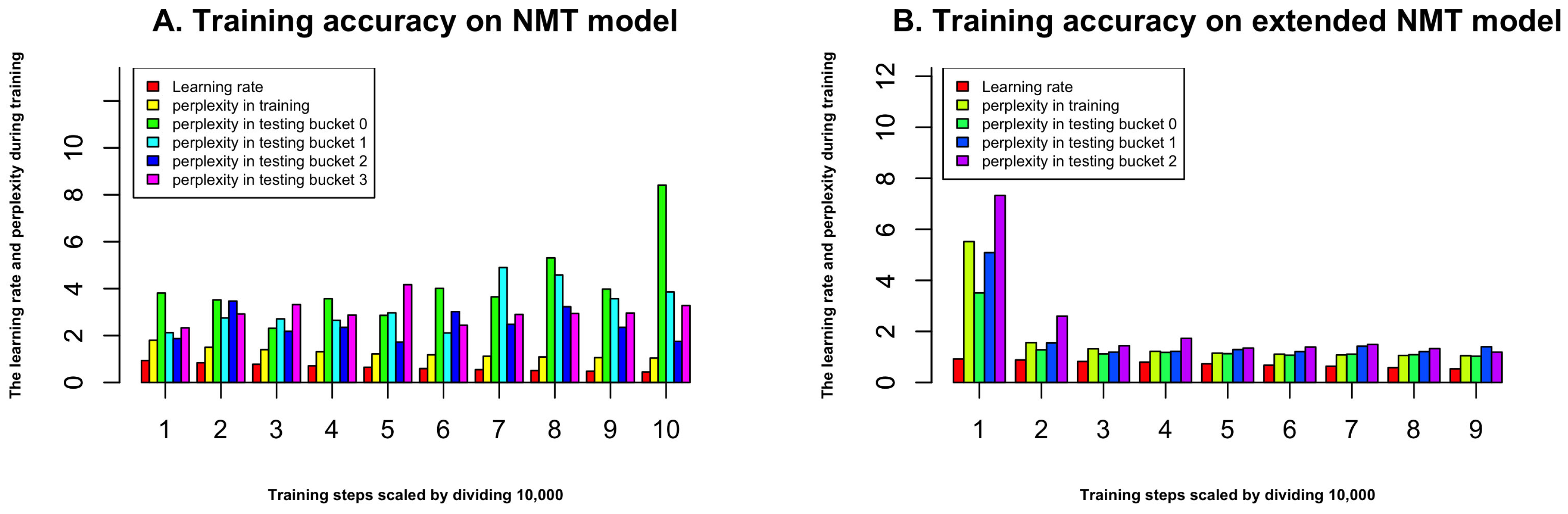
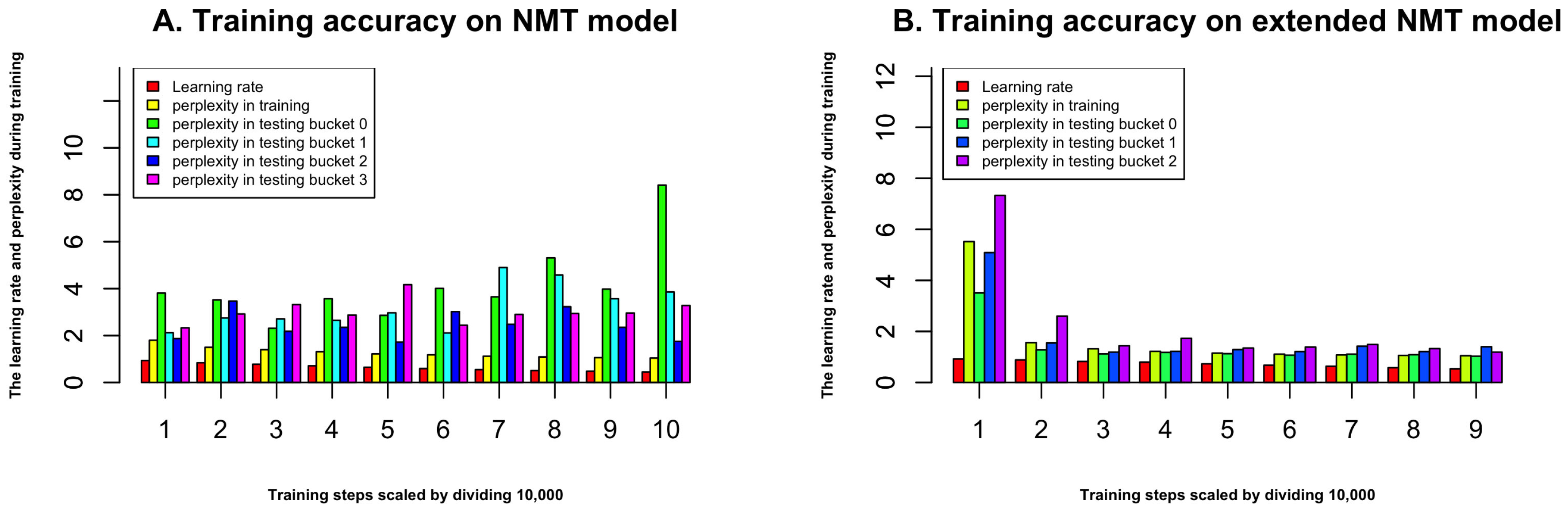
2.1. Performance during Training of Neural Machine Translation Model
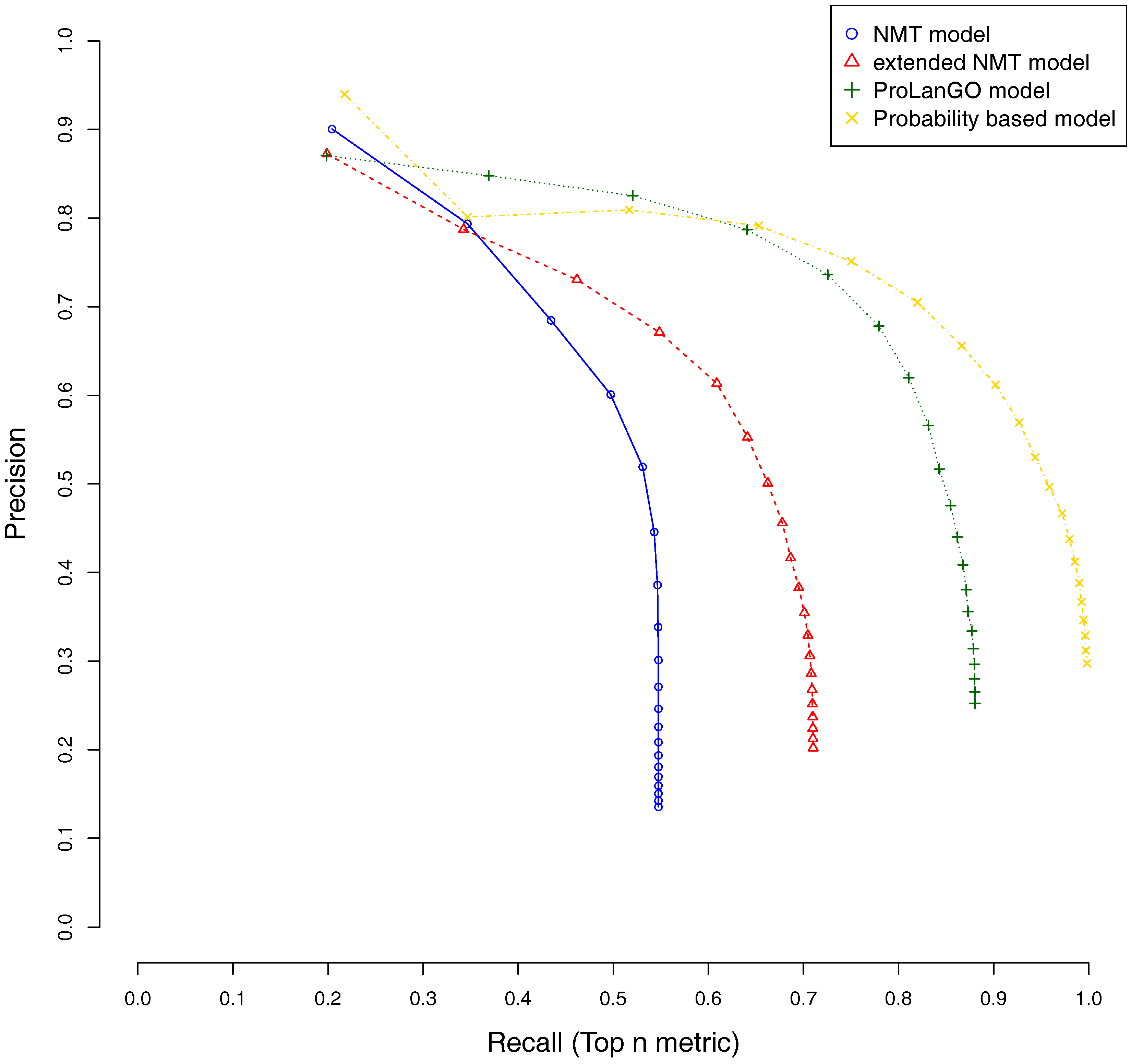
2.2. Performance on Validation Dataset
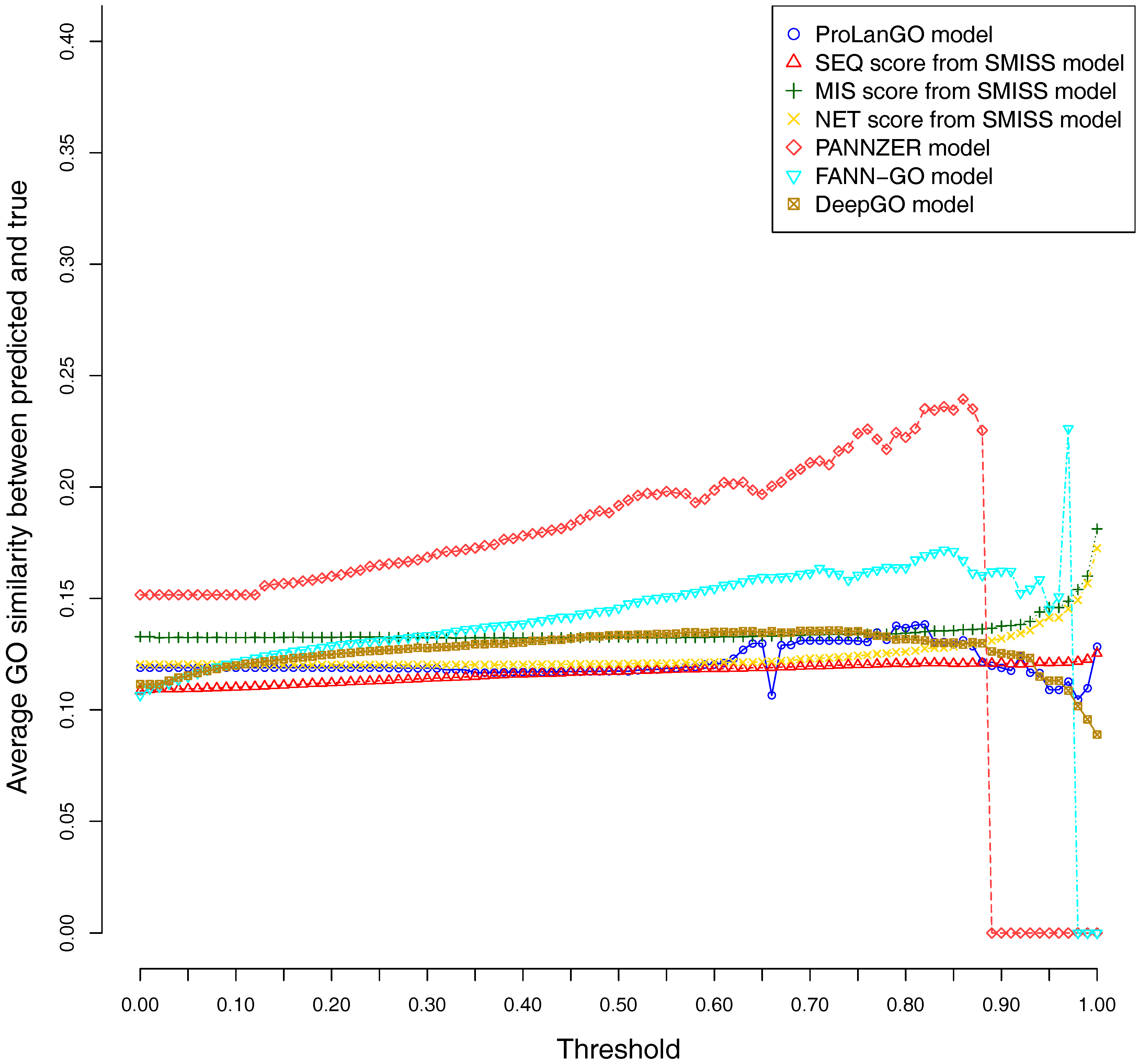
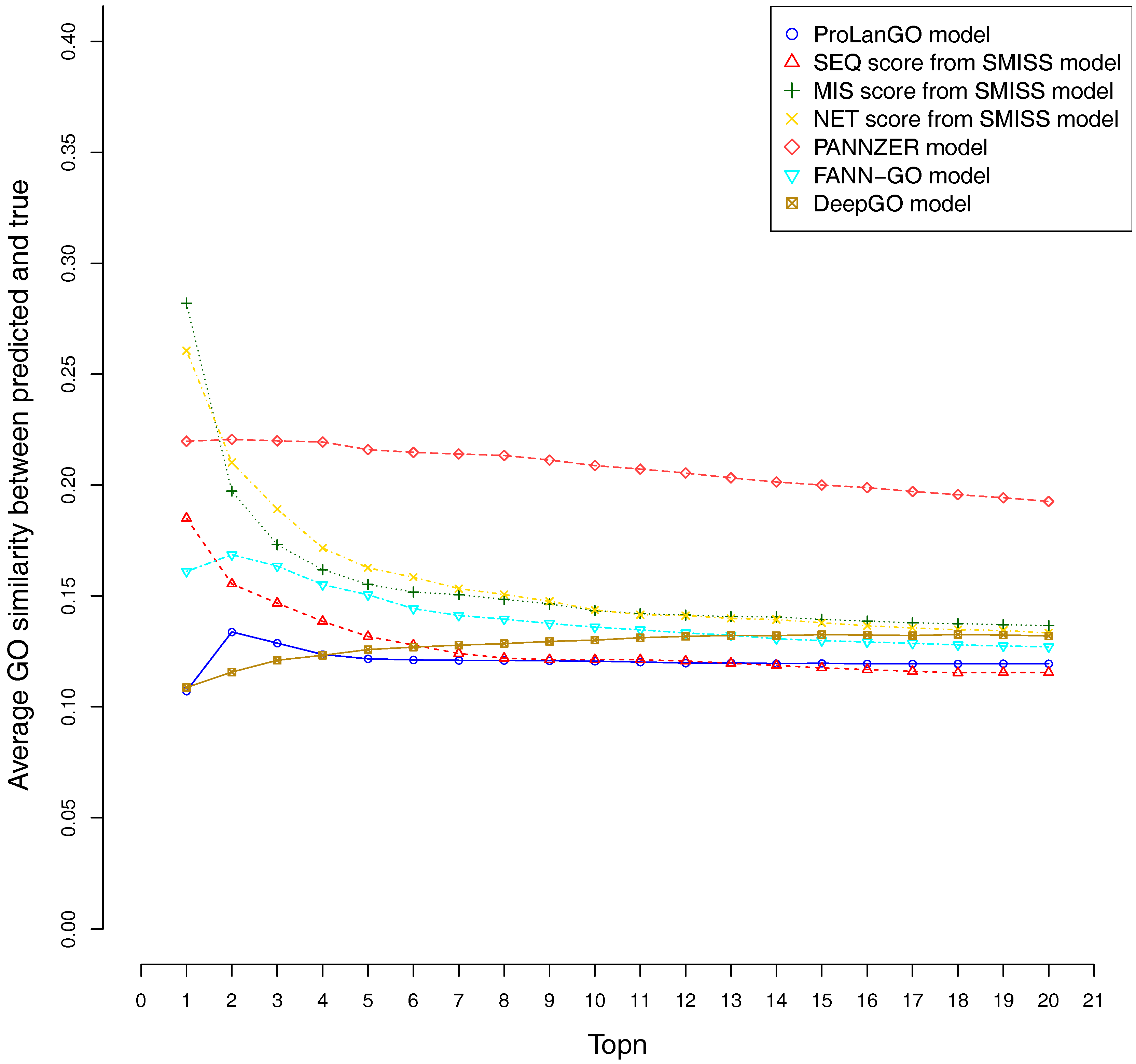
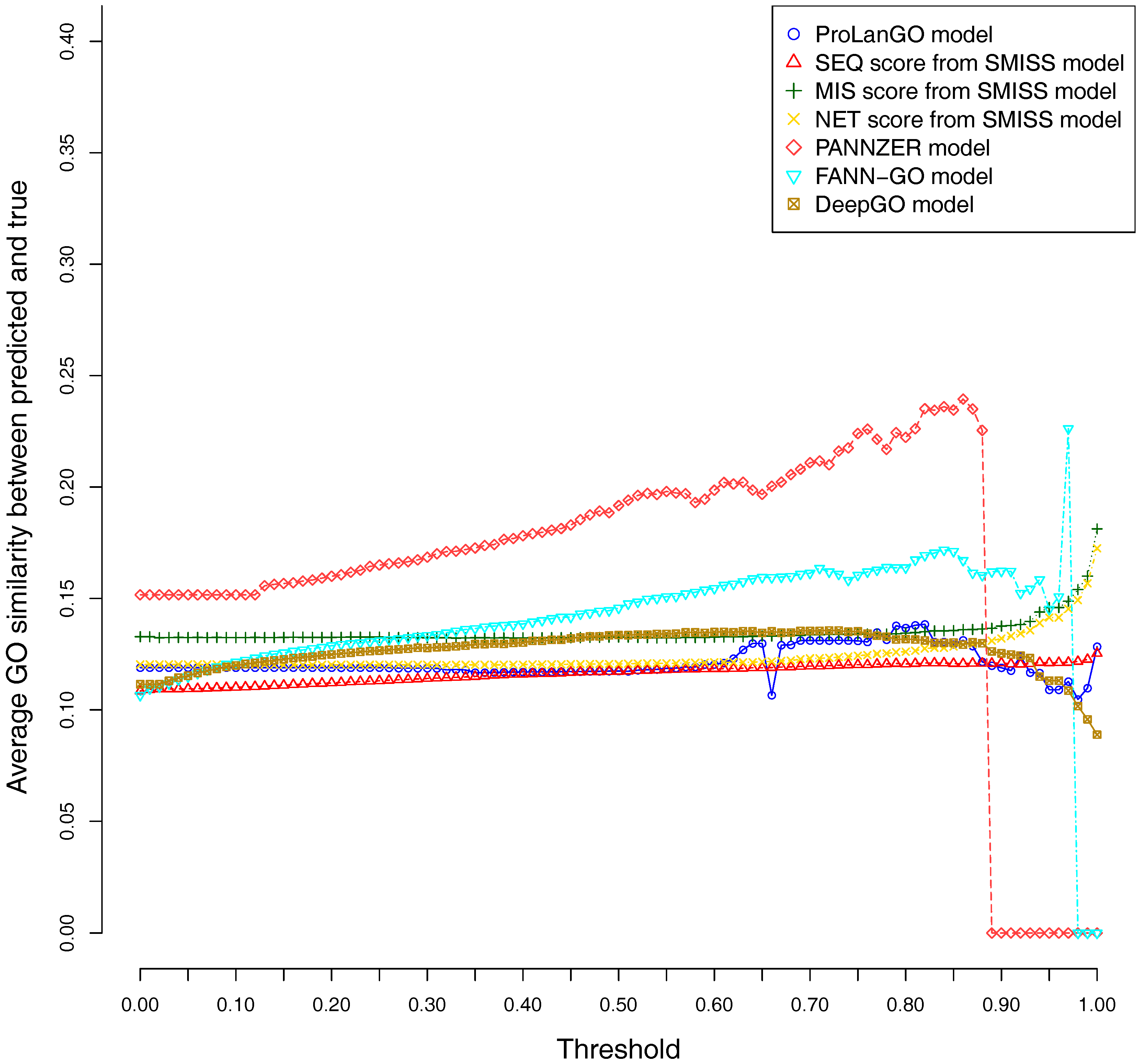
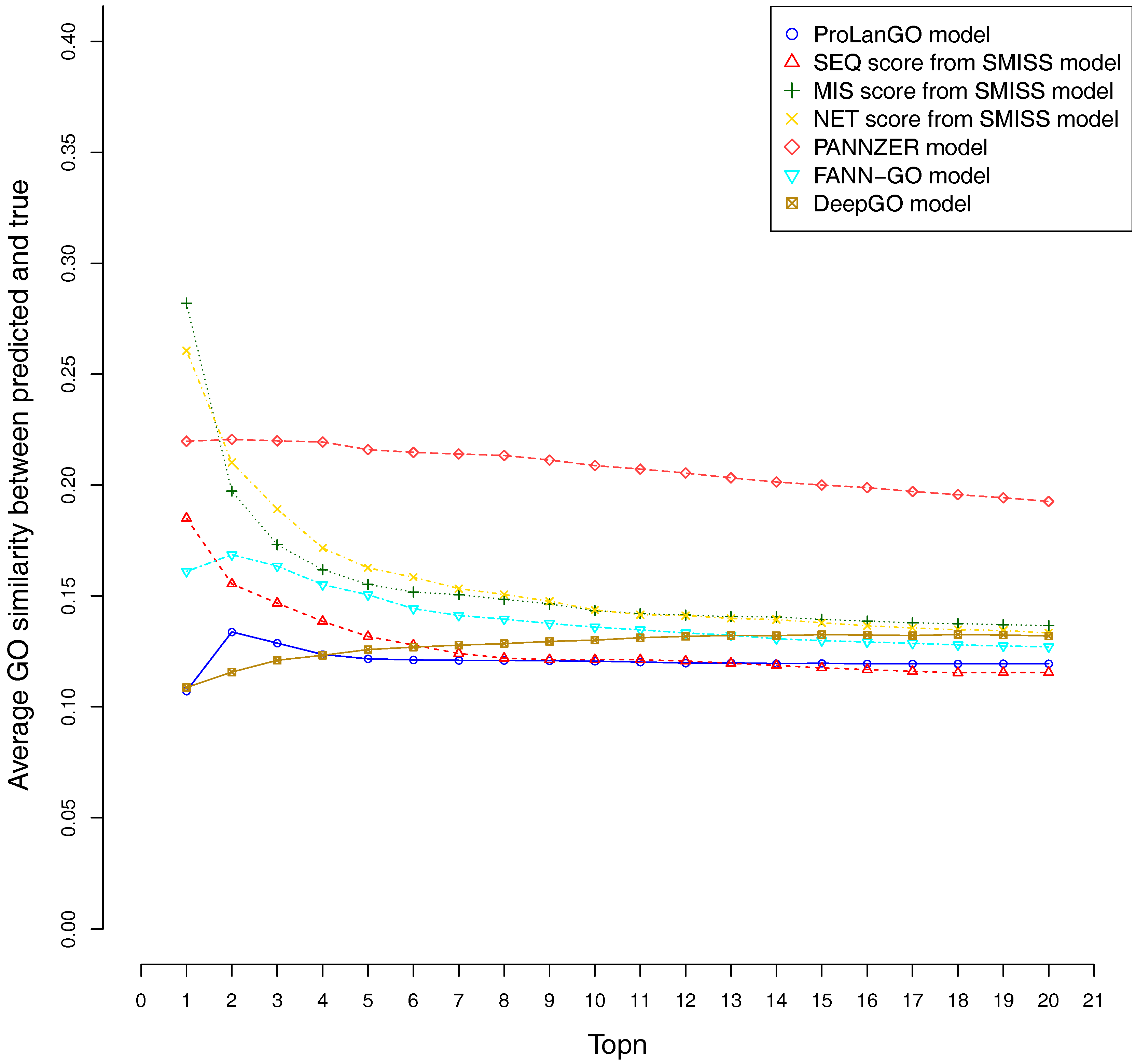
2.3. Comparison with Other Selected Methods on CAFA3
3. Methods
3.1. Data Preparation
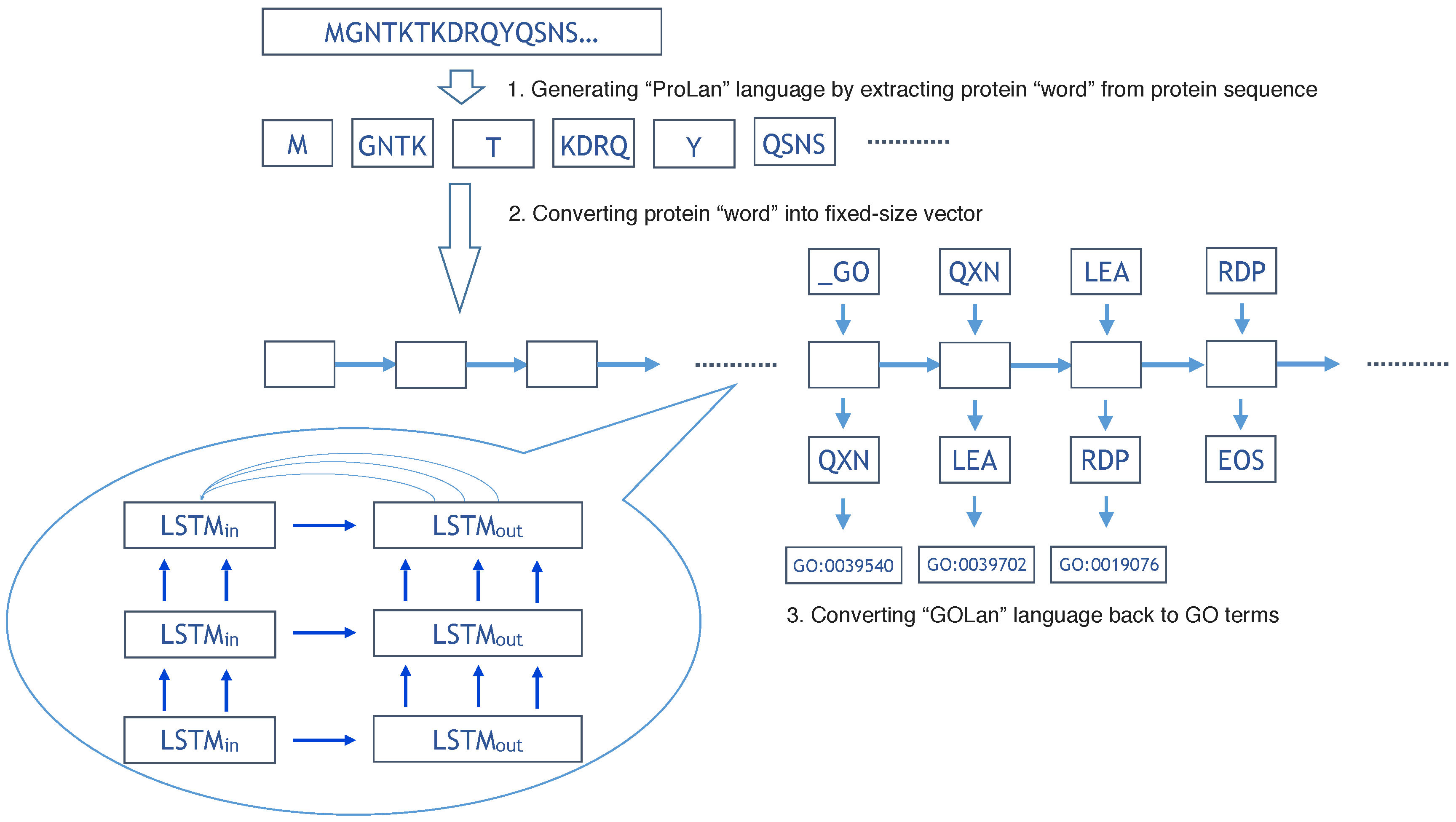
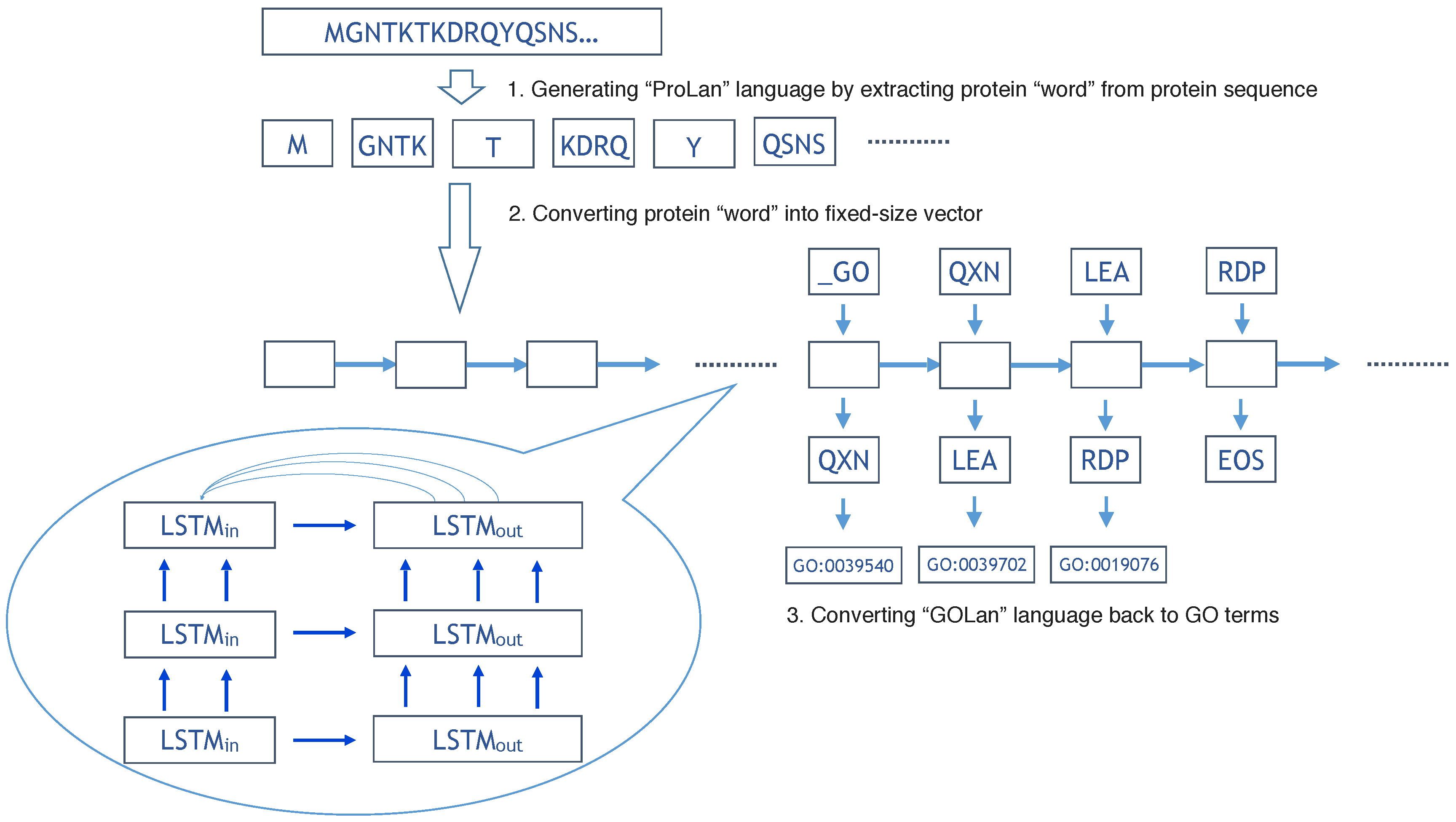
3.2. Understanding Protein Sequence as a Language
3.3. Understanding Gene Ontology Terms as a Language
3.4. ProLanGO Model by Neural Machine Translation Based on Recurrent Neural Network
3.5. Baseline Method on Probability Theory
3.6. Evaluation Method
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cao, R.; Bhattacharya, D.; Hou, J.; Cheng, J. DeepQA: Improving the estimation of single protein model quality with deep belief networks. BMC Bioinform. 2016, 17, 495. [Google Scholar] [CrossRef] [PubMed]
- Radivojac, P.; Clark, W.T.; Oron, T.R.; Schnoes, A.M.; Wittkop, T.; Sokolov, A.; Graim, K.; Funk, C.; Verspoor, K.; Ben-Hur, A.; et al. A large-scale evaluation of computational protein function prediction. Nat. Methods 2013, 10, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Cheng, J. Integrated protein function prediction by mining function associations, sequences, and protein–protein and gene–gene interaction networks. Methods 2016, 93, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Liolios, K.; Chen, I.M.A.; Mavromatis, K.; Tavernarakis, N.; Hugenholtz, P.; Markowitz, V.M.; Kyrpides, N.C. The Genomes On Line Database (GOLD) in 2009: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2009, 38, D346–D354. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [PubMed]
