Screening a Natural Product-Based Library against Kinetoplastid Parasites


 and
and
Abstract
1. Introduction
2. Results
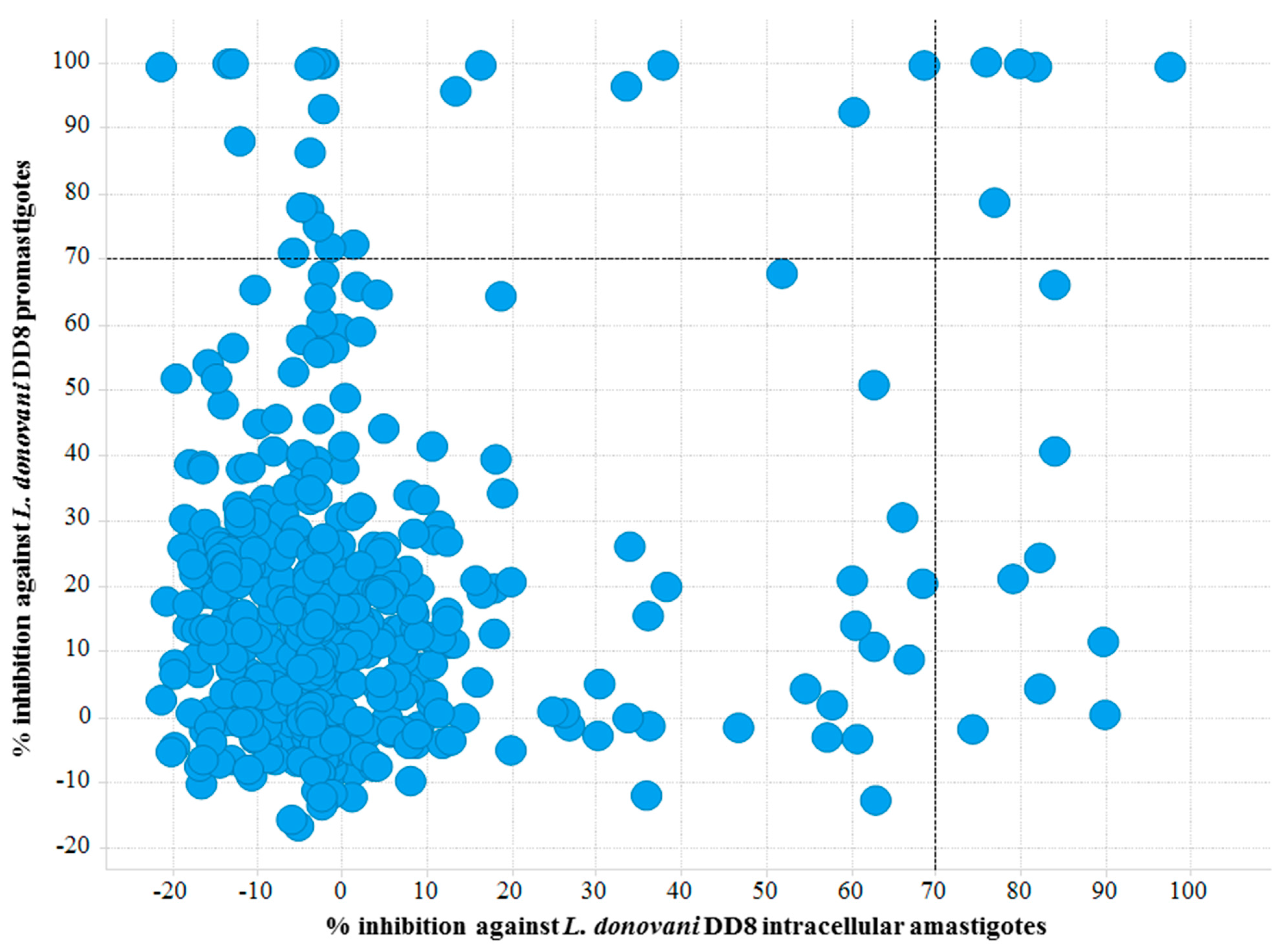
2.1. Screening Campaigns and Hit Identification
2.1.1. L. donovani DD8 Promastigote and Intracellular Amastigote Screening
2.1.2. T. b. brucei Screening
2.1.3. T. cruzi Intracellular Amastigote Screening
2.1.4. Common Activity Against Kinetoplastids
3. Discussion
4. Materials and Methods
4.1. Maintenance of Mammalian Cells Lines
4.2. Maintainanace of Parasites
4.3. Open Access Compound Library and Assay Plate Preparation
4.4. Phenotypic Assays
4.4.1. L. donovani DD8 Intracellular Amastigote Assay
4.4.2. L. donovani DD8 Promastigote Viability Assay
4.4.3. T. b. brucei Resazurin Viability Assay
4.4.4. T. cruzi Intracellular Amastigote Assay
4.4.5. HEK-293 Resazurin Viability Assay
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Investing to Overcome the Global Impact of Neglected Tropical Diseases: Third Who Report on negLected Tropical Diseases 2015; World Health Organization: Geneva, Switzerland, 2015; Volume 3. [Google Scholar]
- Molyneux, D.H.; Savioli, L.; Engel, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef]
- Borghi, S.M.; Fattori, V.; Conchon-Costa, I.; Pinge-Filho, P.; Pavanelli, W.R.; Verri, W.A., Jr. Leishmania infection: Painful or painless? Parasitol. Res. 2017, 116, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Herwaldt, B.L. Leishmaniasis. Lancet 1999, 354, 1191–1199. [Google Scholar] [CrossRef]
- Van Den Abbeele, J.; Claes, Y.; Van Bockstaele, D.; Le Ray, D.; Coosemans, M. Trypanosoma brucei spp. Development in the tsetse fly: Characterization of the post-mesocyclic stages in the foregut and proboscis. Parasitology 1999, 118, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Azambuja, P. Development and interactions of Trypanosoma cruzi within the insect vector. Parasitol. Today 1991, 7, 240–244. [Google Scholar] [CrossRef]
- Handman, E.; Bullen, D.V. Interaction of Leishmania with the host macrophage. Trends Parasitol. 2002, 18, 332–334. [Google Scholar] [CrossRef]
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.; Yukich, J.O.; Sutherland, C.S.; Woods, G.; Tediosi, F. Human african trypanosomiasis prevention, treatment and control costs: A systematic review. Acta Trop. 2015, 150, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Bonney, K.M.; Engman, D.M. Autoimmune pathogenesis of chagas heart disease: Looking back, looking ahead. Am. J. Pathol. 2015, 185, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- McCall, L.-I.; McKerrow, J.H. Determinants of disease phenotype in trypanosomatid parasites. Trends Parasitol. 2014, 30, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.; Avery, V.M. Future treatment options for human african trypanosomiasis. Expert Rev. Anti-Infect. Ther. 2015, 13, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, J.; Davies, C.; Simonazzi, A.; Real, J.P.; Palma, S. Current drug therapy and pharmaceutical challenges for chagas disease. Acta Trop. 2016, 156, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mondelaers, A.; Sanchez-Cañete, M.P.; Hendrickx, S.; Eberhardt, E.; Garcia-Hernandez, R.; Lachaud, L.; Cotton, J.; Sanders, M.; Cuypers, B.; Imamura, H. Genomic and molecular characterization of miltefosine resistance in Leishmania infantum strains with either natural or acquired resistance through experimental selection of intracellular amastigotes. PLoS ONE 2016, 11, e0154101. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human african trypanosomiasis. Future Med. 2011, 6, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.C.O.; Leon, L.L.; Taylor, M.C.; Kelly, J.M. Benznidazole-resistance in trypanosoma cruzi: Evidence that distinct mechanisms can act in concert. Mol. Biochem. Parasitol. 2014, 193, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Don, R.; Ioset, J.R. Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology 2014, 141, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human african trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
- Aronson, N.E. Addressing a clinical challenge: Guidelines for the diagnosis and treatment of leishmaniasis. BMC Med. 2017, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Bhuniya, D.; Mukkavilli, R.; Shivahare, R.; Launay, D.; Dere, R.T.; Deshpande, A.; Verma, A.; Vishwakarma, P.; Moger, M.; Pradhan, A.; et al. Aminothiazoles: Hit to lead development to identify antileishmanial agents. Eur. J. Med. Chem. 2015, 102, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, C.E.; Braillard, S.; Speed, W.; Glossop, P.A.; Whitlock, G.A.; Gibson, K.R.; Mills, J.E.; Brown, A.D.; Gardner, J.M.; Cao, Y.; et al. Novel amino-pyrazole ureas with potent in vitro and in vivo antileishmanial activity. J. Med. Chem. 2015, 58, 9615–9624. [Google Scholar] [CrossRef] [PubMed]
- Zulfiqar, B.; Shelper, T.B.; Avery, V.M. Leishmaniasis drug discovery: Recent progress and challenges in assay development. Drug Discov. Today 2017. [Google Scholar] [CrossRef] [PubMed]
- Torreele, E.; Trunz, B.B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazue, G.; Bray, M.A.; Pecoul, B. Fexinidazole—A new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T. Scyx-7158, an orally-active benzoxaborole for the treatment of stage 2 human african trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Nwaka, S.; Besson, D.; Ramirez, B.; Maes, L.; Matheeussen, A.; Bickle, Q.; Mansour, N.R.; Yousif, F.; Townson, S.; Gokool, S.; et al. Integrated dataset of screening hits against multiple neglected disease pathogens. PLoS Negl. Trop. Dis. 2011, 5, e1412. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.R.; Ramos Ade, S.; Machado, M.; de Moura, D.F.; Neto, Z.; Canto-Cavalheiro, M.M.; Figueiredo, P.; do Rosario, V.E.; Amaral, A.C.; Lopes, D. A review of antimalarial plants used in traditional medicine in communities in portuguese-speaking countries: Brazil, mozambique, cape verde, guinea-bissau, sao tome and principe and angola. Mem. Inst. Oswaldo Cruz 2011, 106, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.B.; Malyszek, C.; Kaiser, M.; Brun, R.; Schmidt, T.J. Alkamides from Anacyclus pyrethrum l. and their in vitro antiprotozoal activity. Molecules 2017, 22, 796. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Feng, Y.; Duffy, S.; Avery, V.M.; Camp, D.; Quinn, R.J.; Davis, R.A. A new quinoline epoxide from the australian plant Drummondita calida. Planta Med. 2011, 77, 1644–1647. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Sykes, M.; Avery, V.M.; Camp, D.; Quinn, R.J. Convolutamines I and J, antitrypanosomal alkaloids from the bryozoan Amathia tortusa. Bioorg. Med. Chem. 2011, 19, 6615–6619. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Davis, R.A.; Sykes, M.L.; Avery, V.M.; Quinn, R.J. Iotrochamides A and B, antitrypanosomal compounds from the Australian marine sponge Iotrochota sp. Bioorg. Med. Chem. Lett. 2012, 22, 4873–4876. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Duffy, S.; Fletcher, S.; Avery, V.M.; Quinn, R.J. Thiaplakortones A-D: Antimalarial thiazine alkaloids from the Australian marine sponge Plakortis lita. J. Org. Chem. 2013, 78, 9608–9613. [Google Scholar] [CrossRef] [PubMed]
