Synthesis of 11C-Labelled Ureas by Palladium(II)-Mediated Oxidative Carbonylation

 , , ,
, , ,
Abstract
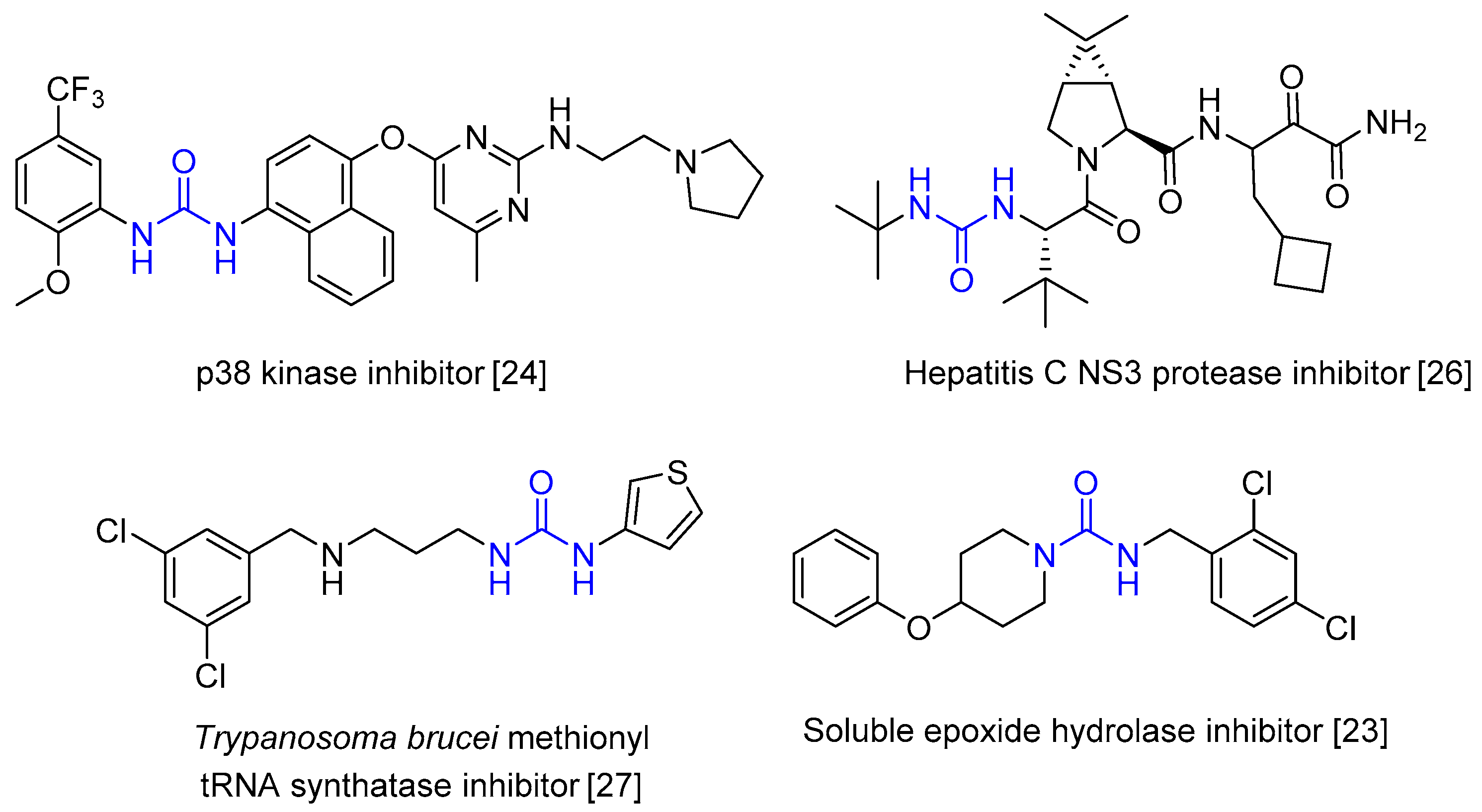
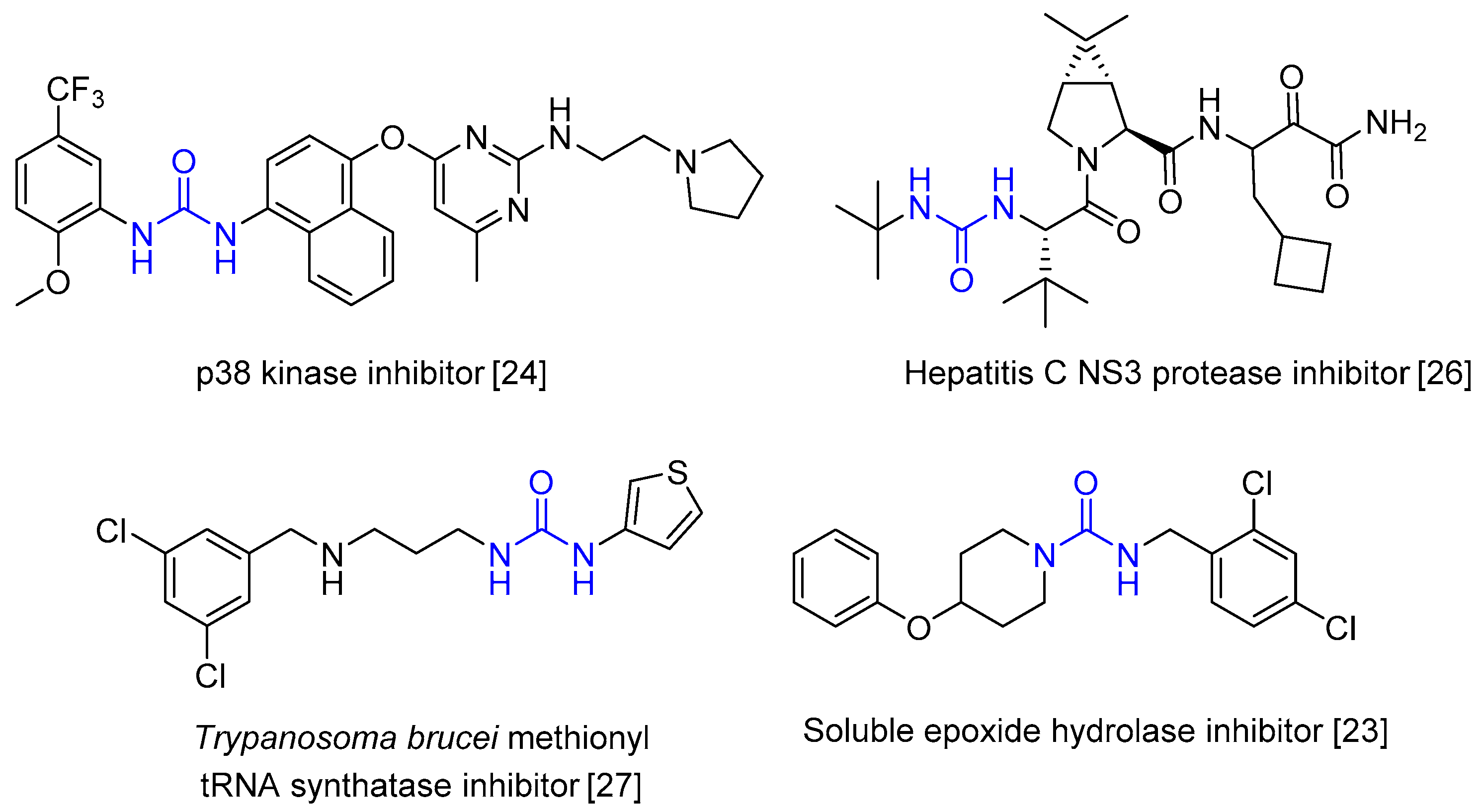
:1. Introduction
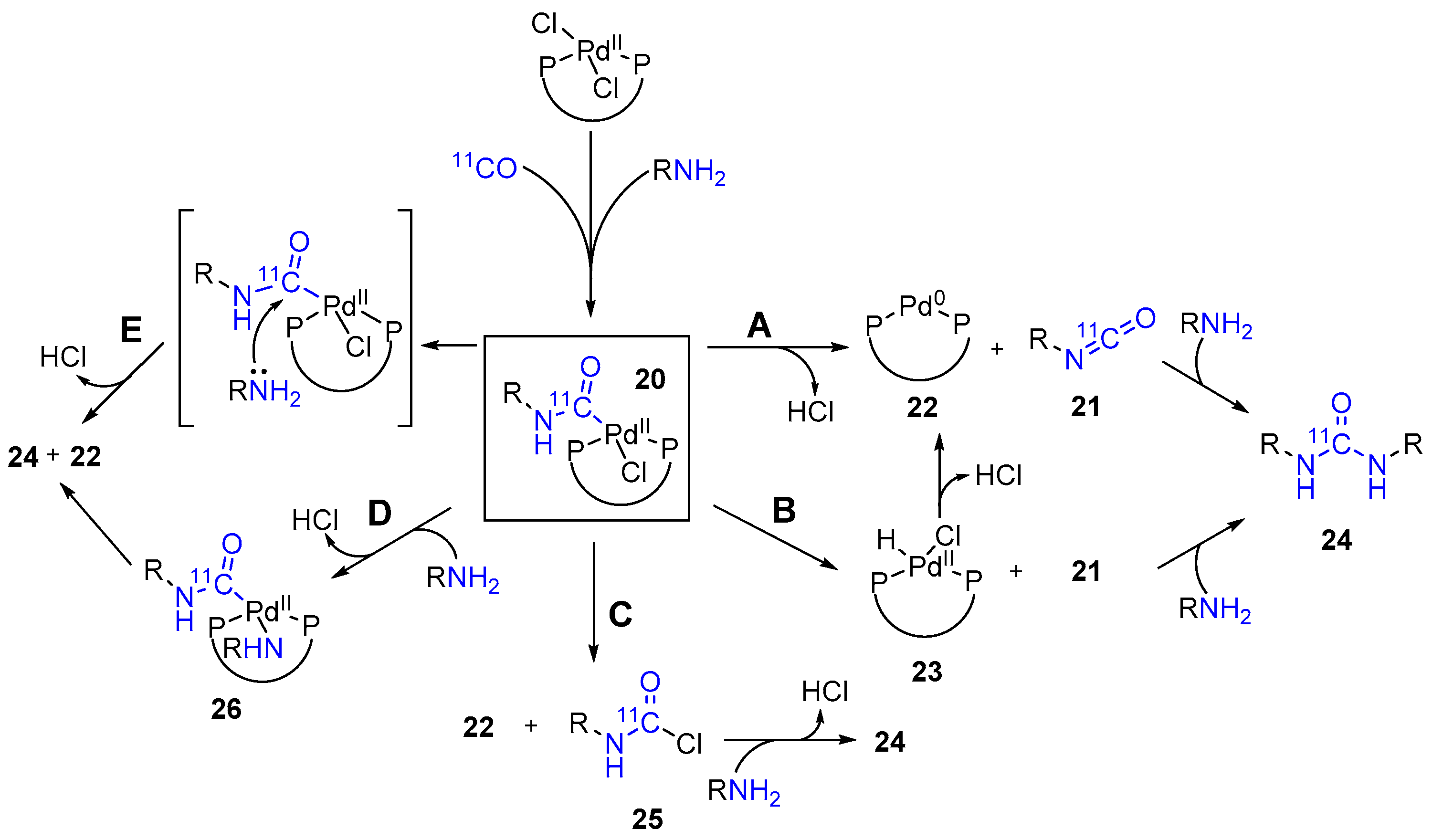
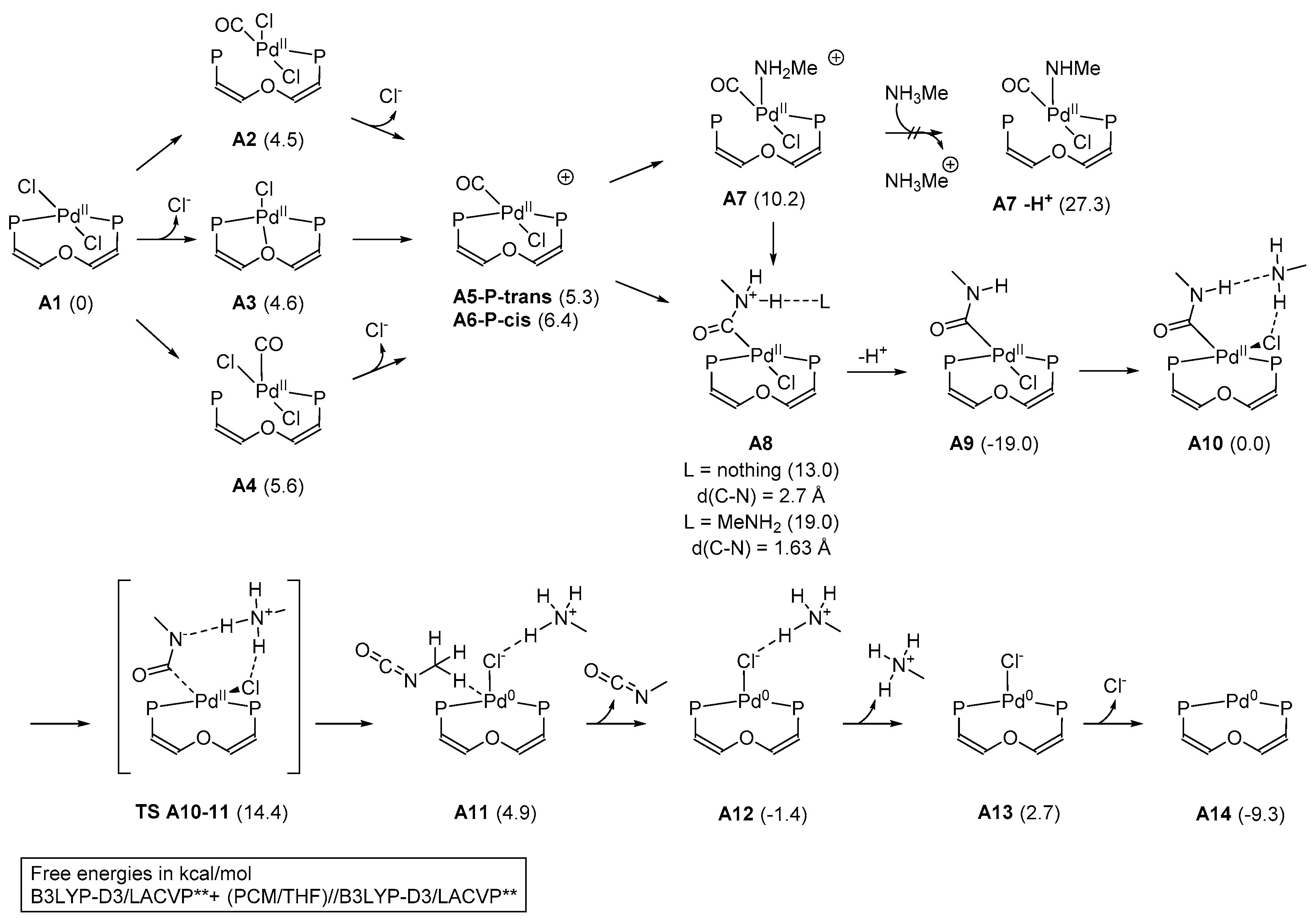
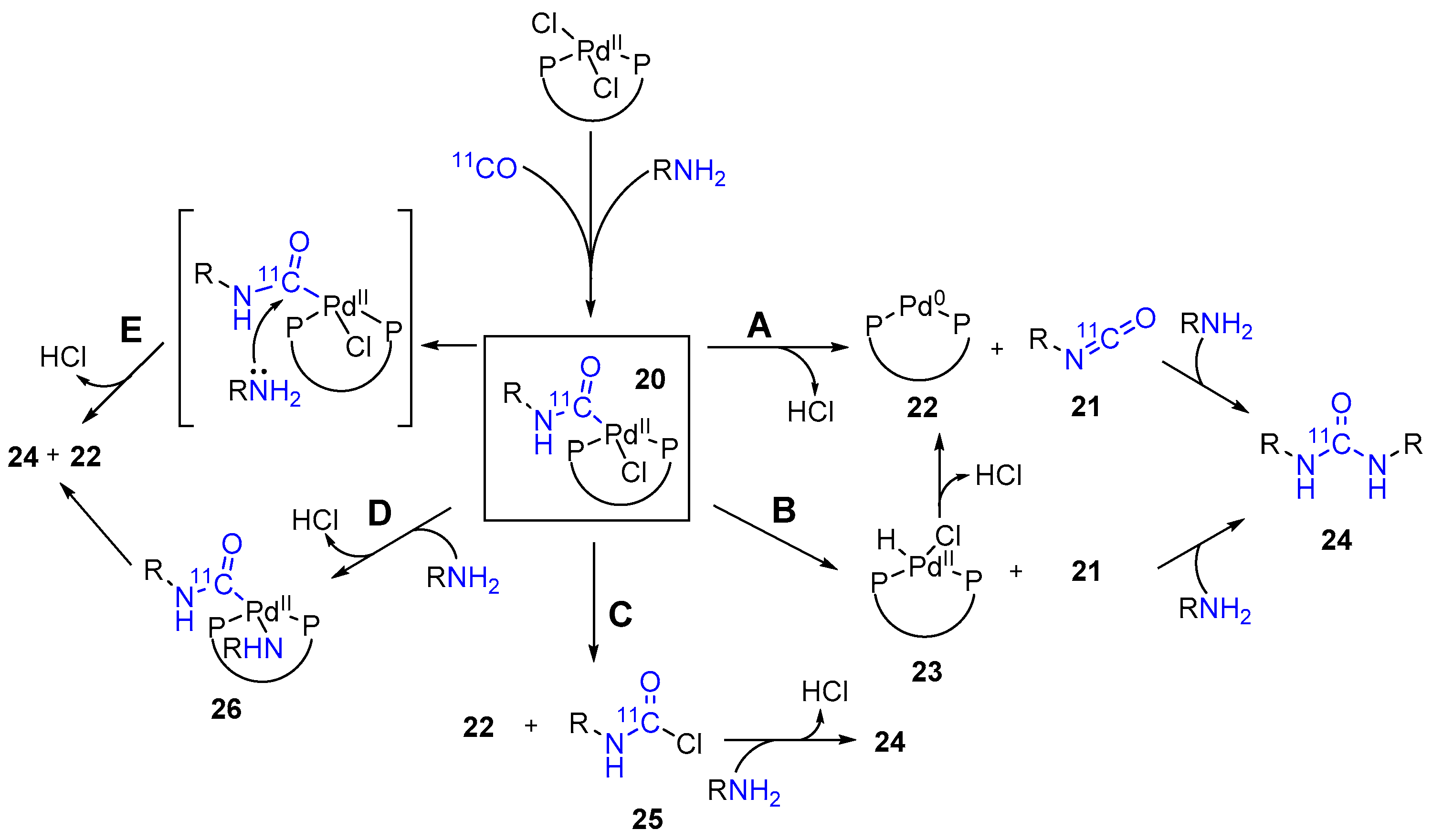
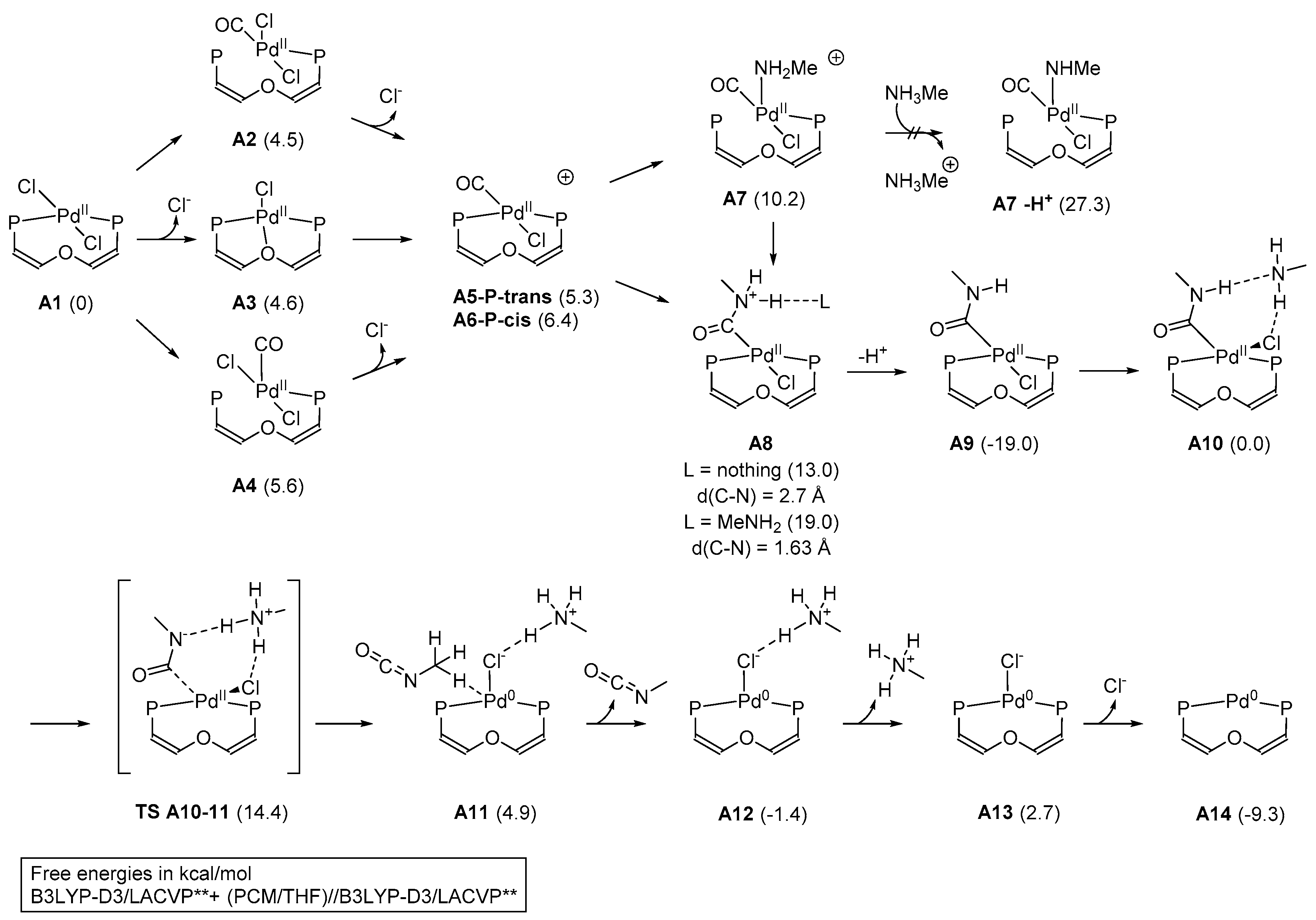
2. Results and Discussion
3. Materials and Methods
3.1. General Chemistry Information
3.2. Synthesis of Starting Materials
3.2.1. tert-Butyl 4-hydroxypiperidine-1-carboxylate [66] CAS: 109384-19-2
3.2.2. tert-Butyl 4-phenoxypiperidine-1-carboxylate [66] CAS: 155989-69-8
3.2.3. 4-Phenoxypiperidine [66] CAS: 3202-33-3
3.3. Synthesis of Reference Compounds
3.3.1. General Procedure for Synthesis of Reference Compounds via an Oxidative Carbonylation
3.3.2. N,N′-Dibenzylurea [69] CAS: 1466-67-7
3.3.3. N-(2-(Pyridin-2-yl)ethyl)piperidine-1-carboxamide CAS: 1710806-84-0
3.3.4. 3,4-Dihydroquinazolin-2(1H)-one [70] CAS: 66655-67-2
3.3.5. 2-Ethylisoindolin-1-one [71] CAS: 23967-95-5
3.3.6. N-(2,4-Dichlorobenzyl)-4-phenoxypiperidine-1-carboxamide CAS: 950645-62-2
3.3.7. General Procedure for the Synthesis of Reference Compounds from an Isocyanate
3.3.8. N-Benzylpiperidine-1-carboxamide [72] CAS: 39531-35-6
3.3.9. N-Butylpiperidine-1-carboxamide CAS: 1461-79-6
3.3.10. N-Isopropylpiperidine-1-carboxamide CAS: 10581-04-1
3.3.11. N-Phenylpiperidine-1-carboxamide [72] CAS: 2645-36-5
3.3.12. N-(4-Methoxyphenyl)piperidine-1-carboxamide CAS: 2645-37-6
3.3.13. N-(4-Fluorophenyl)piperidine-1-carboxamide CAS: 60465-12-5
3.3.14. N-(4-Nitrophenyl)piperidine-1-carboxamide [63] CAS: 2589-20-0
3.3.15. N-Tosylpiperidine-1-carboxamide CAS: 23730-08-7
3.4. General Procedure for the Synthesis and Analysis of 11C-Labelled Ureas
3.4.1. [carbonyl-11C]N,N′-Dibenzylurea 2
3.4.2. [carbonyl-11C]N,N′-Dipropylurea 3
3.4.3. [carbonyl-11C]N,N′-Dicyclohexylurea 4
3.4.4. [carbonyl-11C]N,N′-Diphenylurea 5
3.4.5. [carbonyl-11C]N-Benzylpiperidine-1-carboxamide 7
3.4.6. [carbonyl-11C]N-Butylpiperidine-1-carboxamide 8
3.4.7. [carbonyl-11C]N-(2-(Pyridin-2-yl)ethyl)piperidine-1-carboxamide 9
3.4.8. [carbonyl-11C]N-Isopropylpiperidine-1-carboxamide 10
3.4.9. [carbonyl-11C]N-Phenylpiperidine-1-carboxamide 11
3.4.10. [carbonyl-11C]N-(4-Methoxyphenyl)piperidine-1-carboxamide 12
3.4.11. [carbonyl-11C]N-(4-Fluorophenyl)piperidine-1-carboxamide 13
3.4.12. [carbonyl-11C]N-(4-Nitrophenyl)piperidine-1-carboxamide 14
3.4.13. [carbonyl-11C]3,4-Dihydroquinazolin-2(1H)-one 15
3.4.14. [carbonyl-11C]N-(2,4-Dichlorobenzyl)-4-phenoxypiperidine-1-carboxamide 19
