Design, Modeling and Synthesis of 1,2,3-Triazole-Linked Nucleoside-Amino Acid Conjugates as Potential Antibacterial Agents

 , , ,
, , ,
Abstract
:
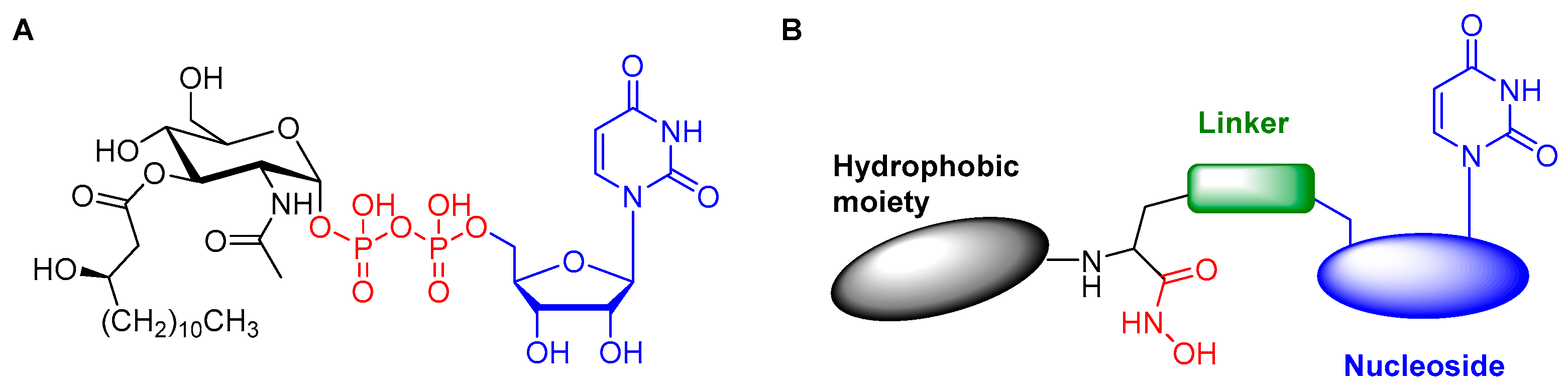
1. Introduction
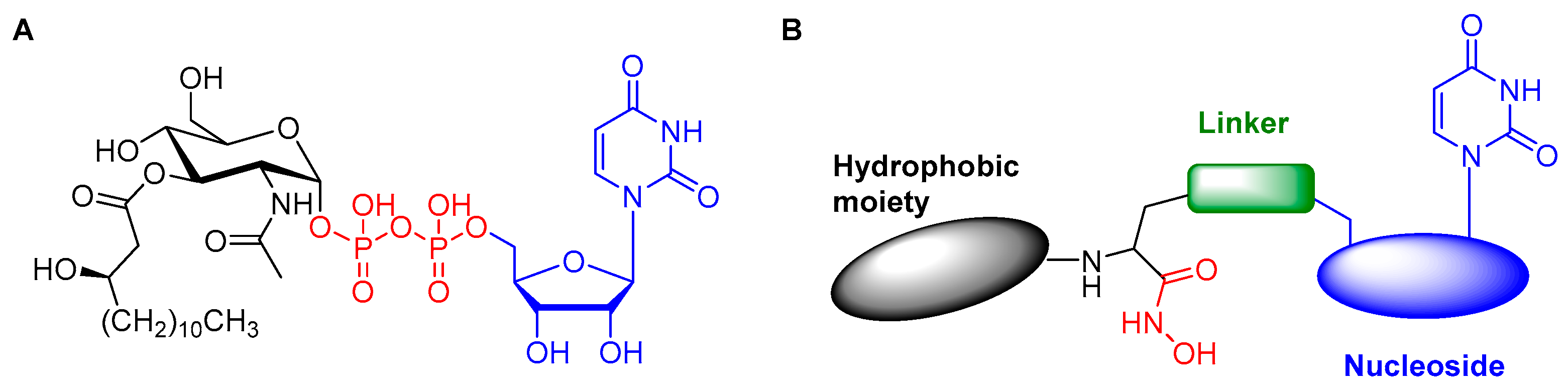