- Rost, B.; Liu, J.; Nair, R.; Wrzeszczynski, K.O.; Ofran, Y. Automatic prediction of protein function. Cell. Mol. Life Sci. 2003, 60, 2637–2650. [Google Scholar] [PubMed]
- Watson, J.D.; Laskowski, R.A.; Thornton, J.M. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 2005, 15, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, I. Automated protein function prediction—The genomic challenge. Brief. Bioinformat. 2006, 7, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Redfern, O.; Orengo, C. Predicting protein function from sequence and structure. Nat. Res. Mol. Cell Biol. 2007, 8, 995. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.C.; Le, M.H.; Xu, D.; Stacey, G.; Cheng, J. A protein domain co-occurrence network approach for predicting protein function and inferring species phylogeny. PLoS ONE 2011, 6, e17906. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, R.; Orengo, C.A. Protein function prediction—The power of multiplicity. Trends Biotechnol. 2009, 27, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Duan, Y.; Zou, Q. HPSLPred: An ensemble multi-label classifier for human protein subcellular location prediction with imbalanced source. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.M.; Berriman, M.; Barton, G.J. GOtcha: A new method for prediction of protein function assessed by the annotation of seven genomes. BMC Bioinform. 2004, 5, 178. [Google Scholar]
- Zehetner, G. OntoBlast function: From sequence similarities directly to potential functional annotations by ontology terms. Nucleic Acids Res. 2003, 31, 3799–3803. [Google Scholar] [CrossRef] [PubMed]
