Electrostatic Self-Assembled Chitosan-Pectin Nano- and Microparticles for Insulin Delivery



 and
and
Abstract
:1. Introduction
2. Results and Discussion
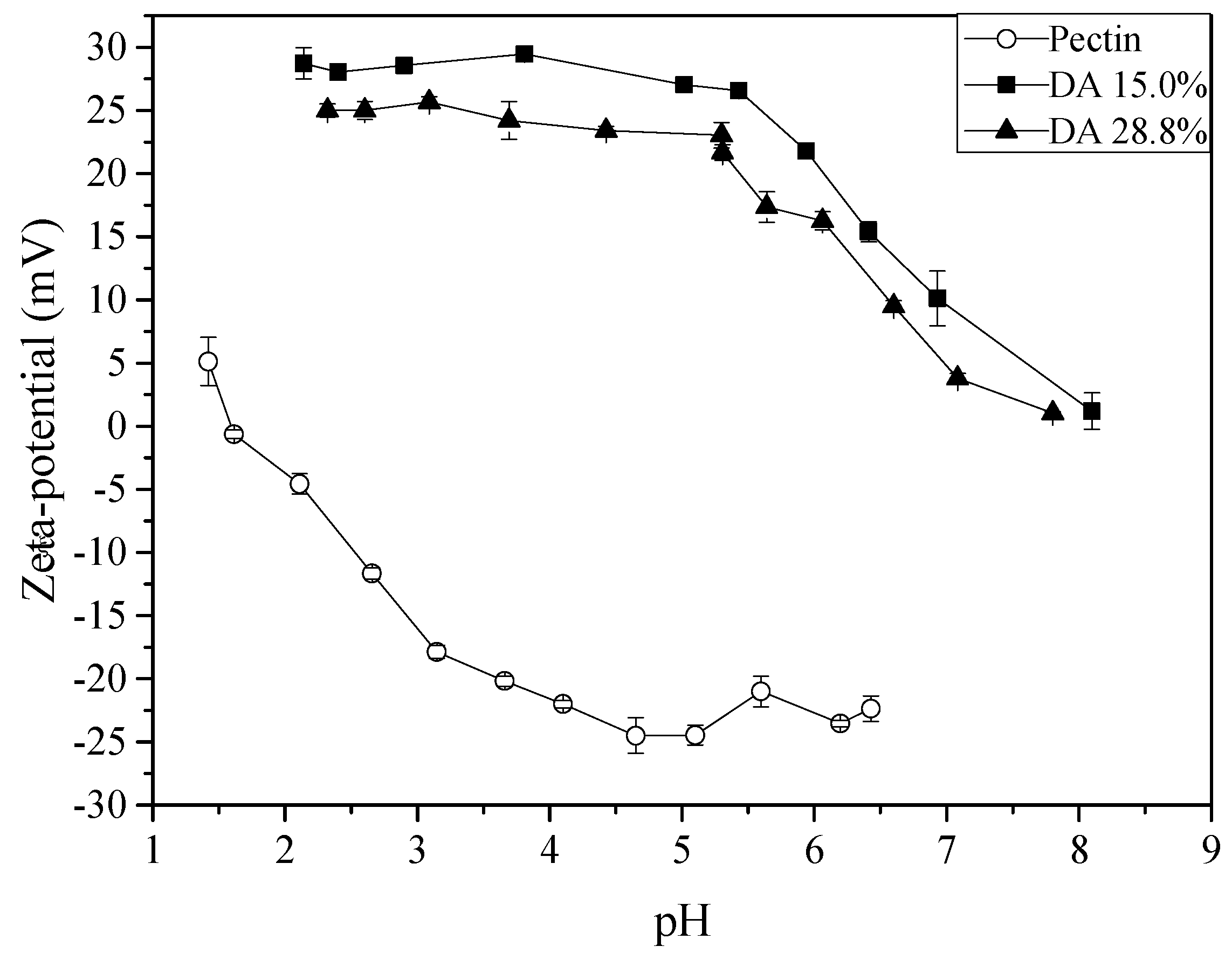
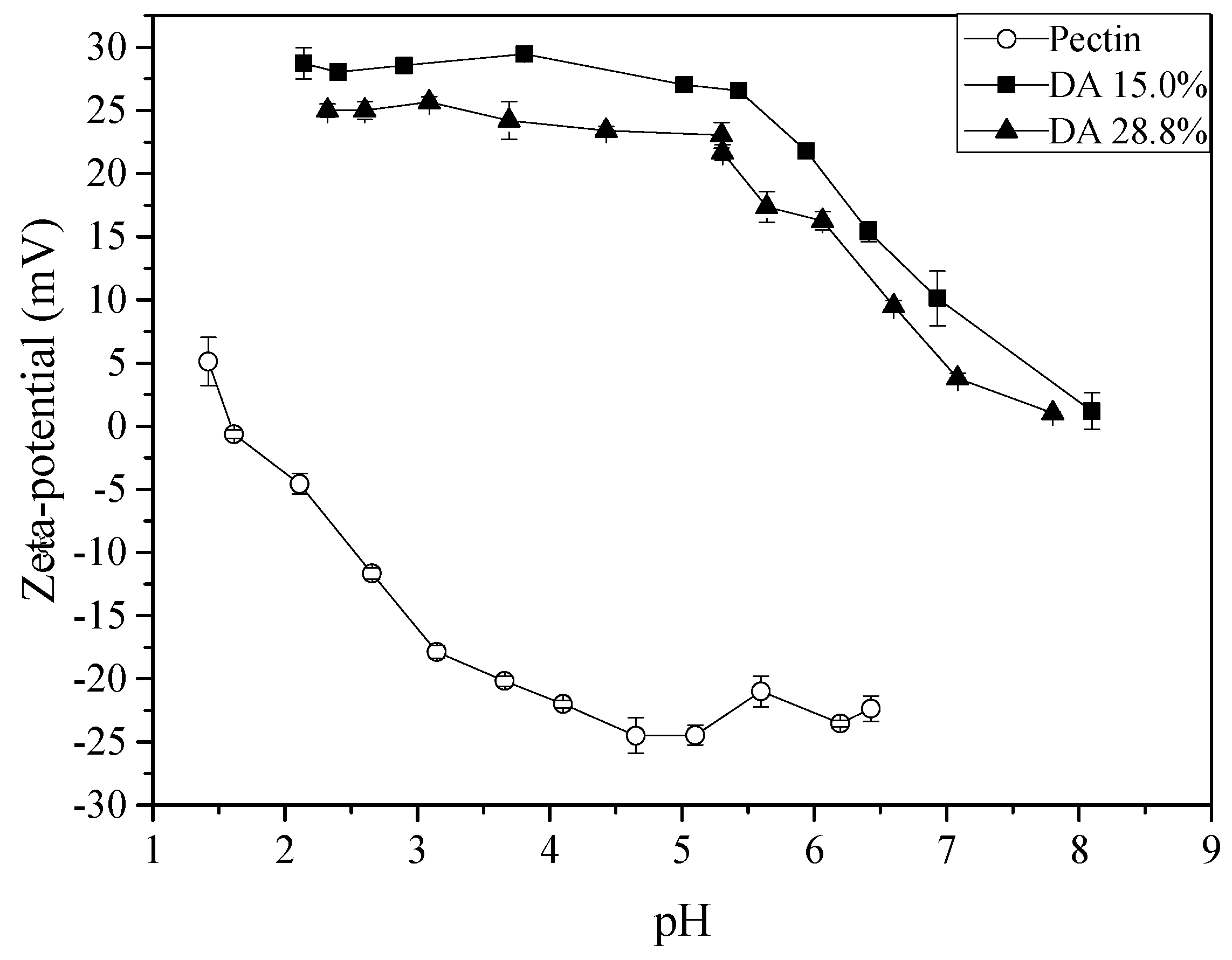
2.1. Chitosan and Pectin Characterisation
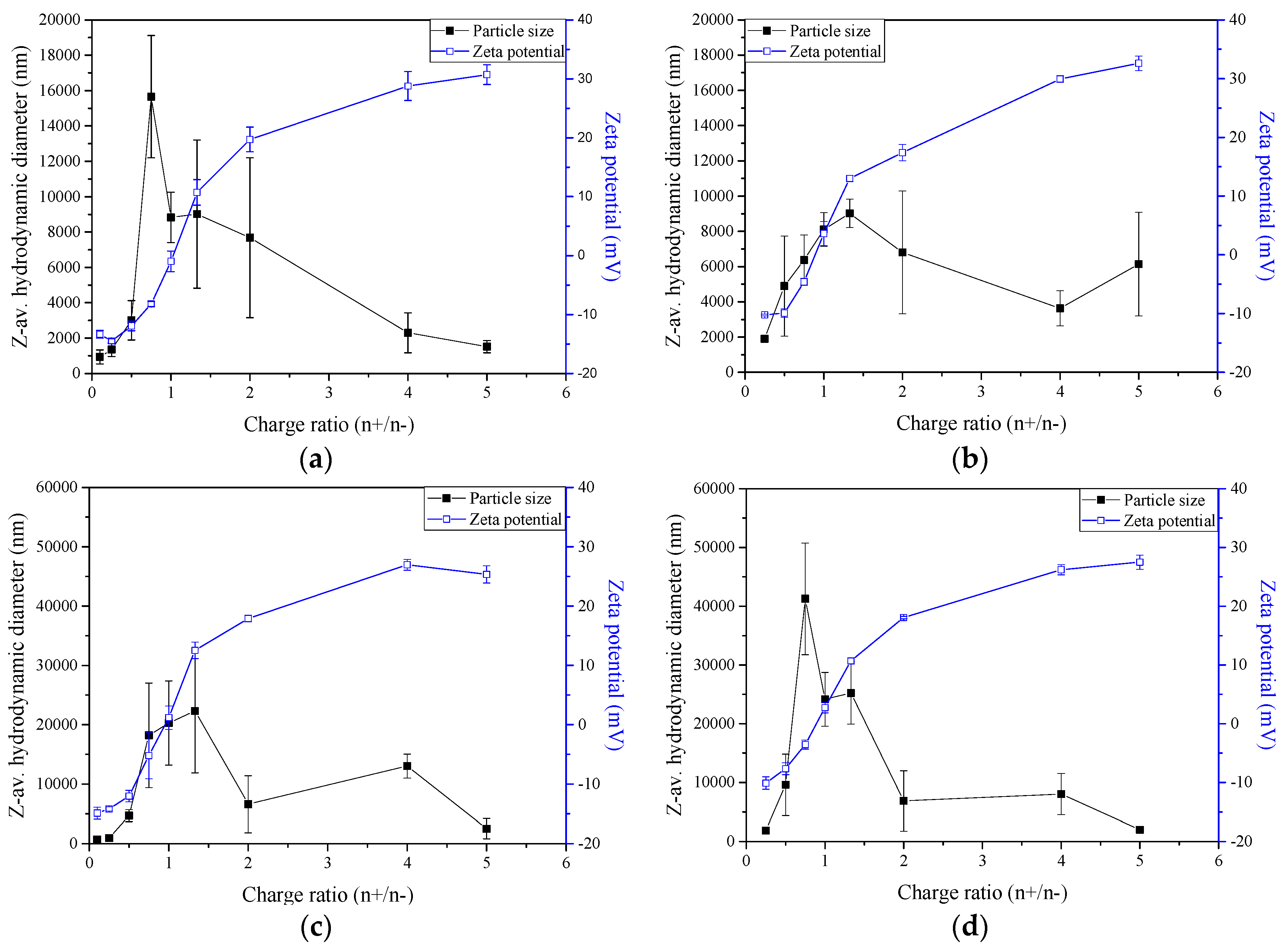
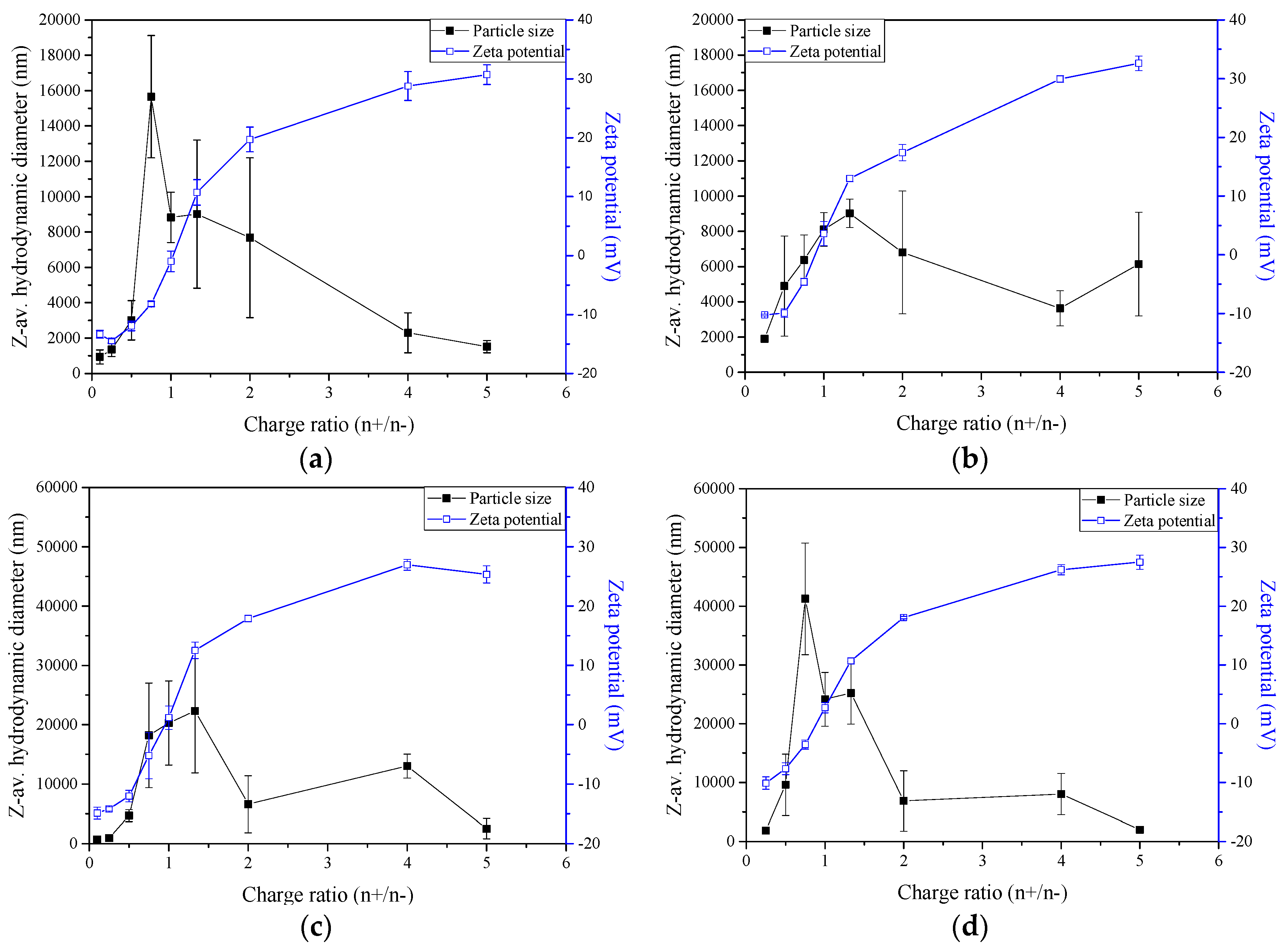
2.2. Nano- and Microparticles Characterisation
2.3. Zeta-Potential and Particle Size Distribution Analysis
2.4. Production Yield of Nano- and Microparticles
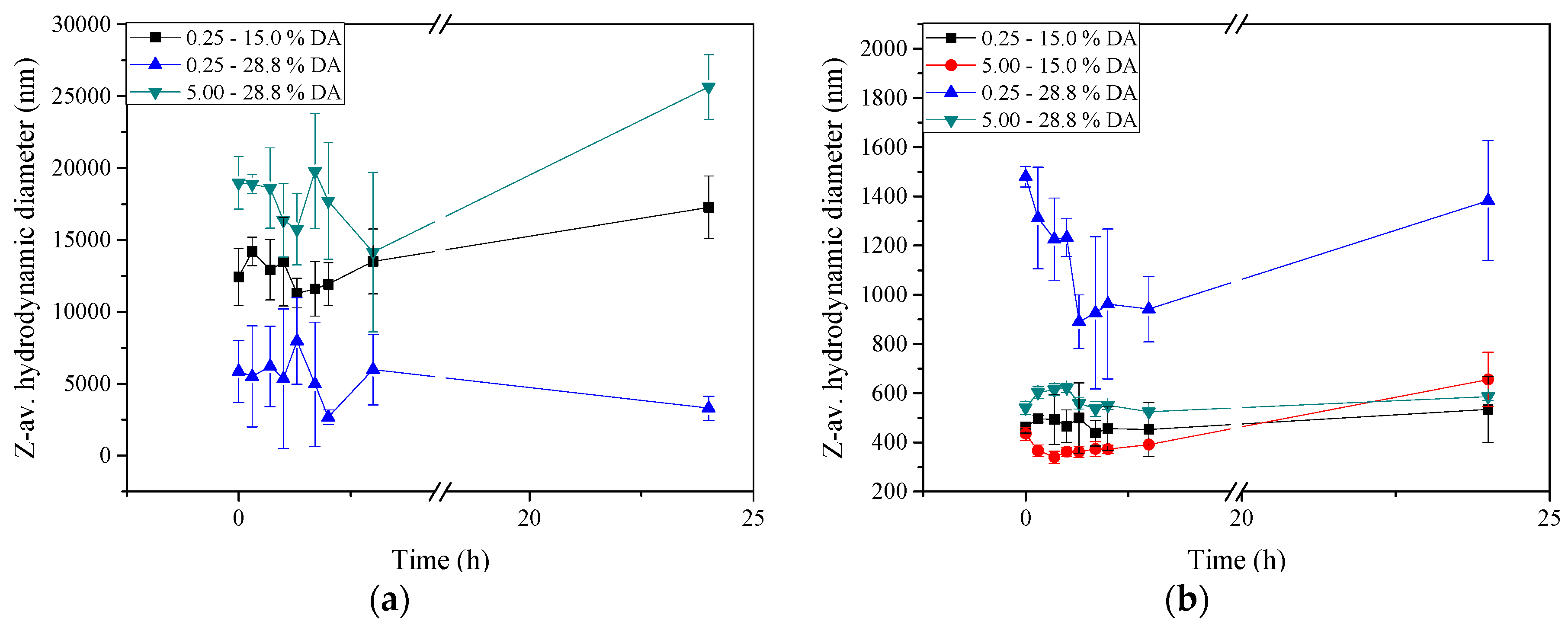
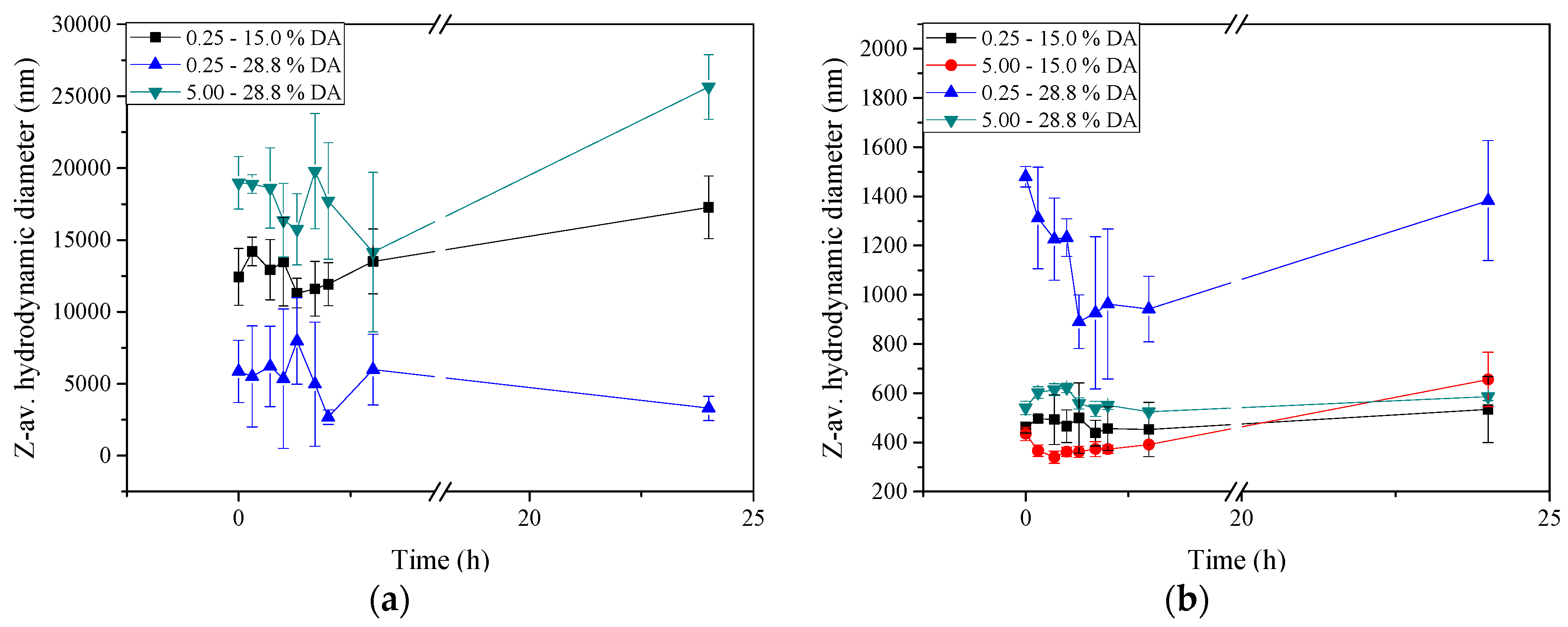
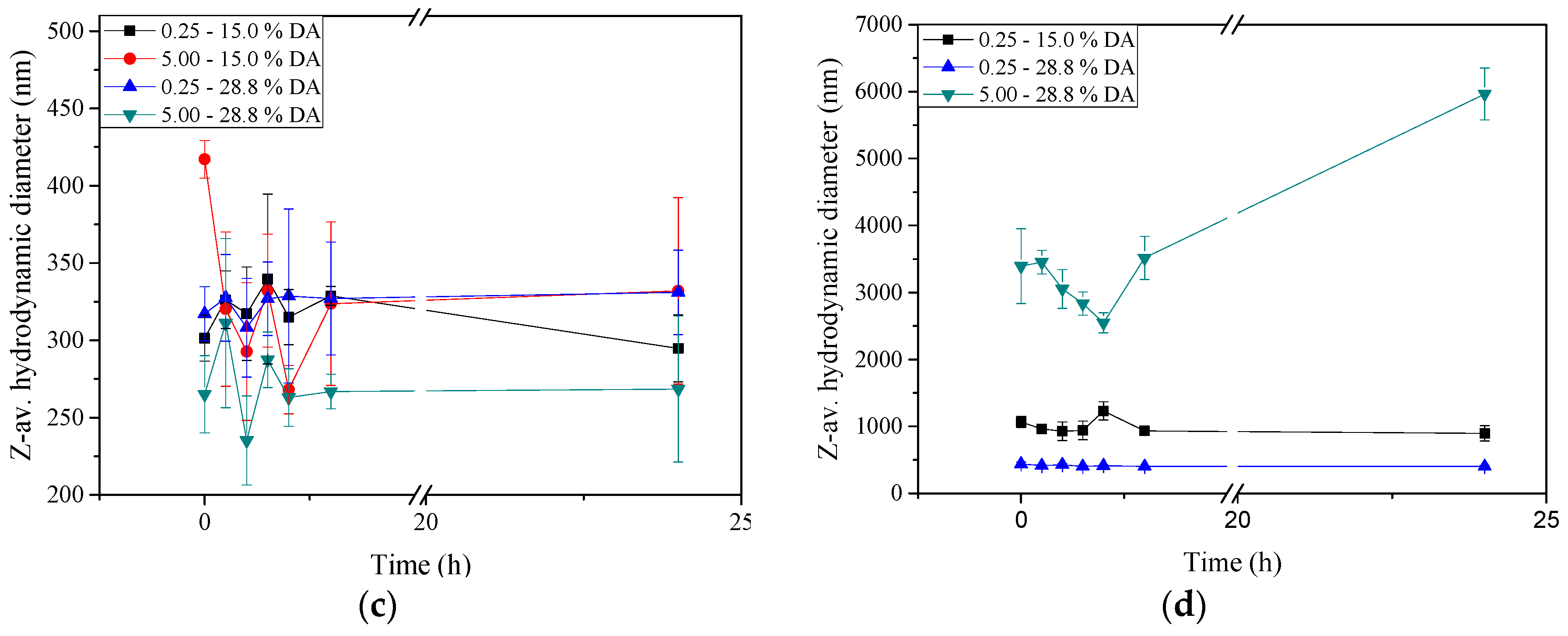
2.5. Stability of Insulin-Loaded Nano- and Microparticles
2.6. Insulin Encapsulation Efficiency (EE)
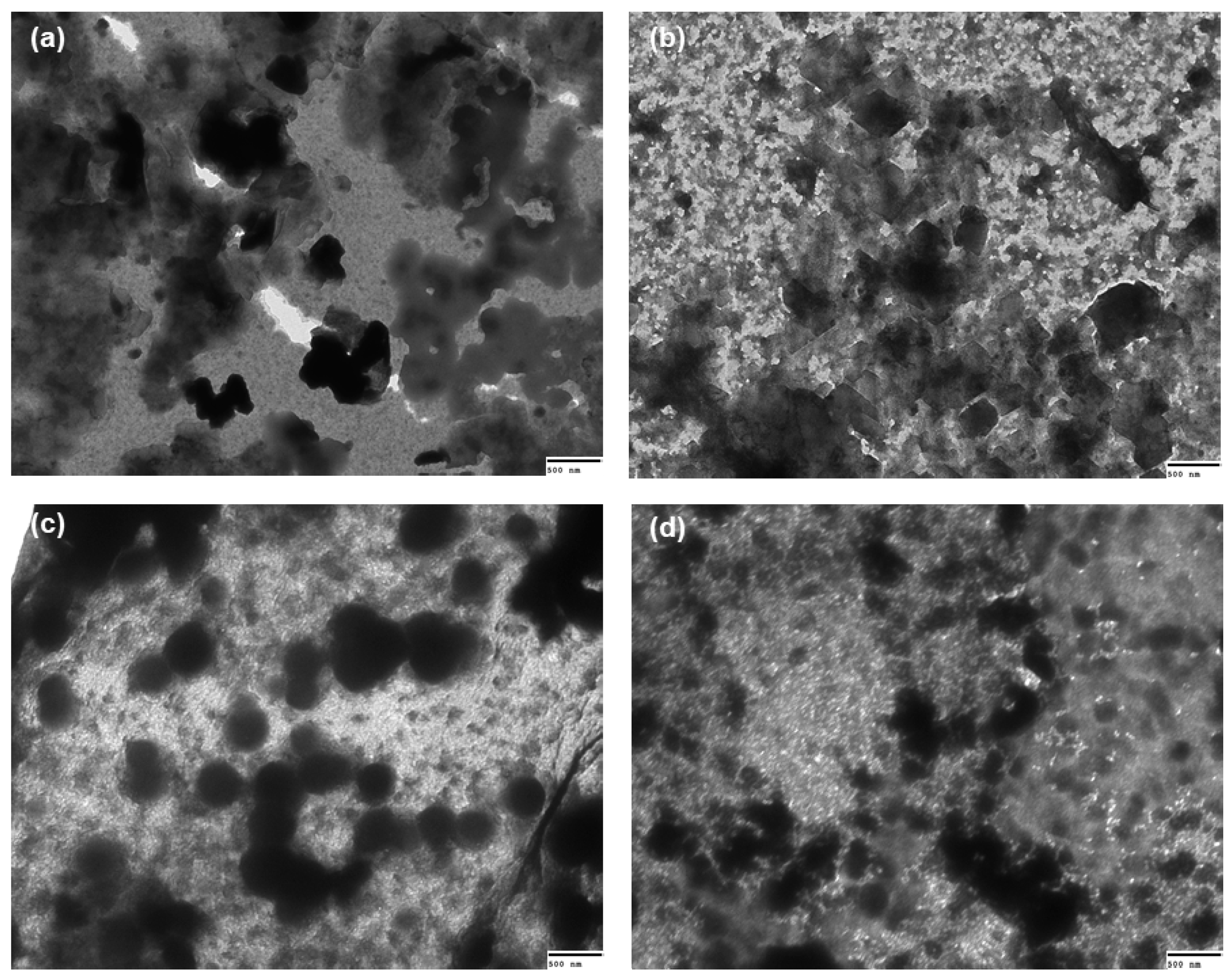
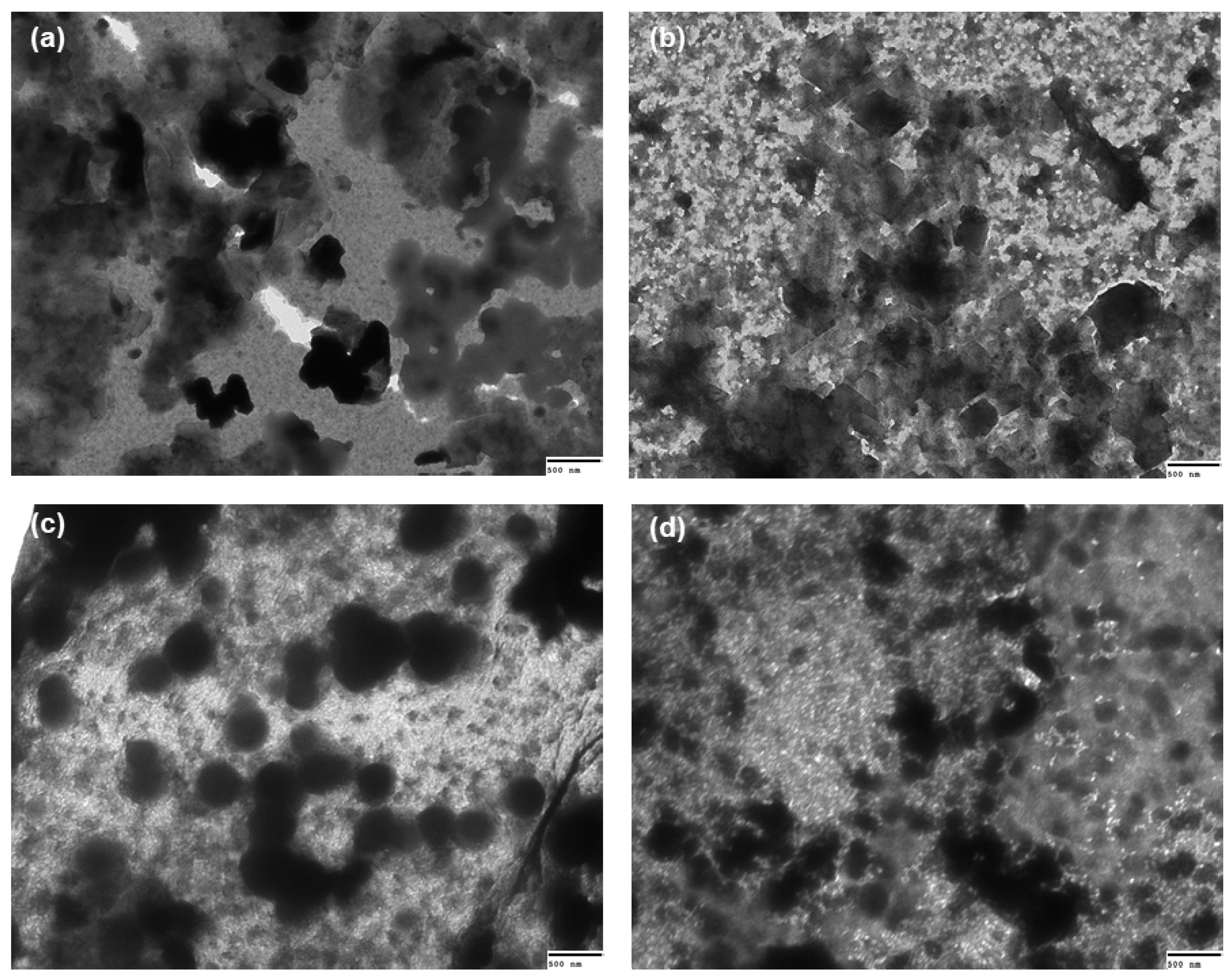
2.7. Transmission Electron Microscopy
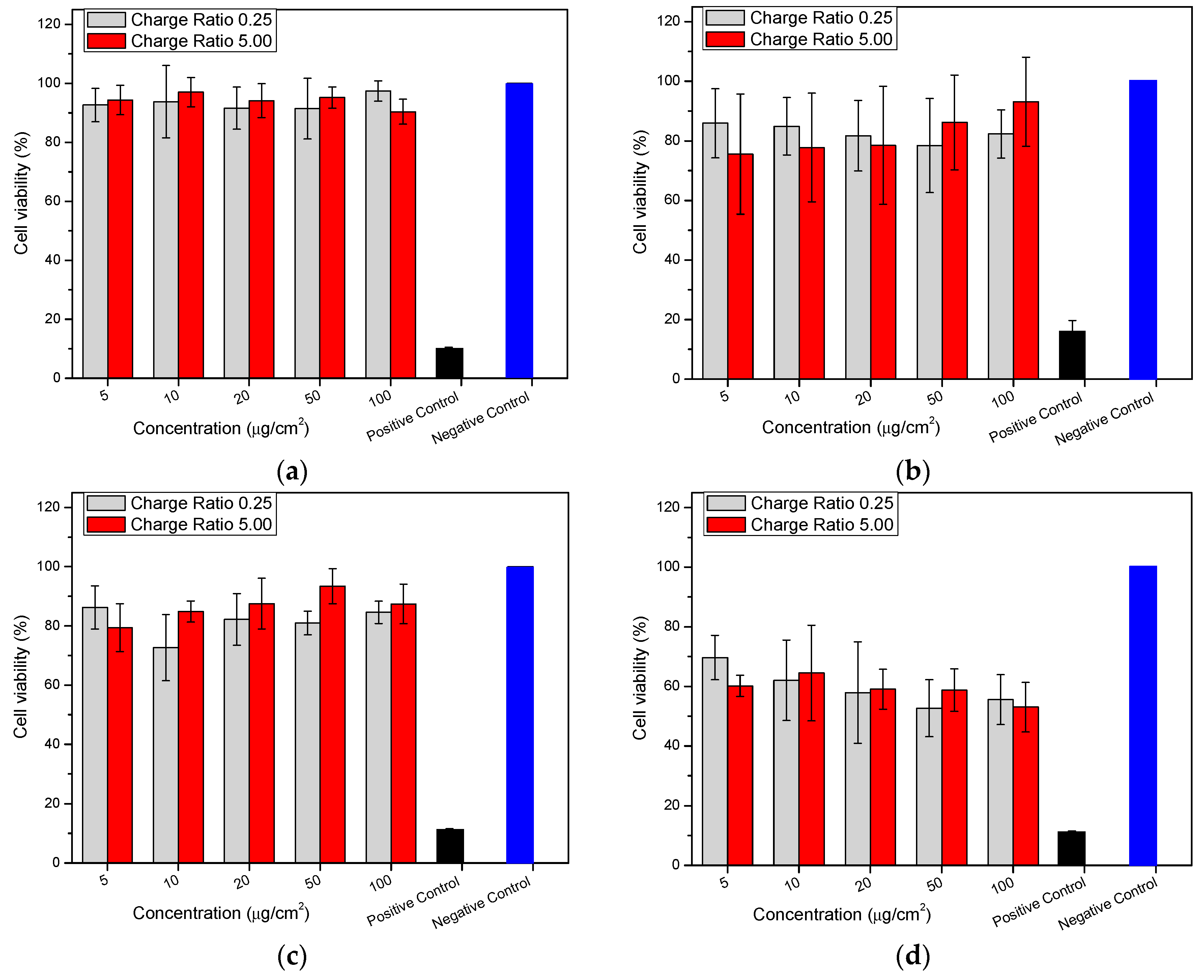
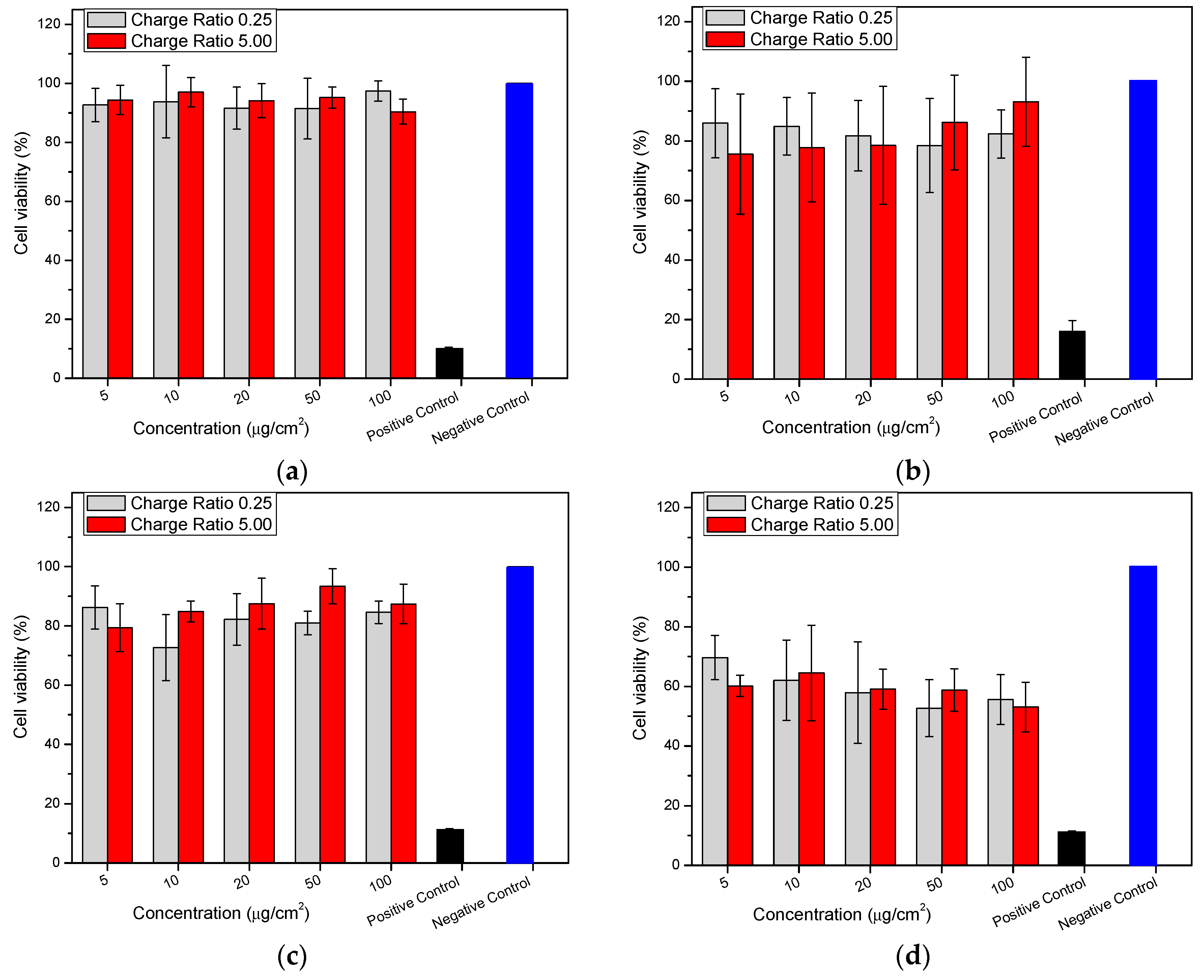
2.8. Cytotoxicity (MTT (3-(4,5-Dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) Assay)
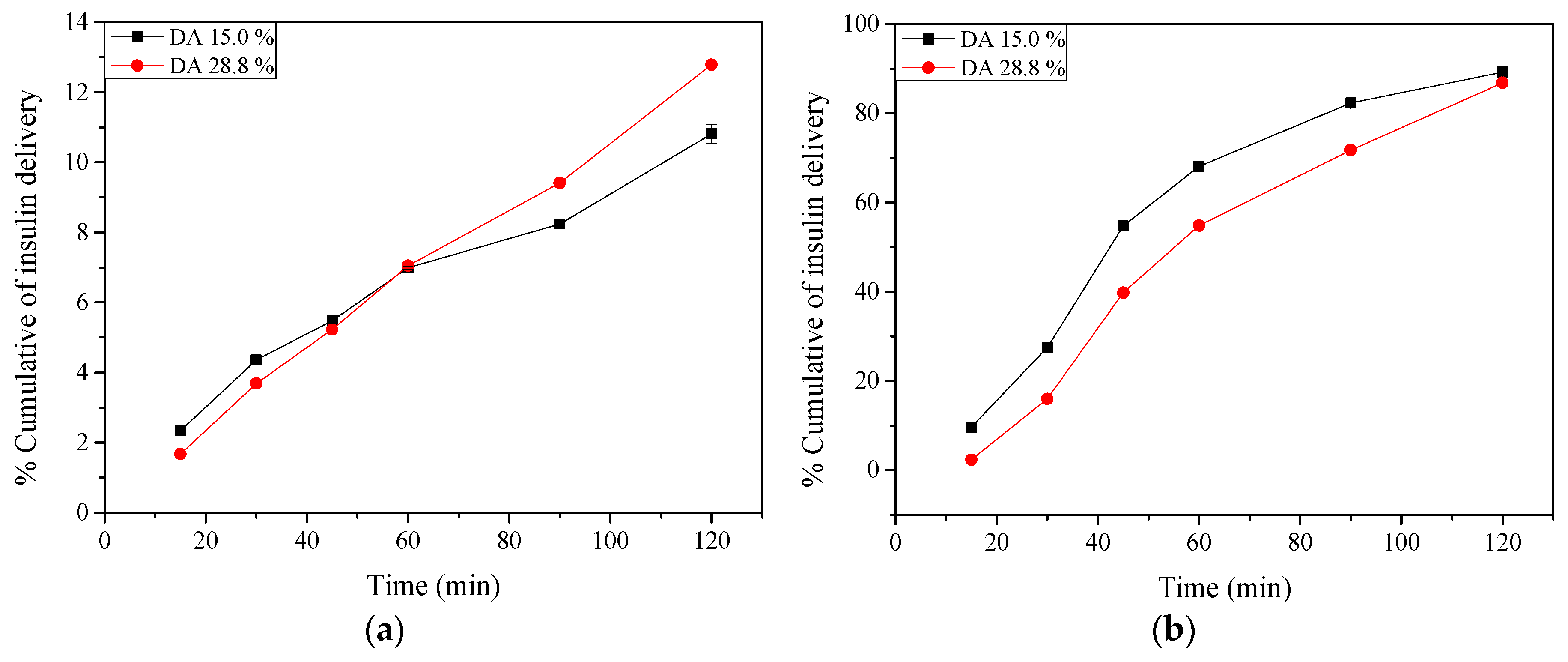
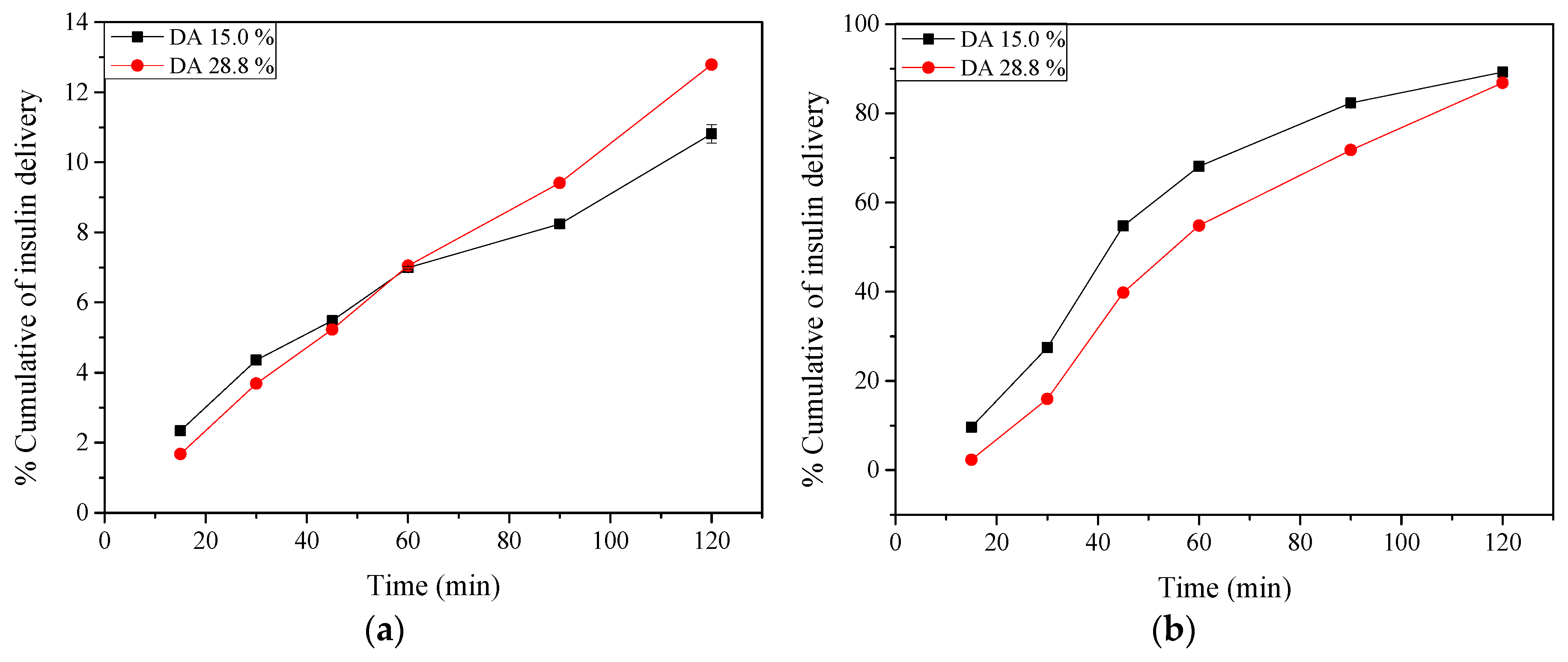
2.9. Insulin Release: In Vitro Tests
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Determination of the Degree of Acetylation of Chitosan
3.2.2. Determination of Intrinsic Viscosity ([η]) and Molar Mass Estimation of Chitosans
3.2.3. Determination of the Degree of Esterification of Pectin
3.2.4. Determination of Intrinsic Viscosity ([η]) and Molar Mass Estimation of Pectin
3.2.5. Preparation of Nano- and Microparticles
Chitosan Solutions
Pectin Solutions
Pectin Purification
3.2.6. Preparation of Nano- and Microparticles
3.2.7. Zeta-potential and Particle Size Determination of Nano- and Microparticles
3.2.8. Production Yield
3.2.9. Stability Tests
3.2.10. Insulin Encapsulation Efficiency
3.2.11. Transmission Electron Microscopy
3.2.12. Cytotoxicity (MTT Assay)
Cell Culture
MTT Assay
3.2.13. Insulin Release: In Vitro Tests
3.3. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ghaffari, A.; Navaee, K.; Oskoui, M.; Bayati, K.; Rafiee-Tehrani, M. Preparation and characterization of free mixed-film of pectin/chitosan/Eudragit® RS intended for sigmoidal drug delivery. Eur. J. Pharm. Biopharm. 2007, 67, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Rashidova, S.S.; Milusheva, R.Y.; Semenova, L.N.; Mukhamedjanova, M.Y.; Voropaeva, N.L.; Vasilyeva, S.; Faizieva, R.; Ruban, I.N. Characteristics of interactions in the pectin–chitosan system. Chromatographia 2004, 59, 779–782. [Google Scholar] [CrossRef]