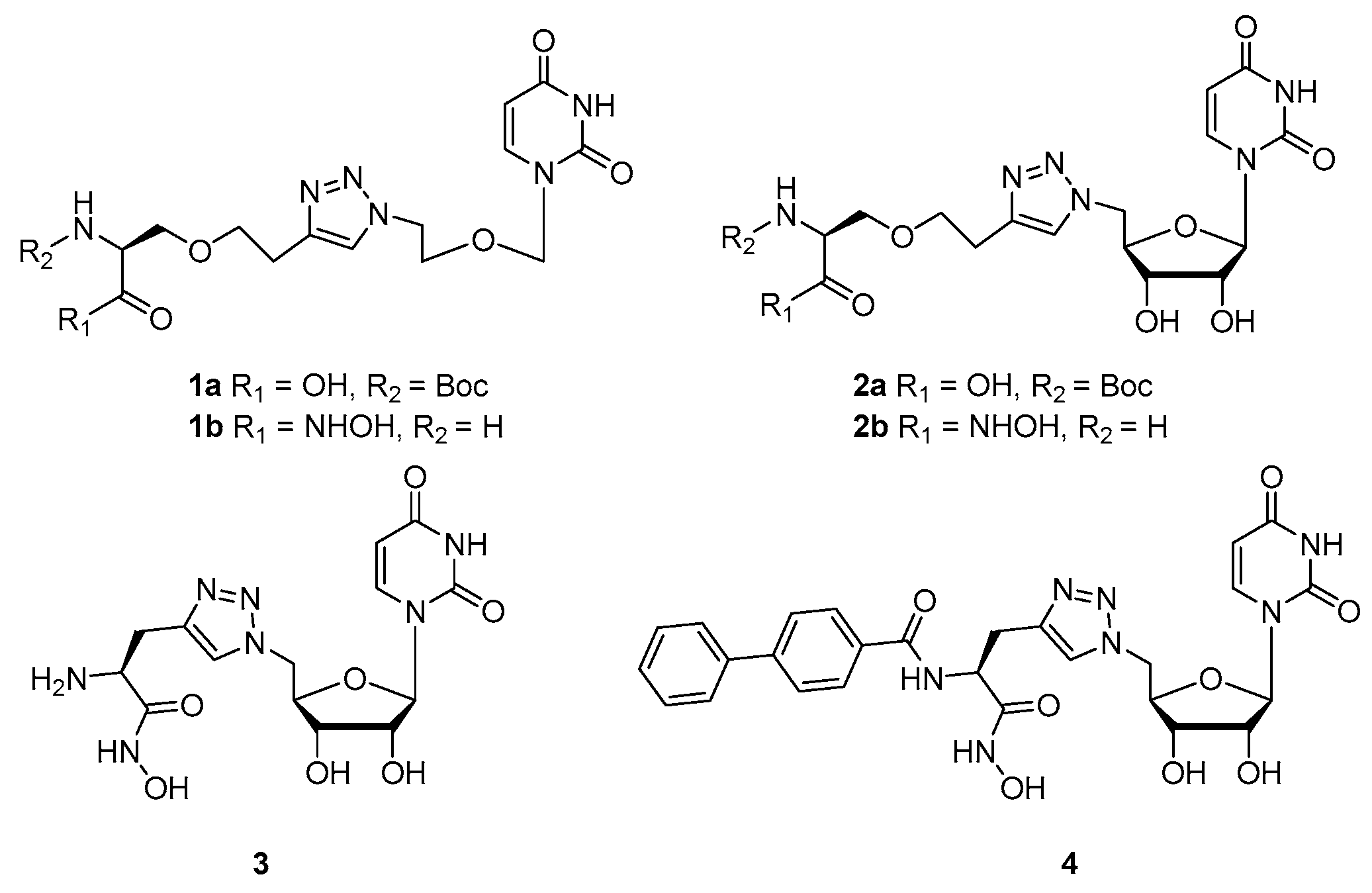
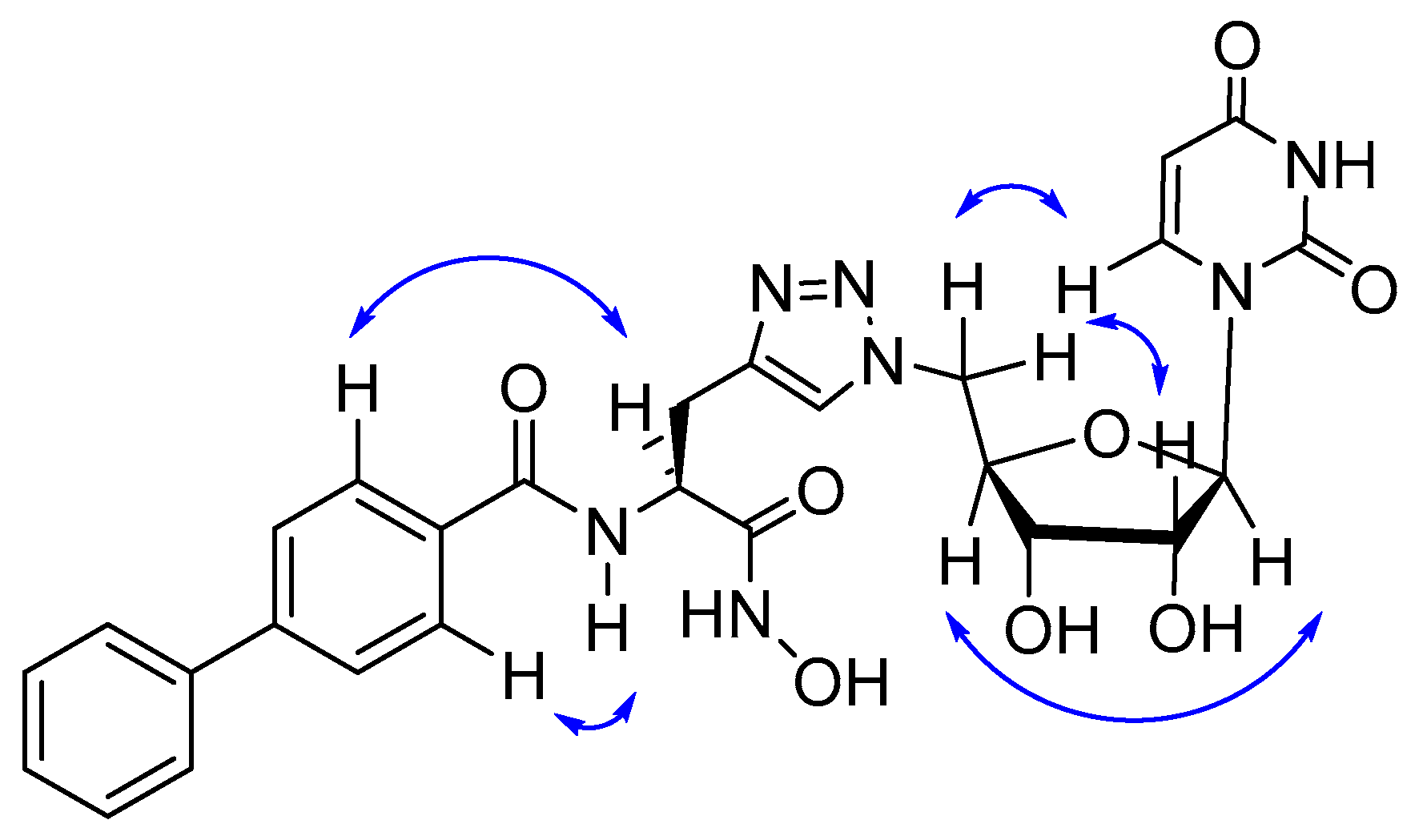