- Choomuenwai, V.; Beattie, K.D.; Healy, P.C.; Andrews, K.T.; Fechner, N.; Davis, R.A. Entonalactams A–C: Isoindolinone derivatives from an Australian rainforest fungus belonging to the genus Entonaema. Phytochemistry 2015, 117, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Siqueira-Neto, J.L.; Moon, S.; Jang, J.; Yang, G.; Lee, C.; Moon, H.K.; Chatelain, E.; Genovesio, A.; Cechetto, J.; Freitas-Junior, L.H. An image-based high-content screening assay for compounds targeting intracellular Leishmania donovani amastigotes in human macrophages. PLoS Negl. Trop. Dis. 2012, 6, e1671. [Google Scholar] [CrossRef] [PubMed]
- Sykes, M.L.; Avery, V.M. Development of an alamar blue viability assay in 384-well format for high throughput whole cell screening of Trypanosoma brucei brucei bloodstream form strain 427. Am. J. Trop. Med. Hyg. 2009, 81, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.; Kaiser, M.; Avery, V.M. Identification and characterization of fty720 for the treatment of human african trypanosomiasis. Antimicrob. Agents Chemother. 2015, 60, 1859–1861. [Google Scholar] [CrossRef] [PubMed]
- Sykes, M.L.; Avery, V.M. Development and application of a sensitive, phenotypic, high-throughput image-based assay to identify compound activity against Trypanosoma cruzi amastigotes. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.; McLeod, R.; Rice, D.; Ginger, M.; Chance, M.L.; Goad, L.J. Fatty acid and sterol metabolism: Potential antimicrobial targets in apicomplexan and trypanosomatid parasitic protozoa. Mol. Biochem. Parasitol. 2003, 126, 129–142. [Google Scholar] [CrossRef]
- Freitas-Junior, L.H.; Chatelain, E.; Kim, H.A.; Siqueira-Neto, J.L. Visceral leishmaniasis treatment: What do we have, what do we need and how to deliver it? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Luque-Ortega, J.R.; Rivas, L. Miltefosine (hexadecylphosphocholine) inhibits cytochrome c oxidase in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 2007, 51, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Boykin, D.W.; Brun, R.; Tidwell, R.R. Human african trypanosomiasis: Pharmacological re-engagement with a neglected disease. Br. J. Pharmacol. 2007, 152, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Rajao, M.A.; Furtado, C.; Alves, C.L.; Passos-Silva, D.G.; de Moura, M.B.; Schamber-Reis, B.L.; Kunrath-Lima, M.; Zuma, A.A.; Vieira-da-Rocha, J.P.; Garcia, J.B.F.; et al. Unveiling benznidazole’s mechanism of action through overexpression of DNA repair proteins in Trypanosoma cruzi. Environ. Mol. Mutagen. 2014, 55, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Piacenza, L.; Hernández, P.; Boiani, L.; Cerecetto, H.; González, M.; Denicola, A. Mode of action of nifurtimox and n-oxide-containing heterocycles against Trypanosoma cruzi: Is oxidative stress involved? Biochem. Pharmacol. 2010, 79, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Annang, F.; Pérez-Moreno, G.; García-Hernández, R.; Cordon-Obras, C.; Martín, J.; Tormo, J.; Rodríguez, L.; De Pedro, N.; Gómez-Pérez, V.; Valente, M. High-throughput screening platform for natural product–based drug discovery against 3 neglected tropical diseases: Human african trypanosomiasis, leishmaniasis, and chagas disease. J. Biomol. Screen. 