- Maciel, V.B.V.; Yoshida, C.M.P.; Franco, T.T. Chitosan/pectin polyelectrolyte complex as a pH indicator. Carbohydr. Polym. 2015, 132, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Jardim, K.V.; Joanitti, G.A.; Azevedo, R.B.; Parize, A.L. Physico-chemical characterization and cytotoxicity evaluation of curcumin loaded in chitosan/chondroitin sulfate nanoparticles. Mater. Sci. Eng. C 2015, 56, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Mi, F.L.; Liao, Z.X.; Hsiao, C.W.; Sonaje, K.; Chung, M.F.; Hsu, L.W.; Sung, H.W. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv. Drug Deliv. Rev. 2013, 65, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Sonia, T.A.; Sharma, C.P. An overview of natural polymers for oral insulin delivery. Drug Discov. Today 2012, 17, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.P.; Geckeler, K.E. Polymer nanoparticles: Preparation techniques and size-control parameters. Prog. Polym. Sci. 2011, 36, 887–913. [Google Scholar] [CrossRef]
- Kreuter, J. Nanoparticles. In Colloidal Drug Delivery Systems, 1st ed.; Kreuter, J., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1994; pp. 219–342. [Google Scholar]
- Ninan, N.; Muthiah, M.; Park, I.K.; Elain, A.; Thomas, S.; Grohens, Y. Pectin/carboxymethyl cellulose/microfibrillated cellulose composite scaffolds for tissue engineering. Carbohydr. Polym. 2013, 98, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Fonte, P.; Araújo, F.; Silva, C.; Pereira, C.; Reis, S.; Santos, H.A.; Sarmento, B. Polymer-based nanoparticles for oral insulin delivery: Revisited approaches. Biotechnol. Adv. 2015, 33, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Herrero, E.P.; Alonso, M.J.; Csaba, N. Polymer-based oral peptide nanomedicines. Ther. Deliv. 2012, 3, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Alai, M.S.; Lin, W.J.; Pingale, S.S. Application of polymeric nanoparticles and micelles in insulin oral delivery. J. Food Drug Anal. 2015, 23, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, E.-S.; Morishita, M.; Onuki, Y.; Takayama, K. Current challenges in non-invasive insulin delivery systems: A comparative review. Adv. Drug Deliv. Rev. 2007, 59, 1521–1546. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Sarkar, K.; Chakraborty, M.; Bhattacharya, S.; Mishra, R.; Kundu, P.P. Oral insulin delivery by self-assembled chitosan nanoparticles: In vitro and in vivo studies in diabetic animal model. Mater. Sci. Eng. C 2013, 33, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Hosseininasab, S.; Pashaei-Asl, R.; Khandaghi, A.A.; Nasrabadi, H.T.; Nejati-Koshki, K.; Akbarzadeh, A.; Joo, S.W.; Hanifehpour, Y.; Davaran, S. Synthesis, characterization, and