3.5. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Wood, K.A.; Hoskin, P.J.; Saunders, M.I. Positron emission tomography in oncology: A review. Clin. Oncol. 2007, 19, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, L.; Niccolini, F.; Politis, M. Recent imaging advances in neurology. J. Neurol. 2015, 262, 2182–2194. [Google Scholar] [CrossRef] [PubMed]
- Boutagy, N.E.; Sinusas, A.J. Recent advances and clinical applications of PET cardiac autonomic nervous system imaging. Curr. Cardiol. Rep. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Langer, O. Use of PET Imaging to evaluate transporter-mediated drug-drug interactions. J. Clin. Pharmacol. 2016, 56, S143–S156. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, L.; Smith-Jones, P.M.; Sarwar, C.M.S.; Marti, C.N.; Yaddanapudi, K.; Skopicki, H.A.; Gheorghiade, M.; Parsey, R.; Butler, J. Utility of positron emission tomography for drug development for heart failure. Am. Heart J. 2016, 175, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Declercq, L.D.; Vandenberghe, R.; Van Laere, K.; Verbruggen, A.; Bormans, G. Drug development in Alzheimer’s disease: The contribution of PET and SPECT. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Colquhoun, H.M.; Thompson, D.J.; Twigg, M.W. Carbonylation—Direct Synthesis of Carbonyl Compounds; Springer Science + Business Media: New York, NY, USA, 1991; pp. 1–281. [Google Scholar]
- Rahman, O. Carbon monoxide in labeling chemistry and positron emission tomography tracer development: Scope and limitations. J. Label. Compd. Radiopharm. 2015, 58, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Purwanto; Deshpande, R.M.; Chaudhari, R.V.; Delmas, H. Solubility of hydrogen, carbon monoxide, and 1-octene in various solvents and solvent mixtures. J. Chem. Eng. Data 1996, 41, 1414–1417. [Google Scholar]
- Kihlberg, T.; Långström, B. Method and Apparatus for Production and Use of [11C]Carbon Monoxide in Labeling Synthesis. Patent WO 02/102711 A1, 2002. [Google Scholar]
- Hostetler, E.D.; Burns, H.D. A remote-controlled high pressure reactor for radiotracer synthesis with [11C]carbon monoxide. Nucl. Med. Biol. 2002, 29, 845–848. [Google Scholar] [CrossRef]
- Kihlberg, T.; Långström, B. Biologically active 11C-labeled amides using palladium-mediated reactions with aryl halides and [11C]carbon monoxide. J. Org. Chem. 1999, 64, 9201–9205. [Google Scholar] [CrossRef]
- Dahl, K.; Schou, M.; Amini, N.; Halldin, C. Palladium-mediated [11C]carbonylation at atmospheric pressure: A general method using xantphos as supporting ligand. Eur. J. Org. Chem. 2013, 1228–1231. [Google Scholar] [CrossRef]