2015, 20, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.; Grkovic, T.; Sykes, M.L.; Avery, V.M. Trypanocidal activity of marine natural products. Mar. Drugs 2013, 11, 4058–4082. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gurtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Poce, G.; Alfonso, S.; Cocozza, M.; Porretta, G.C.; Colotti, G.; Biava, M.; Moraca, F.; Botta, M.; Yardley, V.; et al. Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds: A comparative analysis with its physiological substrate by x-ray crystallography. ChemMedChem 2013, 8, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Corona, P.; Gibellini, F.; Cavalli, A.; Saxena, P.; Carta, A.; Loriga, M.; Luciani, R.; Paglietti, G.; Guerrieri, D.; Nerini, E.; et al. Structure-based selectivity optimization of piperidine-pteridine derivatives as potent Leishmania pteridine reductase inhibitors. J. Med. Chem. 2012, 55, 8318–8329. [Google Scholar] [CrossRef] [PubMed]
- Schroder, J.; Noack, S.; Marhofer, R.J.; Mottram, J.C.; Coombs, G.H.; Selzer, P.M. Identification of semicarbazones, thiosemicarbazones and triazine nitriles as inhibitors of Leishmania mexicana cysteine protease cpb. PLoS ONE 2013, 8, e77460. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Lizzi, F.; Bongarzone, S.; Brun, R.; Luise Krauth-Siegel, R.; Bolognesi, M.L. Privileged structure-guided synthesis of quinazoline derivatives as inhibitors of trypanothione reductase. Bioorg. Med. Chem. Lett. 2009, 19, 3031–3035. [Google Scholar] [CrossRef] [PubMed]
- Mpamhanga, C.P.; Spinks, D.; Tulloch, L.B.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Fairlamb, A.H.; Wyatt, P.G.; Frearson, J.A.; Hunter, W.N.; et al. One scaffold, three binding modes: Novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. [Google Scholar] [CrossRef] [PubMed]
- Breuning, A.; Degel, B.; Schulz, F.; Buchold, C.; Stempka, M.; Machon, U.; Heppner, S.; Gelhaus, C.; Leippe, M.; Leyh, M.; et al. Michael acceptor based antiplasmodial and antitrypanosomal cysteine protease inhibitors with unusual amino acids. J. Med. Chem. 2010, 53, 1951–1963. [Google Scholar] [CrossRef] [PubMed]
- DNDi. Fexinidazole (hat). Available online: Http://www.Dndi.Org/diseases-projects/portfolio/fexinidazole/ (accessed on 17 May 2017).
- Wyllie, S.; Patterson, S.; Stojanovski, L.; Simeons, F.R.; Norval, S.; Kime, R.; Read, K.D.; Fairlamb, A.H. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. [Google Scholar] [CrossRef] [PubMed]
- Marr, J.; Muller, M. Biochemistry and Molecular Biology of Parasites; Academic Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Davis, R.A.; Sandoval, I.T.; Concepcion, G.P.; da Rocha, R.M.; Ireland, C.M. Lissoclinotoxins e and f, novel cytotoxic alkaloids from a Philippine Didemnid ascidian. Tetrahedron 2003, 59, 2855–2859. [Google Scholar] [CrossRef]
- Buchanan, M.S.; Carroll, A.R.; Fechner, G.A.; Boyle, A.; Simpson, M.M.; Addepalli, R.; Avery, V.M.; Hooper, J.N.; Su, N.; Chen, H.; et al. Spermatinamine, the first natural product inhibitor of isoprenylcysteine carboxyl methyltransferase, a new cancer target. Bioorg. Med. Chem. Lett. 2007, 17, 6860–6863. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Davis, R.A.; Shelper, T.; Sykes, M.L.; Avery, V.M.; Elofsson, M.; Sundin, C.; Quinn, R.J. Pseudoceramines A–D, new antibacterial bromotyrosine alkaloids from the marine sponge Pseudoceratina sp. Org. Biomol. Chem. 2011, 9, 6755–6760. [Google Scholar] [CrossRef] [PubMed]