in vitro studies of PLGA–PEG nanoparticles for oral insulin delivery. Chem. Biol. Drug Des. 2014, 84, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Morishita, M.; Peppas, N.A. Is the oral route possible for peptide and protein drug delivery? Drug Discov. Today 2006, 11, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Al-Azi, S.O.S.M.; Tan, Y.T.F.; Wong, T.W. Transforming large molecular weight pectin and chitosan into oral protein drug nanoparticulate carrier. React. Funct. Polym. 2014, 84, 45–52. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Chakraborty, S.; Bhattacharya, S.; Mishra, R.; Kundu, P.P. pH-sensitive chitosan/alginate core-shell nanoparticles for efficient and safe oral insulin delivery. Int. J. Biol. Macromol. 2015, 72, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Andreani, T.; Miziara, L.; Lorenzon, E.N.; Souza, A.L.R.; Kiill, C.P.; Fangueiro, J.F.; Garcia, M.L.; Gremiao, P.D.; Silva, A.M.; Souto, E.B. Effect of mucoadhesive polymers on the in vitro performance of insulin-loaded silica nanoparticles: Interactions with mucin and biomembrane models. Eur. J. Pharm. Biopharm. 2015, 93, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulou, L.; Nocca, G.; Castagnola, M.; Paludetti, G.; Ortaggi, G.; Sciubba, F.; Bevilacqua, M.; Lupi, A.; Gambarini, G.; Palocci, C. Chitosan based nanoparticles functionalized with peptidomimetic derivatives for oral drug delivery. New Biotechnol. 2016, 33, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Ni, C.; Zhang, Y.; Shi, G. Preparation of polyelectrolyte complex nanoparticles of chitosan and poly(2-acry1amido-2-methylpropanesulfonic acid) for doxorubicin release. Mater. Sci. Eng. C 2016, 58, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Kleine-Brüggeney, H.; Zorzi, G.K.; El-Gueddari, N.E.; Moerschbacher, B.M.; Goycoolea, F.M. A rational approach towards the design of chitosan-based nanoparticles obtained by ionotropic gelation. Colloid Surf. B 2015, 135, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ahuja, M. Carboxymethyl gum kondagogu–chitosan polyelectrolyte complex nanoparticles: Preparation and characterization. Int. J. Biol. Macromol. 2013, 62, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, P.; Ferreira, P.; Sousa, H.C.; Batista, P.; Rodrigues, M.A.; Correia, I.J.; Gil, M.H. Preparation and chemical and biological characterization of a pectin/chitosan polyelectrolyte complex scaffold for possible bone tissue engineering applications. Int. J. Biol. Macromol. 2011, 48, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Vaarum, K.M.; Smidsrod, O. Structure-property relationship in chitosans. In Polysaccharides: Structural Diversity and Functional Versatility, 2nd ed.; Dumitriu, S., Ed.; Marcel Dekker Inc.: New York, NY, USA, 2005; pp. 625–660. [Google Scholar]