- Kealey, S.; Miller, P.W.; Long, N.J.; Plisson, C.; Martarello, L.; Gee, A.D. Copper(i)scorpionate complexes and their application in palladium-mediated [11C]carbonylation reactions. Chem. Commun. 2009, 25, 3696–3698. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.; Schou, M.; Rahman, O.; Halldin, C. Improved yields for the palladium-mediated C-11-carbonylation reaction using microwave technology. Eur. J. Org. Chem. 2014, 2, 307–310. [Google Scholar] [CrossRef]
- Audrain, H.; Martarello, L.; Gee, A.; Bender, D. Utilisation of [11C]-labelled boron carbonyl complexes in palladium carbonylation reaction. Chem. Commun. 2004, 5, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Nordeman, P.; Friis, S.D.; Andersen, T.L.; Audrain, H.; Larhed, M.; Skrydstrup, T.; Antoni, G. Rapid and Efficient Conversion of 11CO2 to 11CO through silacarboxylic acids: Applications in Pd-mediated carbonylations. Chem. Eur. J. 2015, 21, 17601–17604. [Google Scholar] [CrossRef] [PubMed]
- Taddei, C.; Bongarzone, S.; Dheere, A.K.H.; Gee, A.D. [11C]CO2 to [11C]CO conversion mediated by [11C]silanes: A novel route for [11C]carbonylation reactions. Chem. Commun. 2015, 51, 11795–11797. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.W.; Long, N.J.; De Mello, A.J.; Vilar, R.; Audrain, H.; Bender, D.; Passchier, J.; Gee, A. Rapid multiphase carbonylation reactions by using a microtube reactor: Applications in positron emission tomography 11C-radiolabeling. Angew. Chem. Int. Ed. 2007, 46, 2875–2878. [Google Scholar] [CrossRef] [PubMed]
- Kealey, S.; Plisson, C.; Collier, T.L.; Long, N.J.; Husbands, S.M.; Martarello, L.; Gee, A.D. Microfluidic reactions using [11C]carbon monoxide solutions for the synthesis of a positron emission tomography radiotracer. Org. Biomol. Chem. 2011, 9, 3313–3319. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.; Schou, M.; Ulin, J.; Sjöberg, C.-O.; Farde, L.; Halldin, C. 11C-carbonylation reactions using gas–liquid segmented microfluidics. RSC Adv. 2015, 5, 88886–88889. [Google Scholar] [CrossRef]
- Eriksson, J.; Van Den Hoek, J.; Windhorst, A.D. Transition metal mediated synthesis using [11C]CO at low pressure—A simplified method for 11C-carbonylation. J. Label. Compd. Radiopharm. 2012, 55, 223–228. [Google Scholar] [CrossRef]
- Jagtap, A.D.; Kondekar, N.B.; Sadani, A.A.; Chern, J.-W. Ureas: Applications in drug design and development. Curr. Med. Chem. 2017, 24, 622–651. [Google Scholar] [CrossRef] [PubMed]
- Dumas, J.; Smith, R.A.; Lowinger, T.B. Recent developments in the discovery of protein kinase inhibitors from the urea class. Curr. Opin. Drug Discov. Dev. 2004, 7, 600–616. [Google Scholar]