- Groth, D.; Lehrach, H.; Hennig, S. GOblet: A platform for Gene Ontology annotation of anonymous sequence data. Nucleic Acids Res. 2004, 32, W313–W317. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, T.; Chitale, M.; Luban, S.; Kihara, D. PFP: Automated prediction of gene ontology functional annotations with confidence scores using protein sequence data. Proteins: Struct. Funct. Bioinform. 2009, 74, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Zhang, K.; Mehta, S.; Chen, T.; Sun, F. Prediction of protein function using protein–protein interaction data. J. Comput. Biol. 2003, 10, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Letovsky, S.; Kasif, S. Predicting protein function from protein/protein interaction data: A probabilistic approach. Bioinformatics 2003, 19, i197–i204. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Flammini, A.; Maritan, A.; Vespignani, A. Global protein function prediction in protein-protein interaction networks. Nat. Biotechnol. 2003, 697–770. [Google Scholar] [CrossRef] [PubMed]
- Hishigaki, H.; Nakai, K.; Ono, T.; Tanigami, A.; Takagi, T. Assessment of prediction accuracy of protein function from protein–protein interaction data. Yeast 2001, 18, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.N.; Sung, W.K.; Wong, L. Exploiting indirect neighbours and topological weight to predict protein function from protein–protein interactions. Bioinformatics 2006, 22, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhang, X.; Zou, Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief. Bioinform. 