- Rinaudo, M.; Pavlov, G.; Desbrières, J. Solubilization of chitosan in strong acid medium. Int. J. Polym. Anal. Charact. 1999, 5, 267–276. [Google Scholar] [CrossRef]
- McConaughy, S.D.; Stroud, P.A.; Boudreaux, B.; Hester, R.D.; McCormick, C.L. Structural characterization and solution properties of a galacturonatepolysaccharide derived from Aloe vera capable of in situ gelation. Biomacromolecules 2008, 9, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Abodinar, A.; Smith, A.M.; Morris, G.A. A novel method to estimate the stiffness of carbohydrate polyelectrolyte polymers based on the ionic strength dependence of zeta potential. Carbohydr. Polym. 2014, 112, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Rolin, C. Commercial pectin preparations. In Pectins and their manipulation, 1st ed.; Seymour, C., Knox, P., Eds.; CRC Press: Boca Raton, FL, USA, 2002; pp. 222–241. [Google Scholar]
- Giancone, T.; Torrieri, E.; Masi, P.; Michon, C. Protein–polysaccharide interactions: Phase behaviour of pectin–soy flour mixture. Food Hydrocoll. 2009, 23, 1263–1269. [Google Scholar] [CrossRef]
- Goycoolea, F.M.; Brunel, F.; Gueddari, N.E.; Coggiola, A.; Lollo, G.; Moerschbacher, B.M.; Remuñán-López, C.; Delair, T.; Domard, A.; Alonso, M.J. Physical Properties and Stability of Soft Gelled Chitosan-Based Nanoparticles. Macromol. Biosci. 2016, 16, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Argüelles-Monal, W.; Cabrera, G.; Peniche, C.; Rinaudo, M. Conductimetric study of the interpolyelectrolyte reaction between chitosan and polygalacturonic acid. Polym. 2000, 41, 2373–2378. [Google Scholar] [CrossRef]
- Fábregas, A.; Miñarro, M.; García-Montoya, E.; Pérez-Lozano, P.; Carrillo, C.; Sarrate, R.; Sánchez, N.; Ticó, J.R.; Suné-Negre, J.M. Impact of physical parameters on particle size and reaction yield when using the ionic gelation method to obtain cationic polymeric chitosan–tripolyphosphate nanoparticles. Int. J. Pharm. 2013, 446, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, Y. Preparation, characterization and evaluation of tea polyphenole-Zn complex loaded β-chitosan nanoparticles. Food Hydrocoll. 2015, 48, 260–273. [Google Scholar] [CrossRef]