- Venkatraman, S.; Bogen, L.; Arasappan, A.; Bennett, F.; Chen, K.; Jao, E.; Liu, Y.; Lovey, R.; Hendrata, S.; Huang, Y.; et al. Orally bioavailable hepatitis C virus NS3 protease inhibitor: A potential therapeutic agent for the treatment of hepatitis C infection. J. Med. Chem. 2015, 49, 6074–6086. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Gillespie, J.R.; Ranade, R.M.; Koh, C.Y.; Kim, J.E.; Laydbak, J.U.; Zucker, F.H.; Hol, W.G.J.; Verlinde, C.L.M.J.; Buckner, F.S.; et al. Urea-based inhibitors of trypanosoma brucei methionyl-trna synthetase: Selectivity and in vivo characterization. J. Med. Chem. 2012, 55, 6342–6351. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, C.; Ogawa, M.; Fujinaga, M.; Kumata, K.; Xie, L.; Yamasaki, T.; Yui, J.; Fukumura, T.; Zhang, M.R. Utilization of [11C]phosgene for radiosynthesis of N-(2-{3-[3,5-bis(trifluoromethyl)]phenyl[11C]ureido}ethyl) glycyrrhetinamide, an inhibitory agent for proteasome and kinase in tumors. Bioorg. Med. Chem. Lett. 2012, 22, 3594–3597. [Google Scholar] [CrossRef] [PubMed]
- Dollé, F.; Martarello, L.; Bramoullé, Y.; Bottlaender, M.; Gee, A.D. Radiosynthesis of carbon-11-labelled GI181771, a new selective CCK-A agonist. J. Label. Compd. Radiopharm. 2005, 48, 501–513. [Google Scholar] [CrossRef]
- Roeda, D.; Westera, G. The Synthesis of Some 11C-labelled antiepileptic drugs with potential utility as radiopharmaceuticals: Hydantoins and Barbiturates. Int. J. Appl. Radiat. Isot. 1981, 32, 843–845. [Google Scholar] [CrossRef]
- Lemoucheux, L.; Rouden, J.; Sobrio, F.; Lasne, M. Debenzylation of tertiary amines using phosgene or triphosgene: An efficient and rapid procedure for the preparation of carbamoyl chlorides and unsymmetrical ureas. Application of carbon-11 chemistry. J. Org. Chem. 2003, 68, 7289–7297. [Google Scholar] [CrossRef] [PubMed]
- Roeda, D.; Dollé, F. [11C]Phosgene: A versatile reagent for radioactive carbonyl insertion into medicinal radiotracers for positron emission tomography. Curr. Top. Med. Chem. 2010, 10, 1680–1700. [Google Scholar] [CrossRef] [PubMed]
- Emran, A.M.; Boothe, T.E.; Finn, R.D.; Vora, M.M.; Kothari, P.J. Preparation of 11C-urea from no-carrier-added 11C-cyanide. Int. J. Appl. Radiat. Isot. 1983, 34, 1013–1014. [Google Scholar] [CrossRef]
- Boothe, T.E.; Emran, A.L.M.; Kothari, J. Use of 11C as a tracer for studying the synthesis of [11C]urea from [11C]cyanide. Int. J. Appl. Radiat. Isot. 1985, 36, 141–144. [Google Scholar] [CrossRef]
- Bera, R.K.; Hartman, N.G.; Jay, M. Continuous production of [C-11] urea for medical application. Appl. Radiat. Isot. 1991, 42, 407–409. [Google Scholar] [CrossRef]
- Van Tilburg, E.W.; Windhorst, A.D.; Van Der Mey, M.; Herscheid, J.D.M. One-pot synthesis of [11C]ureas via triphenylphosphinimines. J. Label. Compd. Radiopharm. 2006, 49, 321–330. [Google Scholar] [CrossRef]
- Schirbel, A.; Holschbach, M.H.; Coenen, H.H. N.C.A.[11C]CO2 as a safe substitute for phosgene in the carbonylation of primary amines. J. Label. Compd. Radiopharm. 1999, 42, 537–551. [Google Scholar] [CrossRef]