2015, 17, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Cheng, J. Deciphering the association between gene function and spatial gene-gene interactions in 3D human genome conformation. BMC Genom. 2015, 16, 880. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Eisenberg, D. Inference of protein function from protein structure. Structure 2005, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Pazos, F.; Sternberg, M.J. Automated prediction of protein function and detection of functional sites from structure. Proc. Natl. Acad. Sci. USA 2004, 101, 14754–14759. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. Protein function prediction using local 3D templates. J. Mol. Biol. 2005, 351, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Hibbs, M.; Myers, C.; Troyanskaya, O.G. A scalable method for integration and functional analysis of multiple microarray datasets. Bioinformatics 2006, 22, 2890–2897. [Google Scholar] [CrossRef] [PubMed]
- Troyanskaya, O.G.; Dolinski, K.; Owen, A.B.; Altman, R.B.; Botstein, D. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc. Natl. Acad. Sci. USA 2003, 100, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Date, S.V.; Adai, A.T.; Marcotte, E.M. A probabilistic functional network of yeast genes. Science 2004, 306, 1555–1558. [Google Scholar] [CrossRef] [PubMed]
- Kourmpetis, Y.A.; Van Dijk, A.D.; Bink, M.C.; van Ham, R.C.; ter Braak, C.J. Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS ONE 2010, 5, e9293. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.; Ben-Hur, A. Hierarchical classification of gene ontology terms using the GOstruct method. J. Bioinform. Comput. Biol. 2010, 8, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Tang, H.; Li, W.C.; Lin, H.; Chen, W.; Chou, K.C. iOri-Human: Identify human origin of replication by incorporating dinucleotide physicochemical properties into pseudo nucleotide composition. Oncotarget 2016, 7, 69783–69793. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, D.; Deng, L. Accurate prediction of functional effects for variants by combining gradient tree boosting with optimal neighborhood properties. PLoS ONE 2017, 12, e0179314. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Tang, J.; Zou, Q. PhosPred-RF: A novel sequence-based predictor for phosphorylation sites using sequential information only. IEEE Trans. Nano Biosci. 2017, 16, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Xing, P.; Su, R.; Shi, G.; Ma, Z.S.; Zou, Q. CPPred-RF: A sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J. Proteome Res. 2017, 16, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Djuric, N.; Guo, Y.; Vucetic, S. MS-k NN: Protein function prediction by integrating multiple data sources. BMC Bioinform. 2013, 14, S8. [Google Scholar]
- Cai, C.; Han, L.; Ji, Z.L.; Chen, X.; Chen, Y.Z. SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003, 31, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lin, Y.S.; Lin, C.J.; Hwang, J.K. Prediction of the bonding states of cysteines using the support vector machines based on multiple feature vectors and cysteine state sequences. Proteins: Struct. Funct. Bioinform. 2004, 55, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wang, W.; Sun, L.; Chen, Y. Protein function classification via support vector machine approach. Math. Biosci. 2003, 185, 111–122. [Google Scholar] [CrossRef]
- Halperin, I.; Glazer, D.S.; Wu, S.; Altman, R.B. The FEATURE framework for protein function annotation: Modeling new functions, improving performance, and extending to novel applications. BMC Genom. 2008, 9, S2. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, A.M.; Snitkin, E.S.; Parker, S.C.; DeLisi, C.; Kasif, S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genom. 2006, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J. SVMQA: Support–vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496–2503. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Lee, J.; Lee, J. Random Forest-Based Protein Model Quality Assessment (RFMQA) Using Structural Features and Potential Energy. PLoS ONE 2014, 9, e106542. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Adhikari, B.; Bhattacharya, D.; Sun, M.; Hou, J.; Cheng, J. QAcon: Single model quality assessment using protein structural and contact information with machine learning techniques. Bioinformatics 2017, 33, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Han, T.X.; Liu, M.C.; Khodayari-Rostamabad, A. Multiple Instance Learning Convolutional Neural Networks for Object Recognition. In Proceedings of the 2016 23rd International Conference on Pattern Recognition (ICPR), Cancun, Mexico, 4–8 December 2016; pp. 3270–3275. [Google Scholar]
- Zou, W.Y.; Wang, X.; Sun, M.; Lin, Y. Generic object detection with dense neural patterns and regionlets. arXiv 2014, arXiv:1404.4316. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. Tensorflow: Large-scale machine learning on heterogeneous distributed systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Ofer, D.; Linial, M. ProFET: Feature engineering captures high-level protein