- Cerchiara, T.; Abruzzo, A.; Di Cagno, M.; Bigucci, F.; Bauer-Brandl, A.; Parolin, C.; Vitali, B.; Gallucci, M.C.; Luppi, B. Chitosan based micro- and nanoparticles for colon-targeted delivery of vancomycin prepared by alternative processing methods. Eur. J. Pharm. Biopharm. 2015, 92, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Woitiski, C.B.; Carvalho, R.A.; Ribeiro, A.J.; Neufeld, R.J.; Veiga, F. Strategies toward the improved oral delivery of insulin nanoparticles via gastrointestinal uptake and translocation. BioDrugs 2015, 22, 223–237. [Google Scholar] [CrossRef]
- Soliman, G.M.; Zhang, Y.L.; Merle, G.; Cerruti, M.; Barralet, J. Hydrocaffeic acid–chitosan nanoparticles with enhanced stability, mucoadhesion and permeation properties. Eur. J. Pharm. Biopharm. 2014, 88, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Gao, H.; Li, C.; Yang, Y.W.; Wang, Y.; Fan, Y.; Wu, G.; Ma, J. Polyelectrolyte complex nanoparticles of amino poly(glycerol methacrylate)s and insulin. Int. J. Pharm. 2012, 423, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Goycoolea, F.M.; Valle-Gallego, A.; Stefani, R.; Menchicchi, B.; David, L.; Rochas, C.; Santander-Ortega, M.J.; Alonso, M.J. Chitosan-based nanocapsules: Physical characterization stability in biological media and capsaicin encapsulation. Colloid Polym. Sci. 2012, 290, 1423–1434. [Google Scholar] [CrossRef]
- Russo, E.; Gaglianone, N.; Baldassari, S.; Parodi, B.; Cafaggi, S.; Zibana, C.; Donalisio, M.; Cagno, V.; Lembo, D.; Caviglioli, G. Preparation, characterization and in vitro antiviral activity evaluation of foscarnet-chitosan nanoparticles. Colloid Surf. B 2014, 118, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Bagre, A.O.; Jain, K.; Jain, N.K. Alginate coated chitosan core shell nanoparticles for oral delivery of enoxaparin: In vitro and in vivo assessment. Int. J. Pharm. 2013, 456, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Teng, Z.; Li, Y.; Wang, Q. Solid lipid nanoparticles for oral drug delivery: Chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr. Polym. 2015, 122, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Ubaidulla, U.; Khar, R.K.; Ahmad, F.J.; Sultana, Y.; Panda, A.K. Development and characterization of chitosan succinate microspheres for the improved oral bioavailability of insulin. J. Pharm. Sci. 2007, 96, 3010–3023. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Dorkoosh, F.A.; Dehpour, A.R.; Moezi, L.; Larijani, B.; Junginger, H.E.; Rafiee-Tehrani, M. Nanoparticles of quaternized chitosan derivatives as a carrier for colon delivery of insulin: Ex vivo and in vivo studies. Int. J. Pharm. 