- Hicks, J.W.; Wilson, A.A.; Rubie, E.A.; Woodgett, J.R.; Houle, S.; Vasdev, N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: Radiosynthesis of [11C-carbonyl] AR-A014418 by [11C]CO2 fixation. Bioorg. Med. Chem. Lett. 2012, 22, 2099–2101. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.W.; Parkes, J.; Tong, J.; Houle, S.; Vasdev, N.; Wilson, A.A. Radiosynthesis and ex vivo evaluation of [11C-carbonyl]carbamate- and urea-based monoacylglycerol lipase inhibitors. Nucl. Med. Biol. 2014, 41, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Dheere, A.; Bongarzone, S.; Taddei, C.; Yan, R.; Gee, A. Synthesis of 11C-labelled symmetrical ureas via the rapid incorporation of [11C]CO2 into aliphatic and aromatic amines. Synlett 2015, 26, 2257–2260. [Google Scholar]
- Chakraborty, P.K.; Mangner, T.J.; Chugani, H.T. The synthesis of no-carrier-added [11C]urea from [11C]carbon dioxide and application to [11C]uracil synthesis. Appl. Radiat. Isot. 1997, 48, 619–621. [Google Scholar] [CrossRef]
- Dahl, K.; Collier, T.L.; Chang, R.; Zhang, X.; Sadovski, O.; Liang, S.H.; Vasdev, N. “In-Loop” [11C]CO2-fixation: Prototype and proof-of-concept. J. Label. Compd. Radiopharm. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kihlberg, T.; Karimi, F.; Långström, B. [11C]carbon monoxide in selenium-mediated synthesis of 11C-carbamoyl compounds. J. Org. Chem. 2002, 67, 3687–3692. [Google Scholar] [CrossRef] [PubMed]
- Åberg, O.; Långström, B. Synthesis of substituted [11C]ureas and [11C]sulphonylureas by Rh(I)-mediated carbonylation. J. Label. Compd. Radiopharm. 2011, 54, 38–42. [Google Scholar] [CrossRef]
- Ilovich, O.; Åberg, O.; Långström, B.; Mishania, E. Rhodium-mediated [11C]carbonylation: A library of N-phenyl-N′-{4-(4-quinolyloxy)-phenyl}-[11C]urea derivatives as potential PET angiogenic probes. J. Label. Compd. Radiopharm. 2009, 52, 151–157. [Google Scholar] [CrossRef]
- Poot, A.J.; van der Wildt, B.; Stigter-van Walsum, M.; Rongen, M.; Schuit, R.C.; Hendrikse, N.H.; Eriksson, J.; van Dongen, G.A.M.S.; Windhorst, A.D. [11C]Sorafenib: Radiosynthesis and preclinical evaluation in tumor-bearing mice of a new TKI-PET tracer. Nucl. Med. Biol. 2013, 40, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Barletta, J.; Suzuki, M.; Noyori, R.; Watanabe, Y. Synthesis of 11C-labelled N,N′-diphenylurea and ethyl phenylcarbamate by a rhodium-promoted carbonylation via [11C]isocyanatebenzene using phenyl azide and [11C]carbon monoxide. Org. Biomol. Chem. 2004, 2, 3063–3066. [Google Scholar] [CrossRef] [PubMed]
- Barletta, J.; Karimi, F.; Långström, B. Synthesis of [11C-carbonyl]hydroxyureas by a rhodium-mediated carbonylation reaction using [11C]carbon monoxide. J. Label. Compd. Radiopharm. 2006, 49, 429–436. [Google Scholar] [CrossRef]