2. Results and Discussion
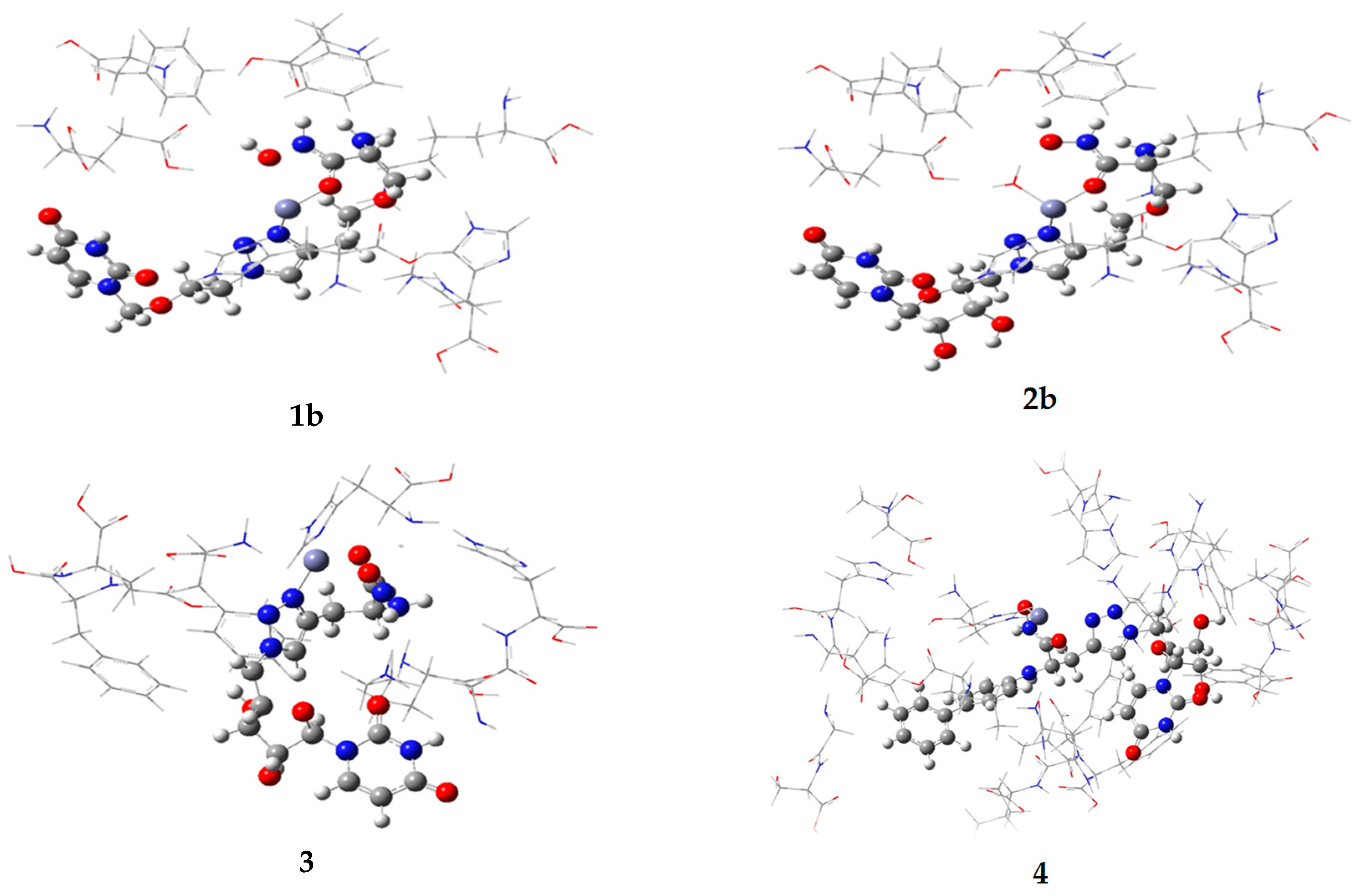