- Choomuenwai, V.; Schwartz, B.D.; Beattie, K.D.; Andrews, K.T.; Khokhar, S.; Davis, R.A. The discovery, synthesis and antimalarial evaluation of natural product-based polyamine alkaloids. Tetrahedron Lett. 2013, 54, 5188–5191. [Google Scholar] [CrossRef]
- Buckner, F.S.; Kateete, D.P.; Lubega, G.W.; Van Voorhis, W.C.; Yokoyama, K. Trypanosoma brucei prenylated-protein carboxyl methyltransferase prefers farnesylated substrates. Biochem. J. 2002, 367, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.D.; Coster, M.J.; Skinner-Adams, T.S.; Andrews, K.T.; White, J.M.; Davis, R.A. Synthesis and antiplasmodial evaluation of analogues based on the tricyclic core of thiaplakortones A-D. Mar. Drugs 2015, 13, 5784–5795. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B.D.; Skinner-Adams, T.S.; Andrews, K.T.; Coster, M.J.; Edstein, M.D.; MacKenzie, D.; Charman, S.A.; Koltun, M.; Blundell, S.; Campbell, A.; et al. Synthesis and antimalarial evaluation of amide and urea derivatives based on the thiaplakortone a natural product scaffold. Org. Biomol. Chem. 2015, 13, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Z.; Kasibhatla, S.; Wang, Y.; Herich, J.; Guastella, J.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery, characterization and sar of gambogic acid as a potent apoptosis inducer by a hts assay. Bioorg. Med. Chem. 2004, 12, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Mackey, Z.B.; Baca, A.M.; Mallari, J.P.; Apsel, B.; Shelat, A.; Hansell, E.J.; Chiang, P.K.; Wolff, B.; Guy, K.R.; Williams, J.; et al. Discovery of trypanocidal compounds by whole cell hts of Trypanosoma brucei. Chem. Biol. Drug Des. 2006, 67, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Cao, S.; Goh, S.; Hsu, A.; Tan, B.K. Mitochondrial destabilisation and caspase-3 activation are involved in the apoptosis of jurkat cells induced by gaudichaudione a, a cytotoxic xanthone. Planta Med. 2002, 68, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Bessho, T.; Morii, S.; Kusumoto, T.; Shinohara, T.; Noda, M.; Uchiyama, S.; Shuto, S.; Nishimura, S.; Djikeng, A.; Duszenko, M. Characterization of the novel Trypanosoma brucei inosine 5′-monophosphate dehydrogenase. Parasitology 2013, 140, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Aboge, G.O.; Terkawi, M.A.; Zhou, M.; Luo, Y.; Yu, L.; Li, Y.; Goo, Y.; Kamyingkird, K.; Masatani, T. Cloning, characterization and validation of inosine 5′-monophosphate dehydrogenase of Babesia gibsoni as molecular drug target. Parasitol. Int. 2013, 62, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, W.J.; Dixon, S.E.; Li, C.; Striepen, B.; Queener, S.F. Imp dehydrogenase from the protozoan parasite Toxoplasma gondii. Antimicrob. Agents Chemother. 2005, 49, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, K.; Sarwono, A.E.; Mitsuhashi, S.; Jakalski, M.; Okada, T.; Nthatisi, M.; Yamagishi, J.; Ubukata, M.; Inoue, N. Mycophenolic acid and its derivatives as potential chemotherapeutic agents targeting inosine monophosphate dehydrogenase in Trypanosoma congolense. Antimicrob. Agents Chemother. 2016, 60, 4391–4393. [Google Scholar] [CrossRef] [PubMed]
- Ceriotti, G. Narciclasine: An antimitotic substance from narcissus bulbs. Nature 1967, 213, 595–596. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.; Fresno, M.; Vazquez, D. Narciclasine: An antitumour alkaloid which blocks peptide bond formation by eukaryotic ribosomes. FEBS Lett. 1975, 52, 236–239. [Google Scholar] [CrossRef]