- Dahl, K.; Itsenko, O.; Rahman, O.; Ulin, J.; Sjöberg, C.O.; Sandblom, P.; Larsson, L.A.; Schou, M.; Halldin, C. An evaluation of a high-pressure 11CO carbonylation apparatus. J. Label. Compd. Radiopharm. 2015, 58, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Kealey, S.; Husbands, S.M.; Bennacef, I.; Gee, A.D.; Passchier, J. Palladium-mediated oxidative carbonylation reactions for the synthesis of 11C-radiolabelled ureas. J. Label. Compd. Radiopharm. 2014, 57, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.A.; Garcia, A.; Houle, S.; Sadovski, O.; Vasdev, N. Synthesis and application of isocyanates radiolabeled with carbon-11. Chem. Eur. J. 2011, 17, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Gómez-vallejo, V.; Gaja, V.; Koziorowski, J.; Llop, J. Positron Emission Tomography—Current Clinical and Research Aspects; Hsieh, C.-H., Ed.; InTech: Rijeka, Croatia, 2012; p. 183. [Google Scholar]
- Henriksen, G.; Drzezga, A. Small Animal Imaging; Kiessling, F., Pichler, B.J., Eds.; Springer: Berlin, Germany, 2011; pp. 499–513. [Google Scholar]
- Roslin, S.; Rosa, M.D.; Deuther-Conrad, W.; Eriksson, J.; Odell, L.R.; Antoni, G.; Brust, P.; Larhed, M. Synthesis and in vitro evaluation of 5-substituted benzovesamicol analogs containing N-substituted amides as potential positron emission tomography tracers for the vesicular acetylcholine transporter. Bioorg. Med. Chem. 2017, 25, 5095–5106. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.Y.; Odell, L.R.; Eriksson, J. Low-pressure radical 11C-aminocarbonylations of alkyl iodides via thermal initiation. Eur. J. Org. Chem. 2016, 2016, 5980–5989. [Google Scholar] [CrossRef]
- Stevens, M.Y.; Chow, S.Y.; Estrada, S.; Eriksson, J.; Asplund, V.; Orlova, A.; Mitran, B.; Antoni, G.; Larhed, M.; Åberg, O.; et al. Synthesis of 11C-labeled sulfonyl carbamates through a multicomponent reaction employing sulfonyl azides, alcohols, and [11C]CO. ChemistryOpen 2016, 5, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, J.R.; Watson, D.A.; Freckmann, D.M.M.; Barder, T.E.; Buchwald, S.L. Palladium-catalyzed carbonylation reactions of aryl bromides at atmospheric pressure: A general system based on xantphos. J. Org. Chem. 2008, 73, 7102–7107. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.L.; Friis, S.D.; Audrain, H.; Nordeman, P.; Antoni, G.; Skrydstrup, T. Efficient 11C-carbonylation of isolated aryl palladium complexes for PET: Application to challenging radiopharmaceutical synthesis. J. Am. Chem. Soc. 2015, 137, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- For more information, see Supplementary Materials.