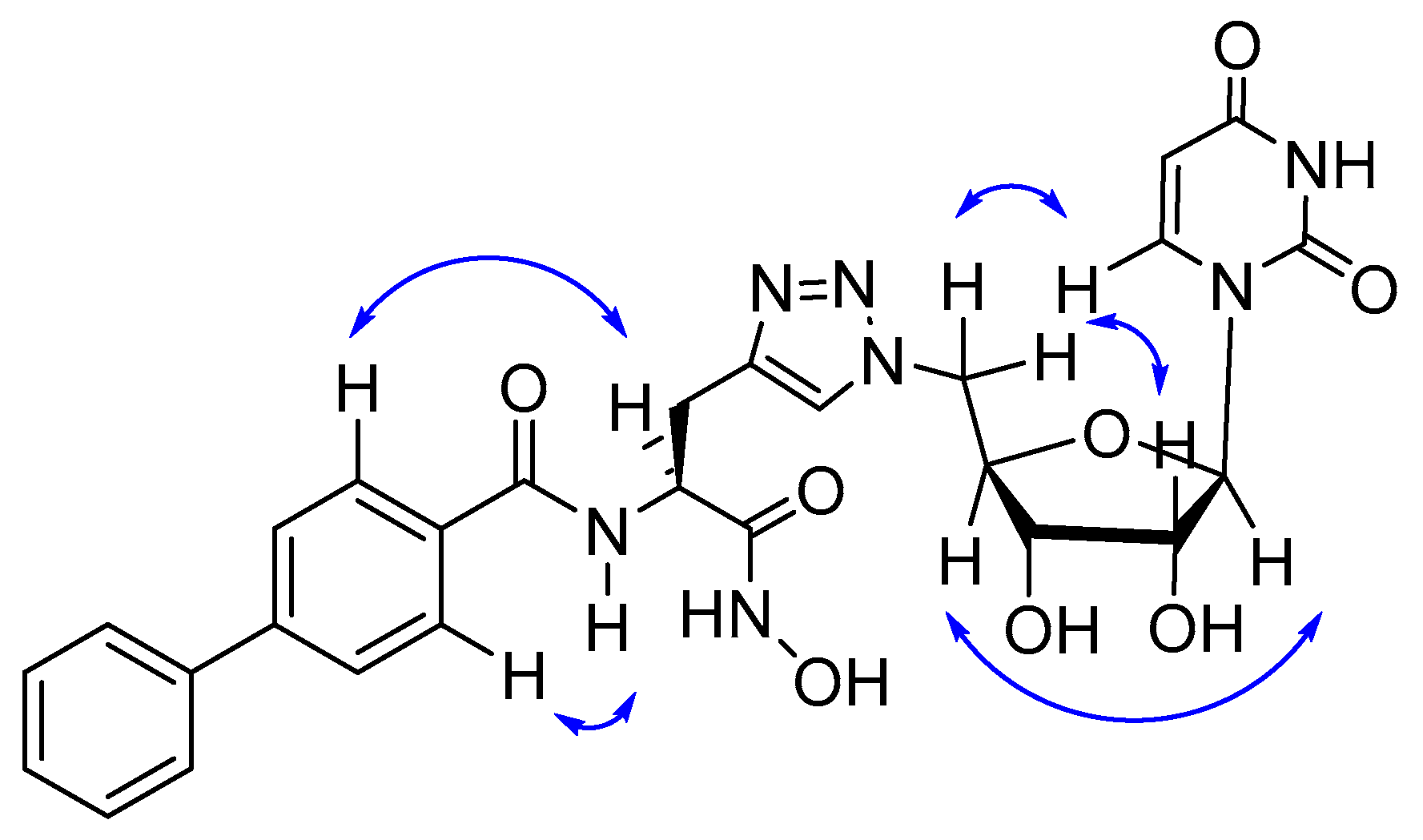
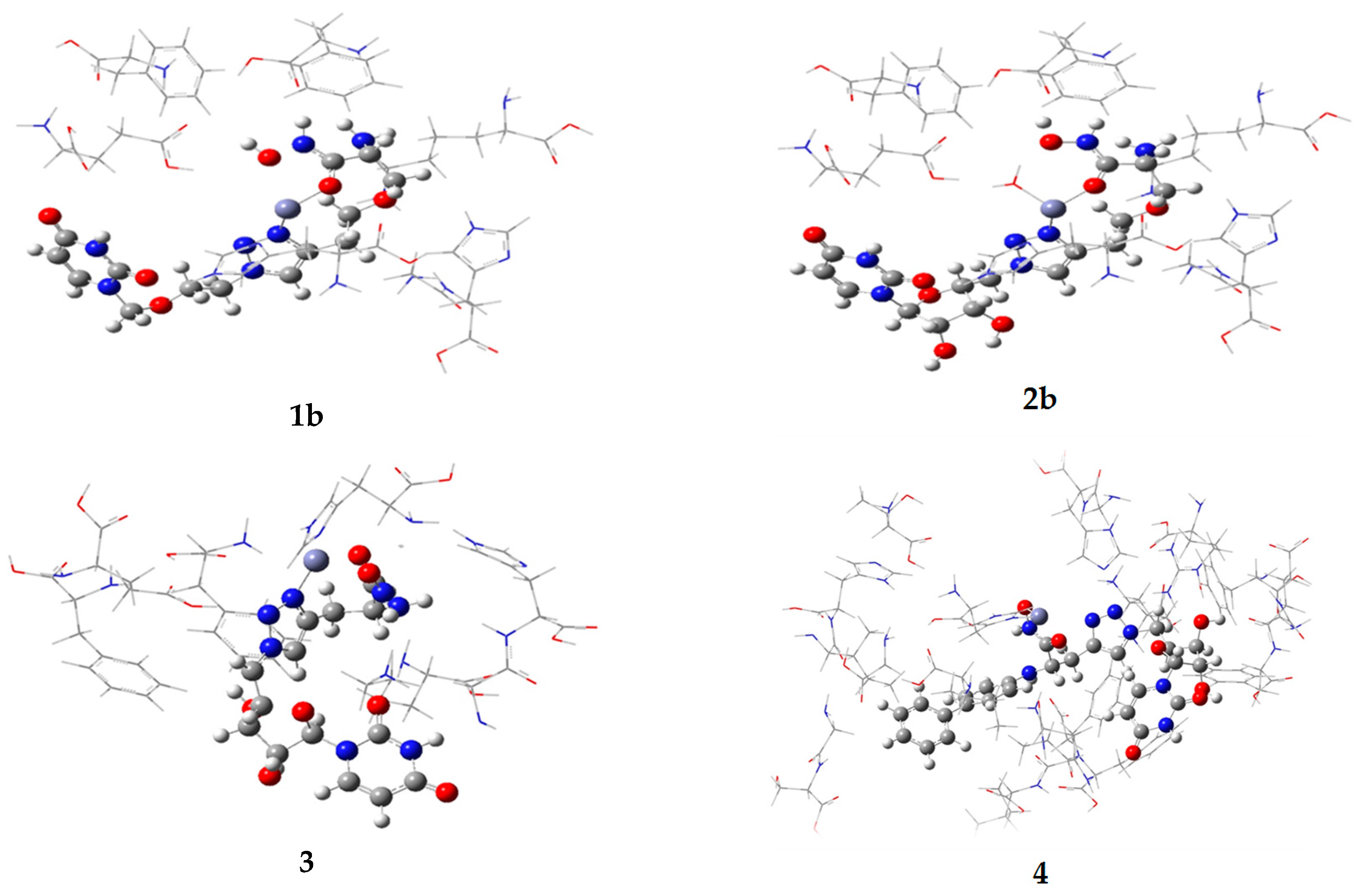