- Gabrielsen, B.; Monath, T.P.; Huggins, J.W.; Kirsi, J.J.; Hollingshead, M.; Shannon, W.M.; Pettit, G.R. Activity of selected Amaryllidaceae constituents and related synthetic substances against medically important RNA viruses. In Natural Products as Antiviral Agents; Springer: New York, NY, USA, 1992; pp. 121–135. [Google Scholar]
- Yang, X.; Davis, R.A.; Buchanan, M.S.; Duffy, S.; Avery, V.M.; Camp, D.; Quinn, R.J. Antimalarial bromotyrosine derivatives from the australian marine sponge Hyattella sp. J. Nat. Prod. 2010, 73, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.M.; Werbovetz, K.A. Antiprotozoal compounds from Psorothamnus polydenius. J. Nat. Prod. 2005, 68, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Yardley, V.; Kendrick, H. Drug sensitivity of Leishmania species: Some unresolved problems. Trans. R. Soc. Trop. Med. Hyg. 2002, 96, S127–S129. [Google Scholar] [CrossRef]
- Neal, R.; Allen, S.; McCoy, N.; Olliaro, P.; Croft, S. The sensitivity of Leishmania species to aminosidine. J. Antimicrob. Chemother. 1995, 35, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Otigbuo, I.N.; Onabanjo, A.O. The in vitro and in vivo effects of mefloquine on Trypanosoma brucei brucei. J. Hyg. Epidemiol. Microbiol. Immunol. 1992, 36, 191–199. [Google Scholar] [PubMed]
- Planer, J.D.; Hulverson, M.A.; Arif, J.A.; Ranade, R.M.; Don, R.; Buckner, F.S. Synergy testing of fda-approved drugs identifies potent drug combinations against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2014, 8, e2977. [Google Scholar] [CrossRef] [PubMed]
- Lasserre, R. Treatment of amoebiasis. Phil. J. Microbiol. Infect. Dis. 1979, 8, 1. [Google Scholar]
- Lemmens-Gruber, R.; Karkhaneh, A.; Studenik, C.; Heistracher, P. Cardiotoxicity of emetine dihydrochloride by calcium channel blockade in isolated preparations and ventricular myocytes of guinea-pig hearts. Br. J. Pharmacol. 1996, 117, 377–383. [Google Scholar] [CrossRef]
- Matthews, H.; Usman-Idris, M.; Khan, F.; Read, M.; Nirmalan, N. Drug repositioning as a route to anti-malarial drug discovery: Preliminary investigation of the in vitro anti-malarial efficacy of emetine dihydrochloride hydrate. Malaria J. 2013, 12, 359. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Hallyburton, I.; Stojanovski, L.; Read, K.D.; Frearson, J.A.; Fairlamb, A.H. Identification of a kappa-opioid agonist as a potent and selective lead for drug development against human african trypanosomiasis. Biochem. Pharmacol. 2010, 80, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Lee, M.C.; Tan, K.O.; Yang, L.K.; Lee, A.S.; Flotow, H.; Fu, N.Y.; Butler, M.S.; Soejarto, D.D.; Buss, A.D.; et al. Identification of chelerythrine as an inhibitor of bclxl function. J. Biol. Chem. 2003, 278, 20453–20456. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, V.; Wink, M. Alkaloids induce programmed cell death in bloodstream forms of trypanosomes (trypanosoma b. Brucei). Molecules 2008, 13, 2462–2473. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Sykes, M.L.; Jones, A.J.; Shelper, T.B.; Simpson, M.; Lang, R.; Poulsen, S.A.; Sleebs, B.E.; Avery, V.M. Screening the mmv pathogen box across multiple pathogens reclassifies starting points for open source drug discovery. Antimicrob. Agents Chemother. 2017, 61, 9. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A. Isolation and structure elucidation of the new fungal metabolite (−)-xylariamide a. J. Nat. Prod. 2005, 68, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Levrier, C.; Balastrier, M.; Beattie, K.D.; Carroll, A.R.; Martin, F.; Choomuenwai, V.; Davis, R.A. Pyridocoumarin, aristolactam and aporphine alkaloids from the Australian rainforest plant Goniothalamus australis. Phytochemistry 2013, 86, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Choomuenwai, V.; Andrews, K.T.; Davis, R.A. Synthesis and antimalarial evaluation of a screening library based on a tetrahydroanthraquinone natural product scaffold. Bioorg. Med. Chem. 2012, 20, 7167–7174. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.C.; Said, N.A.B.; Williams, E.D.; Hooper, J.N.; Davis, R.A. Ecionines A and B, two new cytotoxic pyridoacridine alkaloids from the Australian marine sponge, Ecionemia geodides. Tetrahedron 2010, 66, 283–287. [Google Scholar] [CrossRef]
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | L. donovani DD8 Promastigotes | L. donovani DD8 Intracellular Amastigotes | T. b. brucei | T. cruzi | Mechanism of Action |
|---|---|---|---|---|---|
| IC50 (mean ± SD) (μM) (S.I HEK-293) (S.I THP-1) | IC50 (mean ± SD) (μM) (S.I HEK-293) (S.I THP-1) | IC50 (mean ± SD) (μM) (S.I HEK-293) | IC50 (mean ± SD) (μM) (S.I HEK-293) | ||
| Amphotericin B | 0.12 ± 0.01 (>68.27) a (>16.39) b | 0.20 ± 0.02 (>41.65) a (>9.61) b | - | - | Binds to ergosterol, the principal sterol in fungal cell membranes and Leishmania cells [41]. |
| Miltefosine | 3.48 ± 0.26 (>9.57) a (>5.74) b | 2.54 ± 0.57 (>13.12) a (>7.27) b | - | - | Interacts with lipids (phospholipids and sterols), including membrane lipids, inhibition of cytochrome C oxidase (mitochondrial function), and apoptosis [42,43]. |
| Pentamidine | - | - | 0.002 ± 0.001 (>334.52) a | - | Accumulates in trypanosomes; disrupts mitochondrial processes [44]. |
| Diminazene | - | - | 0.04 ± 0.01 (>962.75) a | - | Interferes with RNA editing and trans-splicing [44]. |
| Puromycin | - | - | 0.03 ± 0.00 (12.96) a | 1.65 ± 0.35 (0.150) a | Protein synthesis inhibitor via premature chain termination during translation in the ribosome [44]. |
| Benznidazole | - | - | - | 3.36 ± 1.52 (>44.84) a | Causes oxidation in the nucleotide pool which in turn causes the formation of breaks in double stranded DNA [45]. |
| Nifurtimox | - | - | - | 0.62 ± 0.10 (> 243.03) a | Unknown. Possibly appears to be due to oxidative stress—potentially from the formation of hydrogen peroxide [46]. |
| Compounds | Anti-Leishmanial Activity | Anti-Trypanosomal Activity | Comments | ||
|---|---|---|---|---|---|
| L. donovani DD8 Promastigotes | L. donovani DD8 Intracellular Amastigotes | T. b. brucei | T. cruzi | ||
| IC50 (mean ± SD) (μM) | IC50 (mean ± SD) (μM) | IC50 (mean ± SD) (μM) | IC50 (mean ± SD) (μM) | ||
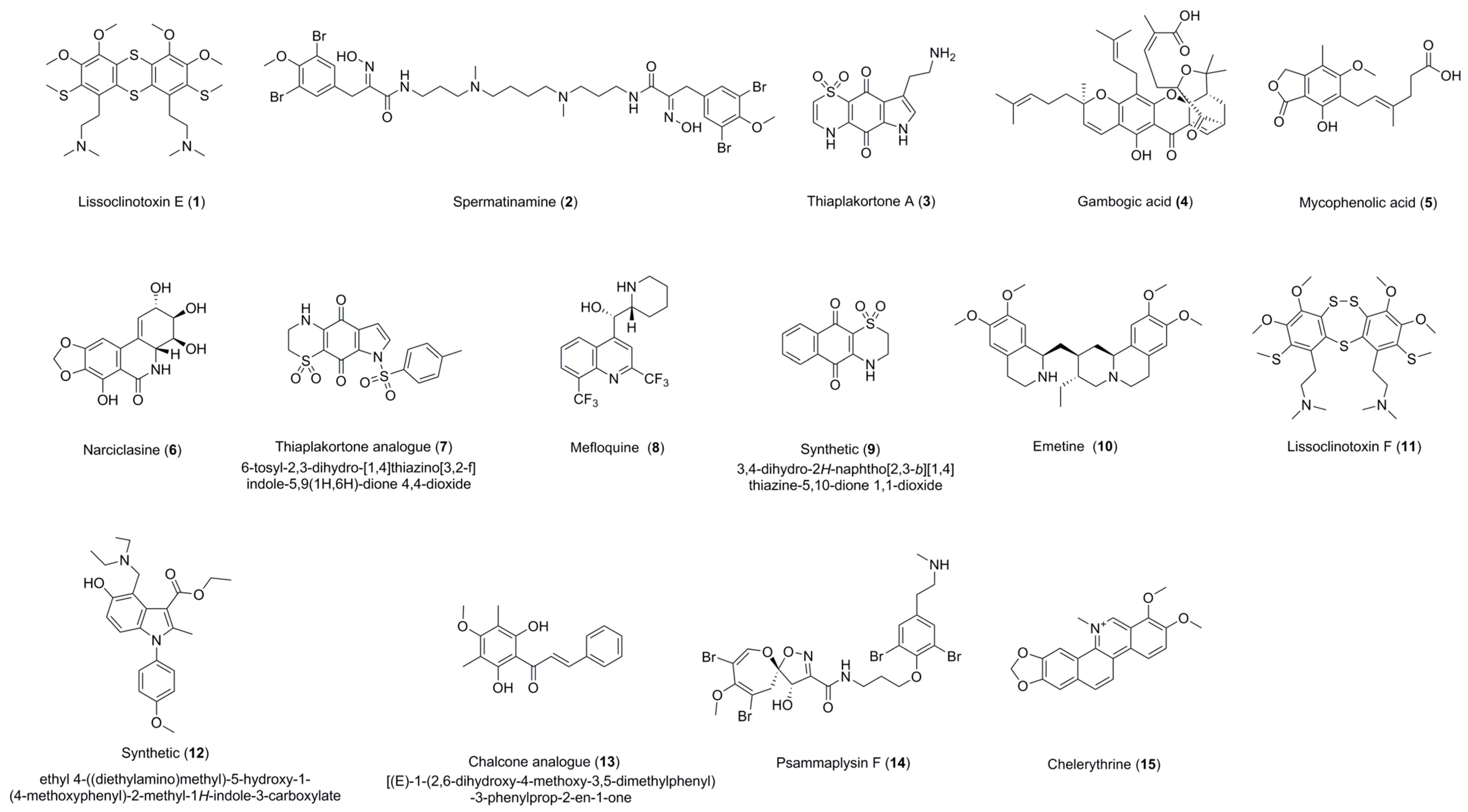
| Lissoclinotoxin E (1) | 0.72 ± 0.16 (>11.72) a (6.88) b | 4.41 ± 0.24 (>2.26) a (1.13) b | 0.57 ± 0.20 (>17.68) a | 3.92 ± 0.38 (>2.52) a(>4.67) c | Active on all three parasites with IC50 < 5 uM. Exhibits high selectivity for T. b. brucei in relation to HEK-293 cells. |
| Spermatinamine (2) | 11.87 ± 0.56 (0.33) a (0.41) b | 6.15 ± 0.05 (0.64) a (0.80) b | 1.00 ± 0.26 (5.89) a | - | Active on L. donovani DD8 and T. b. brucei. Moderate selectivity for T. b. brucei in relation to HEK-293 cells. |