- Lum, R.T.; Cheng, M.; Cristobal, C.P.; Goldfine, I.D.; Evans, J.L.; Keck, J.G.; Macsata, R.W.; Manchem, V.P.; Matsumoto, Y.; Park, S.J.; et al. Design, synthesis, and structure-activity relationships of novel insulin receptor tyrosine kinase activators. J. Med. Chem. 2008, 51, 6173–6187. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Yadav, N.; King, R.W.; Swanson, M.S.; Weinstein, E.J.; Bedford, M.T. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 2004, 279, 23892–23899. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Hammock, B.D. Discovery of inhibitors of soluble epoxide hydrolase: A target with multiple potential therapeutic indications discovery of inhibitors of soluble epoxide hydrolase: A target with multiple potential therapeutic indications RY800-C114 department of medici. J. Med. Chem. 2012, 55, 1789–1808. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Kay, S.A. XLVIII—Velocity of urea formation in aqueous alcogol. J. Chem. Soc. Trans. 1897, 71, 489–508. [Google Scholar] [CrossRef]
- Orito, K.; Miyazawa, M.; Nakamura, T.; Horibata, A.; Ushito, H.; Nagasaki, H.; Yuguchi, M.; Yamashita, S.; Yamazaki, T.; Tokuda, M. Pd(OAc)2-Catalyzed carbonylation of amines. J. Org. Chem. 2006, 71, 5951–5958. [Google Scholar] [CrossRef] [PubMed]
- Hiwatari, K.; Kayaki, Y.; Okita, K.; Ukai, T.; Shimizu, I.; Yamamoto, A. Selective oxidative carbonylation of amines to oxamides and ureas catalyzed by palladium complexes. Bull. Chem. Soc. Jpn. 2004, 77, 2237–2250. [Google Scholar] [CrossRef]
- Aresta, M.; Giannoccaro, P.; Tommasi, I.; Dibenedetto, A.; Maria, A.; Lanfredi, M.; Ugozzoli, F.; Lanfredi, A.M.M.; Ugozzoli, F. Synthesis and solid state and solution characterization of mono- and di-(η 1-C) carbamoyl-palladium complexes. New efficient palladium-catalyzed routes to carbamoyl chlorides: Key intermediates to isocyanates, carbamic esters, and ureas. Organometallics 2000, 19, 3879–3889. [Google Scholar] [CrossRef]
- Kiesewetter, D.O.; Eckelman, W.C. Utility of azetidinium methanesulfonates for radiosynthesis of 3-[18F]fluoropropyl amines. J. Label. Compd. Radiopharm. 2004, 47, 953–969. [Google Scholar] [CrossRef]
- Hermange, P.; Lindhardt, A.T.; Taaning, R.H.; Bjerglund, K.; Lupp, D.; Skrydstrup, T. Ex situ generation of stoichiometric and substoichiometric 12CO and 13CO and its efficient incorporation in palladium catalyzed aminocarbonylations. J. Am. Chem. Soc. 2011, 133, 6061–6071. [Google Scholar] [CrossRef] [PubMed]
- Nordeman, P.; Odell, L.R.; Larhed, M. Aminocarbonylations employing Mo(CO)6 and a bridged two-vial system: Allowing the use of nitro group substituted aryl iodides and aryl bromides. J. Org. Chem. 2012, 77, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.H.; Lei, H.; Chen, M.; Ren, Z.H.; Bai, Y.; Wang, Y.Y. Palladium-catalyzed carbonylation of amines: Switchable approaches to carbamates and N,N′-disubstituted ureas. Adv. Synth. Catal. 2012, 354, 489–496. [Google Scholar] [CrossRef]
- Paz, J.; Pérez-Balado, C.; Iglesias, B.; Muñoz, L. Carbon dioxide as a carbonylating agent in the synthesis of 2-oxazolidinones, 2-oxazinones, and cyclic ureas: Scope and limitations. J. Org. Chem. 2010, 75, 3037–3046. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Addis, D.; Knöpke, L.R.; Bentrup, U.; Junge, K.; Brückner, A.; Beller, M. Selective catalytic monoreduction of phthalimides and imidazolidine-2,4-diones. Angew. Chem. Int. Ed. 2011, 50, 9180–9184. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Matsushita, H.; Clapham, B.; Janda, K.D. The direct conversion of carbamates to ureas using aluminum amides. Tetrahedron 2004, 60, 3439–3443. [Google Scholar] [CrossRef]
- Matsumura, Y.; Satoh, Y.; Onomura, O.; Maki, T. A New method for synthesis of unsymmetrical ureas using electrochemically prepared trifluoroethyl carbamates. J. Org. Chem. 2000, 65, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W., Jr.; Davies, C.A.; Downey, J.R., Jr.; Frurip, D.J.; McDonald, R.A.; Syverud, A.N. Janaf Thermochemical Tables 3rd ed. J. Phys. Chem. Ref. Data 1985, 14, 718–1856. [Google Scholar]