2008, 356, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Calvo, P.; Remunan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Goycoolea, F.M.; Lollo, G.; Remuñán-López, C.; Quaglia, F.; Alonso, M.J. Chitosan–alginate blended nanoparticles as carriers for transmucosal delivery of macromolecules. Biomacromolecules 2009, 10, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, Y.; Zhao, H.; Zheng, J.M.; Xu, H.; Wei, G.; Hao, J.; Cui, F. Bioadhesive polysaccharide in protein delivery system: Chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int. J. Pharm. 2002, 249, 139–147. [Google Scholar] [CrossRef]
- Loh, J.W.; Saunders, M.; Lim, L.Y. Cytotoxicity of monodispersed chitosan nanoparticles against the Caco-2 cells. Toxicol. Appl. Pharm. 2012, 262, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Adv. Drug Deliv. Rev. 2015, 57, 2215–2237. [Google Scholar] [CrossRef] [PubMed]
- Vihola, H.; Laukkanen, A.; Valtola, L.; Tenhu, H.; Hirvonen, J. Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam). Biomaterials 2005, 26, 3055–3064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, M.; Zheng, A.; Cao, D.; Bi, Y.; Sun, J. Preparation and characterization of insulin-loaded bioadhesive PLGA nanoparticles for oral administration. Eur. J. Pharm. Sci. 2012, 45, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Juan, C.; Perez-Blas, M.; Valeri, A.P.; Aguilera, N.; Arnaiz-Villena, A.; Pacheco-Castro, A.; Martin-Villa, J.M. Cell surface phenotype and cytokine secretion in Caco-2 cell cultures: Increased RANTES production and IL-2 transcription upon stimulation with IL-1β. Tissue Cell 2001, 33, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.; Creed, E.; Brayden, D.; Baird, A.W. Evaluation of the Caco-2 monolayer as a model epithelium for iontophoretic transport. Pharma. Res. 2000, 17, 1181–1188. [Google Scholar] [CrossRef]
- Loretz, B.; Bernkop-Schnurch, A. In vitro cytotoxicty testing of non-thiolated and thiolated chitosan nanoparticles for oral gene delivery. Nanotoxicology 2007, 1, 139–148. [Google Scholar] [CrossRef]
- Biswas, A.; Chattopadhyay, M.; Sen, K.K.; Saha, M.K. Development and characterization of alginate coated low molecular weight chitosan nanoparticles as new carriers for oral vaccine delivery in mice. Carbohydr. Polym. 2015, 121, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Chen, X.; Xu, Y.; Han, X.; Xu, Z. Tracing transport of chitosan nanoparticles and molecules in Caco-2 cells by fluorescent labelling. Carbohydr. Polym. 2009, 78, 323–329. [Google Scholar] [CrossRef]