| Thiaplakortone A (3) | - | - | 3.94 ± 0.78 (0.70) a | 4.26 ± 0.65 (0.53) a (1.06) c | Active on T. b. brucei and T. cruzi with no selectivity. |
| Gambogic acid (4) | 100% inhibition at 16.7 µM | - | 0.27 ± 0.04 (7.11) a | 1.87 ± 0.07 (1.50) a (1.00) c | Active on T .b. brucei and T. cruzi. Moderately selective for T. b. brucei in relation to HEK 293 cells. |
| Mycophenolic acid (5) | - | - | 0.51 ± 0.10 (1.01) a | 1.59 ± 0.03 (0.37) a (0.13) c | Active on T. b. brucei and T. cruzi with no selectivity. |
| Narciclasine (6) | 100% inhibition at 16.7 µM | - | 0.03 ± 0.01 (1.09) a | 0.20 ± 0.01 (0.21) a (3.28) c | Active on T. b. brucei and T. cruzi. Low selectivity for T. cruzi in 3T3 cells. |
| Thiaplakortone analogue (7) | - | - | 0.68 ± 0.01 (4.19) a | 3.55 ± 0.38 (1.46) a (1.33) c | Active on T. b. brucei and T. cruzi. Moderately selective for T. b. brucei in relation to HEK-293 cells. |
| Mefloquine HCl (8) | 100% inhibition at 16.7 µM | - | 0.62 ± 0.06 (7.97) a | 3.96 ± 0.58 (>5.06) a (>1.70) c | Moderate selectivity for T. cruzi and T. b. brucei in relation to HEK 293 cells. |
| 3,4-Dihydro-2H-naphtho[2,3-b]-[1,4] thiazine-5,10-dione 1,1-dioxide (9) | 100% inhibition at 16.7 µM | - | - | 3.81 ± 0.60 (1.11) a (1.24) c | Active on L. donovani DD8 promastigotes and T. cruzi with no selectivity. |
| Emetine dihydrochloride (10) | 100% inhibition at 16.7 µM | - | 0.05 ± 0.01 (1.61) a | 0.09 ± 0.00 (0.92) a (6.04) c | Active on T. b. brucei and T. cruzi with selectivity for T. cruzi in 3T3 cells. |
| Lissoclinotoxin F (11) | 5.51 ± 0.30 (>3.62) a (>1.81) b | 8.31 ± 0.67 (>2.40) a (>1.20) b | - | - | Active on both forms of L. donovani DD8 parasite. Low selectivity for both forms of L. donovani DD8 parasite. |
| Ethyl 4-((diethyl-amino)methyl)-5-hydroxy-1-(4-methoxyphenyl)-2-methyl-1H-indole-3-carboxylate (12) | 100% inhibition at 16.7 µM | 11.09 ± 0.31 (>2.97) a (>1.50) b | - | - | Low selectivity for L. donovani DD8 intracellular amastigotes. |
| Chalcone analogue (13) | 100% inhibition at 16.7 µM | 5.65 ± 0.26 (11.87) a (3.59) b | - | - | Active on L. donovani DD8 intracellular amastigotes. Highly selective for L. donovani DD8 intracellular amastigotes in relation to HEK 293 cells. |
| Psammaplysin F (14) | - | - | - | 5.63 ± 0.76 (>3.51) a (>1.90) c | Moderate Selectivity for T. cruzi in relation to HEK-293 cells. |
| Chelerythrine chloride (15) | - | - | 0.23 ± 0.04 (>18.71) a | - | Only selective on T. b. brucei. Highly selective for T. b. brucei in relation to HEK-293 cells. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zulfiqar, B.; Jones, A.J.; Sykes, M.L.; Shelper, T.B.; Davis, R.A.; Avery, V.M. Screening a Natural Product-Based Library against Kinetoplastid Parasites. Molecules 2017, 22, 1715. https://doi.org/10.3390/molecules22101715
Zulfiqar B, Jones AJ, Sykes ML, Shelper TB, Davis RA, Avery VM. Screening a Natural Product-Based Library against Kinetoplastid Parasites. Molecules. 2017; 22(10):1715. https://doi.org/10.3390/molecules22101715
Chicago/Turabian StyleZulfiqar, Bilal, Amy J. Jones, Melissa L. Sykes, Todd B. Shelper, Rohan A. Davis, and Vicky M. Avery. 2017. "Screening a Natural Product-Based Library against Kinetoplastid Parasites" Molecules 22, no. 10: 1715. https://doi.org/10.3390/molecules22101715
APA StyleZulfiqar, B., Jones, A. J., Sykes, M. L., Shelper, T. B., Davis, R. A., & Avery, V. M. (2017). Screening a Natural Product-Based Library against Kinetoplastid Parasites. Molecules, 22(10), 1715. https://doi.org/10.3390/molecules22101715