- Marten, B.; Kim, K.; Cortis, C.; Friesner, R.A.; Murphy, R.B.; Ringnalda, M.N.; Sitkoff, D.; Honig, B. New model for calculation of solvation free energies: Correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar] [CrossRef]
Sample Availability: Samples of the reference compounds 2, 7–15 and 17–19 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}

| Entry | T (°C) | Time (min) | Conversion a (%) | Product Selectivity b (%) | RCY c (%) |
|---|---|---|---|---|---|
| 1 | 120 | 5 | 56 ± 2.2 | >99 | 55 ± 2.1 (3) |
| 2 | 120 | 10 | 83 ± 3.9 | >99 | 82 ± 3.9 (3) |
| 3 | 150 | 5 | 66 ± 4.5 | 96 ± 2.6 | 63 ± 4.3 (3) |
| 4 | 150 | 10 | 90 ± 2.5 | 97 ± 2.1 | 87 ± 3.4 (3) |
| 5 d | 120 | 10 | 66 ± 1.0 | >99 | 65 ± 1.0 (2) |

| Compound | 11C-Labelled Urea | Conversion a (%) | RCY b (%) | RCP c (%) |
|---|---|---|---|---|
| 2 | 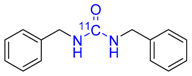 | 81 ± 5 | 65 ± 1 | >99 |
| 41 d | ||||
| 3 |  | 67 ± 4 | 40 ± 6 | >99 |
| 4 | 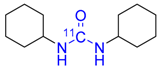 | 71 ± 2 | 48 ± 4 | >99 |
| 5 |  | 15 ± 1 | 4 ± 1 | >99 |
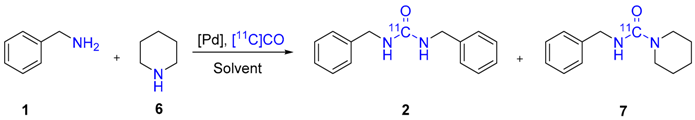
| Entry | Catalyst | T (°C) | 6 (Equiv.) | Conversion a (%) | Product Selectivity b (%) | 2:7 c | RCY d (%) |
|---|---|---|---|---|---|---|---|
| 1 | Pd(Xantphos)Cl2 | 120 | 1 | 53 ± 5.6 | 79 ± 2.9 | 12:88 | 42 ± 5.9 (3) |
| 2 | Pd(PPh3)2Cl2 | 120 | 1 | 69 ± 4.1 | 46 ± 3.6 | 16:84 | 32 ± 3.2 (3) |
| 3 | Pd(OAc)2 + dppf | 120 | 1 | 95 ± 3.5 | 13 ± 3.5 | 11:89 | 12 ± 4.0 (2) |
| 4 | Pd(OAc)2 + dppp | 120 | 1 | 75 ± 2.5 | 21 ± 0.5 | 23:77 | 16 ± 0.5 (2) |
| 5 | Pd(OAc)2 + Xantphos | 120 | 1 | 67 ± 9 | 10 ± 1 | 23:77 | 7 ± 1.5 (2) |
| 6 e | Pd(Xantphos)Cl2 | 120 | 1 | 43 ± 1.7 | 49 ± 3.6 | 16:84 | 21 ± 1.6 (3) |
| 7 | Pd(Xantphos)Cl2 | 80 | 1 | 57 ± 9.2 | 44 ± 6.8 | 9:91 | 26 ± 8.5 (3) |
| 8 | Pd(Xantphos)Cl2 | 150 | 1 | 44 ± 11 | 87 ± 4.3 | 9:91 | 41 ± 6.2 (4) |
| 9 | Pd(Xantphos)Cl2 | 120 | 2 | 46 ± 4.3 | 63 ± 2.2 | 9:91 | 29 ± 3.3 (3) |
| 10 | Pd(Xantphos)Cl2 | 120 | 5 | 58 ± 1.7 | 71 ± 3.6 | 2:98 | 42 ± 3.1 (3) |
| 11 f | Pd(Xantphos)Cl2 | 120 | 1 | 67 ± 1.7 | 89 ± 3.3 | 7:93 | 60 ± 3.4 (3) |
| 12 g | Pd(PPh3)4 | 120 | 1 | 93 | - | - | - |

| Compound | 11C-Labelled Urea | Conversion a (%) | RCY b (%) | RCP c (%) |
|---|---|---|---|---|
| 7 | 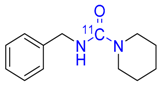 | 65 ± 0 | 41 ± 6 | 98 ± 1 |
| 39 d | 17 d | |||
| 59 e | 31 e | |||
| 8 | 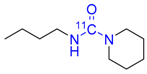 | 55 ± 4 | 23 ± 1 | >99 |
| 60 e | 14 e | |||
| 9 |  | 20 ± 0 | 12 ± 1 | >99 |
| 10 |  | 35 ± 6 | 14 ± 4 | >99 |
| 11 | 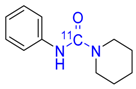 | 60 ± 3 f | 7 ± 2 f | 80 ± 7 f |
| 59 ± 1 | 12 ± 0 g | 97 ± 2 g | ||
| 70 e | 6 e | 88 e | ||
| 12 |  | 66 ± 3 | 9 ± 1 | 99 ± 1 |
| 74 e | 21 e | |||
| 13 | 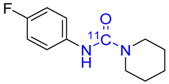 | 67 ± 9 f | 8 ± 1 f | >99 |
| 88 e | 28 e | |||
| 14 |  | 63 ± 0 | 5 ± 1 | >99 |
| 83 e | 6 e | |||
| 15 | 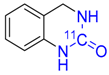 | 27 ± 1 | 1 ± 0 | 90 ± 10 |
| 90 e | Trace e | - | ||
| 16 |  | 12 ± 2 | Trace | - |
| 17 | 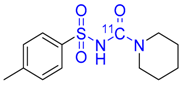 | 51 ± 0 | Trace | - |
| 18 | 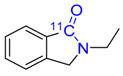 | 66 ± 8 | - | - |
| 19 | 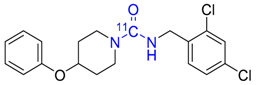 | 74 | 41 ± 7 f | 99 ± 0 f |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roslin, S.; Brandt, P.; Nordeman, P.; Larhed, M.; Odell, L.R.; Eriksson, J. Synthesis of 11C-Labelled Ureas by Palladium(II)-Mediated Oxidative Carbonylation. Molecules 2017, 22, 1688. https://doi.org/10.3390/molecules22101688
Roslin S, Brandt P, Nordeman P, Larhed M, Odell LR, Eriksson J. Synthesis of 11C-Labelled Ureas by Palladium(II)-Mediated Oxidative Carbonylation. Molecules. 2017; 22(10):1688. https://doi.org/10.3390/molecules22101688
Chicago/Turabian StyleRoslin, Sara, Peter Brandt, Patrik Nordeman, Mats Larhed, Luke R. Odell, and Jonas Eriksson. 2017. "Synthesis of 11C-Labelled Ureas by Palladium(II)-Mediated Oxidative Carbonylation" Molecules 22, no. 10: 1688. https://doi.org/10.3390/molecules22101688
APA StyleRoslin, S., Brandt, P., Nordeman, P., Larhed, M., Odell, L. R., & Eriksson, J. (2017). Synthesis of 11C-Labelled Ureas by Palladium(II)-Mediated Oxidative Carbonylation. Molecules, 22(10), 1688. https://doi.org/10.3390/molecules22101688