- Sadeghi, A.M.M.; Dorkoosh, F.A.; Avadi, M.R.; Weinhold, M.; Bayat, A.; Delie, F.; Gurny, R.; Larijani, B.; Rafiee-Tehrani, M.; Junginger, H.E. Permeation enhancer effect of chitosan and chitosan derivatives: Comparison of formulations as soluble polymers and nanoparticulate systems on insulin absorption in Caco-2 cells. Eur. J. Pharm. Biopharm. 2008, 70, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Avadi, M.R.; Sadeghi, A.M.M.; Mohammadpour, N.; Abedin, S.; Atyabi, F.; Dinarvand, R.; Rafiee-Tehrani, M. Preparation and characterization of insulin nanoparticles using chitosan and Arabic gum with ionic gelation method. Nanomedicine 2010, 6, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Lavertu, M.; Xia, Z.; Serreqi, A.N.; Berrada, M.; Rodrigues, A.; Wang, D.; Buschmann, M.D.; Gupta, A. A validated 1H-NMR method for the determination of the degree of deacetylation of chitosan. J. Pharm. Biomed. Anal. 2003, 32, 1149–1158. [Google Scholar] [CrossRef]
- Rinaudo, M.; Milas, M.; Le Dung, P. Characterization of chitosan. Influence of ionic strength and degree of acetylation on chain expansion. Int. J. Biol. Macromol. 1993, 15, 281–285. [Google Scholar] [CrossRef]
- Rosenbohm, C.; Lundt, I.; Christensen, T.M.I.E.; Young, N.W.G. Chemically methylated and reduced pectins: Preparation, characterisation by 1H-NMR spectroscopy, enzymatic degradation, and gelling properties. Carbohydr. Res. 2003, 338, 637–649. [Google Scholar] [CrossRef]
- Anger, H.; Berth, G. Gel permeation chromatography and the Mark-Houwink relation for pectins with different degrees of esterification. Carbohydr. Polym. 1986, 6, 193–202. [Google Scholar] [CrossRef]
- Bernabé, P.; Peniche, C.; Argüelles-Monal, W. Swelling behavior of chitosan/pectin polyelectrolyte complex membranes. Effect of thermal cross-linking. Polym. Bull. 2005, 55, 367–375. [Google Scholar] [CrossRef]
- Fuenzalida, J.P.; Weikert, T.; Hoffmann, S.; Vila-Sanjurjo, C.; Moerschbacher, B.M.; Goycoolea, F.M.; Kolkenbrock, S. Affinity protein-based fret tools for cellular tracking of chitosan nanoparticles and determination of the polymer degree of acetylation. Biomacromolecules 2014, 15, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.; Di Gioia, S.; Ditaranto, N.; Cioffic, N.; Goycoolea, F.M.; Carbone, A.; Garcia-Fuentes, M.; Conese, M.; Alonso, M.J. Systemic heparin delivery by the pulmonary route using chitosan and glycol chitosan nanoparticles. Int. J. Pharm. 2013, 447, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Marschütz, M.K.; Bernkop-Schnürch, A. Oral peptide drug delivery: Polymer-inhibitor conjugates protecting insulin from enzymatic degradation in vitro. Biomaterials 2000, 21, 1499–1507. [Google Scholar] [CrossRef]