functions. Bioinformatics 2015, 31, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Oron, T.R.; Clark, W.T.; Bankapur, A.R.; D’Andrea, D.; Lepore, R.; Funk, C.S.; Kahanda, I.; Verspoor, K.M.; Ben-Hur, A.; et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016, 17, 184. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Zhong, Z.; Cheng, J. SMISS: A protein function prediction server by integrating multiple sources. arXiv 2016, arXiv:1607.01384. [Google Scholar]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.T.; Radivojac, P. Analysis of protein function and its prediction from amino acid sequence. Proteins: Struct. Funct. Bioinform. 2011, 79, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Kulmanov, M.; Khan, M.A.; Hoehndorf, R. DeepGO: Predicting protein functions from sequence and interactions using a deep ontology-aware classifier. arXiv 2017, arXiv:1705.05919. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Chen, X.X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.P.; Li, W.C.; Zhong, Z.J.; Deng, E.Z.; Ding, H.; Chen, W.; Lin, H. Predicting the subcellular localization of mycobacterial proteins by incorporating the optimal tripeptides into the general form of pseudo amino acid composition. Mol. BioSyst. 2015, 11, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Tang, H.; Li, W.C.; Wu, H.; Chen, W.; Ding, H.; Lin, H. Identification of bacterial cell wall lyases via pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 1654623. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, H.; Chen, X.X.; Zhang, C.J.; Zhu, P.P.; Ding, H.; Chen, W.; Lin, H. Identification of secretory proteins in mycobacterium tuberculosis using pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 5413903. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning phrase representations using RNN encoder-decoder for statistical machine translation. arXiv 2014, arXiv:1406.1078. [Google Scholar]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to sequence learning with neural networks. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2014; pp. 3104–3112. [Google Scholar]
- Tsoumakas, G.; Katakis, I. Multi-label classification: An overview. Int. J. Data Warehous. Min. 2006, 3, 1–13. [Google Scholar] [CrossRef]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural machine translation by jointly learning to align and translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- Jean, S.; Cho, K.; Memisevic, R.; Bengio, Y. On using very large target vocabulary for neural machine translation. arXiv 2014, arXiv:1412.2007. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, R.; Freitas, C.; Chan, L.; Sun, M.; Jiang, H.; Chen, Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22, 1732. https://doi.org/10.3390/molecules22101732
Cao R, Freitas C, Chan L, Sun M, Jiang H, Chen Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules. 2017; 22(10):1732. https://doi.org/10.3390/molecules22101732
Chicago/Turabian StyleCao, Renzhi, Colton Freitas, Leong Chan, Miao Sun, Haiqing Jiang, and Zhangxin Chen. 2017. "ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network" Molecules 22, no. 10: 1732. https://doi.org/10.3390/molecules22101732
APA StyleCao, R., Freitas, C., Chan, L., Sun, M., Jiang, H., & Chen, Z. (2017). ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules, 22(10), 1732. https://doi.org/10.3390/molecules22101732