- Krauland, A.H.; Alonso, M.J. Chitosan/cyclodextrin nanoparticles as macromolecular drug delivery system. Int. J. Pharm. 2007, 340, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, N.; Garrigue, J.S.; Razafindratsita, A.; Lambert, G.; Benita, S. Excipient effects on in vitro cytotoxicity of a novel paclitaxel selfemulsifying drug delivery system. J. Pharm. Sci. 2003, 92, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis | Insulin | Chitosan DA 15.0% | Chitosan DA 28.8% | ||
|---|---|---|---|---|---|
| Charge Ratio (n+/n−) | Charge Ratio (n+/n−) | ||||
| 0.25 | 5.00 | 0.25 | 5.00 | ||
| Particles size (d, nm) | Blank | 2530 ± 384 aA | 2618 ± 175 aA | 1875 ± 135 aA | 2011 ± 266 aA |
| Loaded | 1351 ± 384 aB | 1522 ± 346 aB | 964 ± 32 aB | 2510 ± 107 bB | |
| ζ-potential (mV) | Blank | −22.5 ± 0.8 aA | +35.0 ± 1.4 bA | −23.4 ± 0.9 aA | +27.2 ± 1.4 bA |
| Loaded | −22.5 ± 2.5 aA | +33.2 ± 2.3 bA | −22.4 ± 2.4 aA | +28.6 ± 2.0 bA | |
| Production yield (%) | Blank | 23.8 ± 1.1 aA | 18.9 ± 3.0 bA | 22.6 ± 0.6 aA | 23.7 ± 3.3 aA |
| Loaded | 27.2 ± 3.9 aA | 33.8 ± 4.3 aB | 24.3 ± 2.0 aA | 22.4 ± 2.0 aA | |
| Formulation | Charge Ratio (n+/n−) | DA (%) | EE (%) |
|---|---|---|---|
| 1 | 0.25 | 15.0 | 36.6 ± 6.6 a |
| 2 | 5.00 | 15.0 | 62.2 ± 3.1 b |
| 3 | 0.25 | 28.8 | 34.2 ± 8.1 a |
| 4 | 5.00 | 28.8 | 61.9 ± 0.5 b |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciel, V.B.V.; Yoshida, C.M.P.; Pereira, S.M.S.S.; Goycoolea, F.M.; Franco, T.T. Electrostatic Self-Assembled Chitosan-Pectin Nano- and Microparticles for Insulin Delivery. Molecules 2017, 22, 1707. https://doi.org/10.3390/molecules22101707
Maciel VBV, Yoshida CMP, Pereira SMSS, Goycoolea FM, Franco TT. Electrostatic Self-Assembled Chitosan-Pectin Nano- and Microparticles for Insulin Delivery. Molecules. 2017; 22(10):1707. https://doi.org/10.3390/molecules22101707
Chicago/Turabian StyleMaciel, Vinicius B. V., Cristiana M. P. Yoshida, Susana M. S. S. Pereira, Francisco M. Goycoolea, and Telma T. Franco. 2017. "Electrostatic Self-Assembled Chitosan-Pectin Nano- and Microparticles for Insulin Delivery" Molecules 22, no. 10: 1707. https://doi.org/10.3390/molecules22101707
APA StyleMaciel, V. B. V., Yoshida, C. M. P., Pereira, S. M. S. S., Goycoolea, F. M., & Franco, T. T. (2017). Electrostatic Self-Assembled Chitosan-Pectin Nano- and Microparticles for Insulin Delivery. Molecules, 22(10), 1707. https://doi.org/10.3390/molecules22101707