2.1. Computational
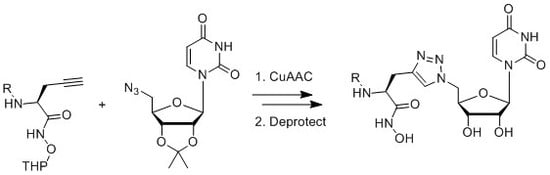
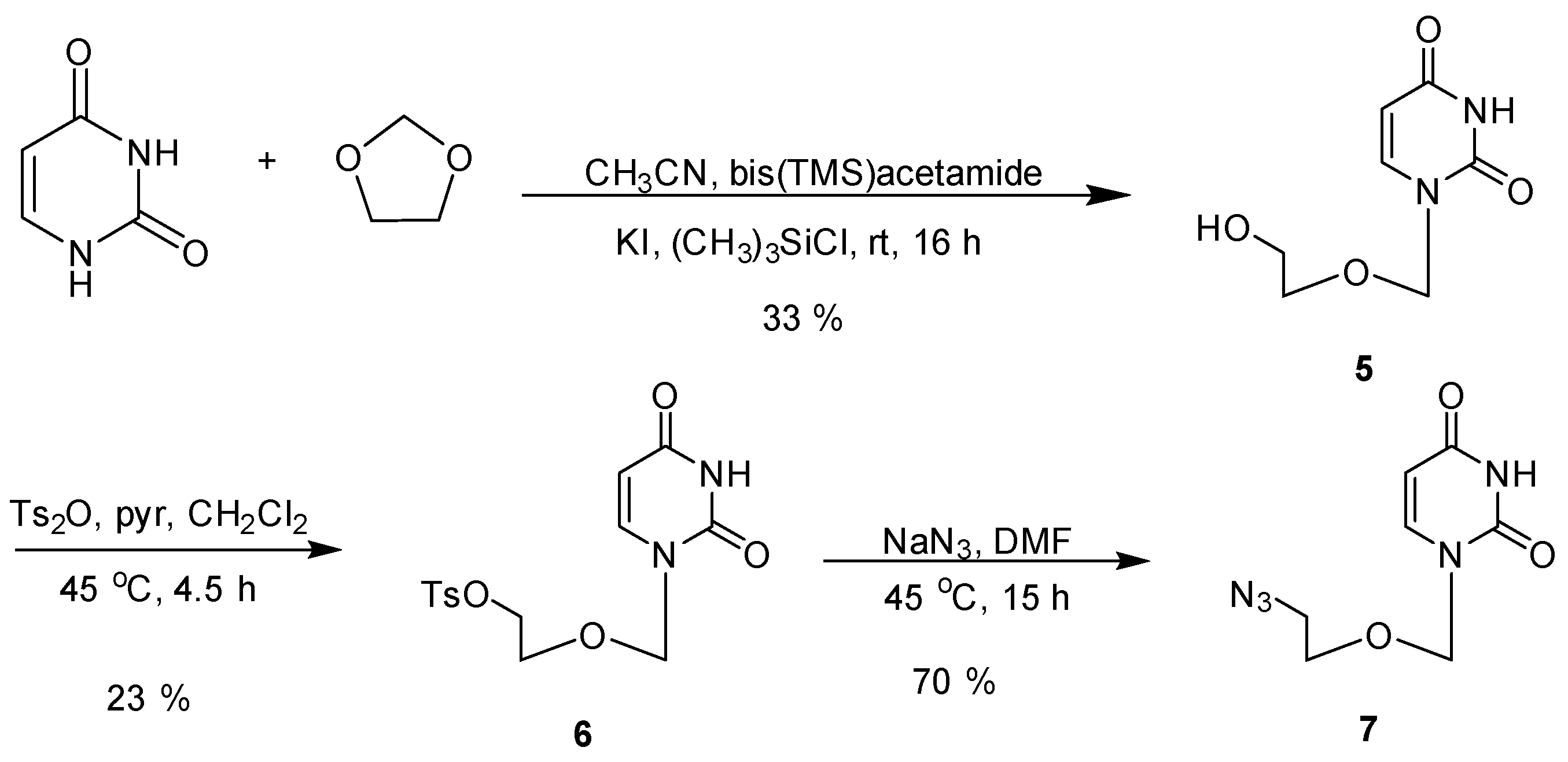
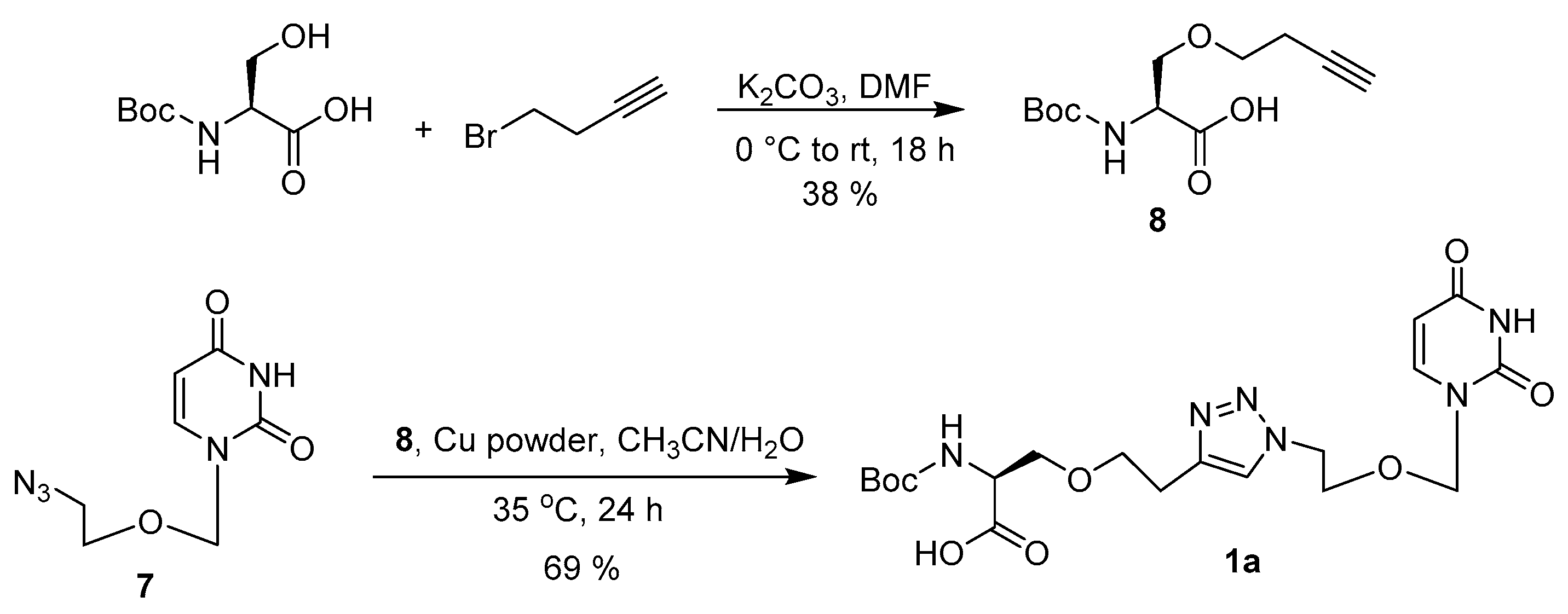
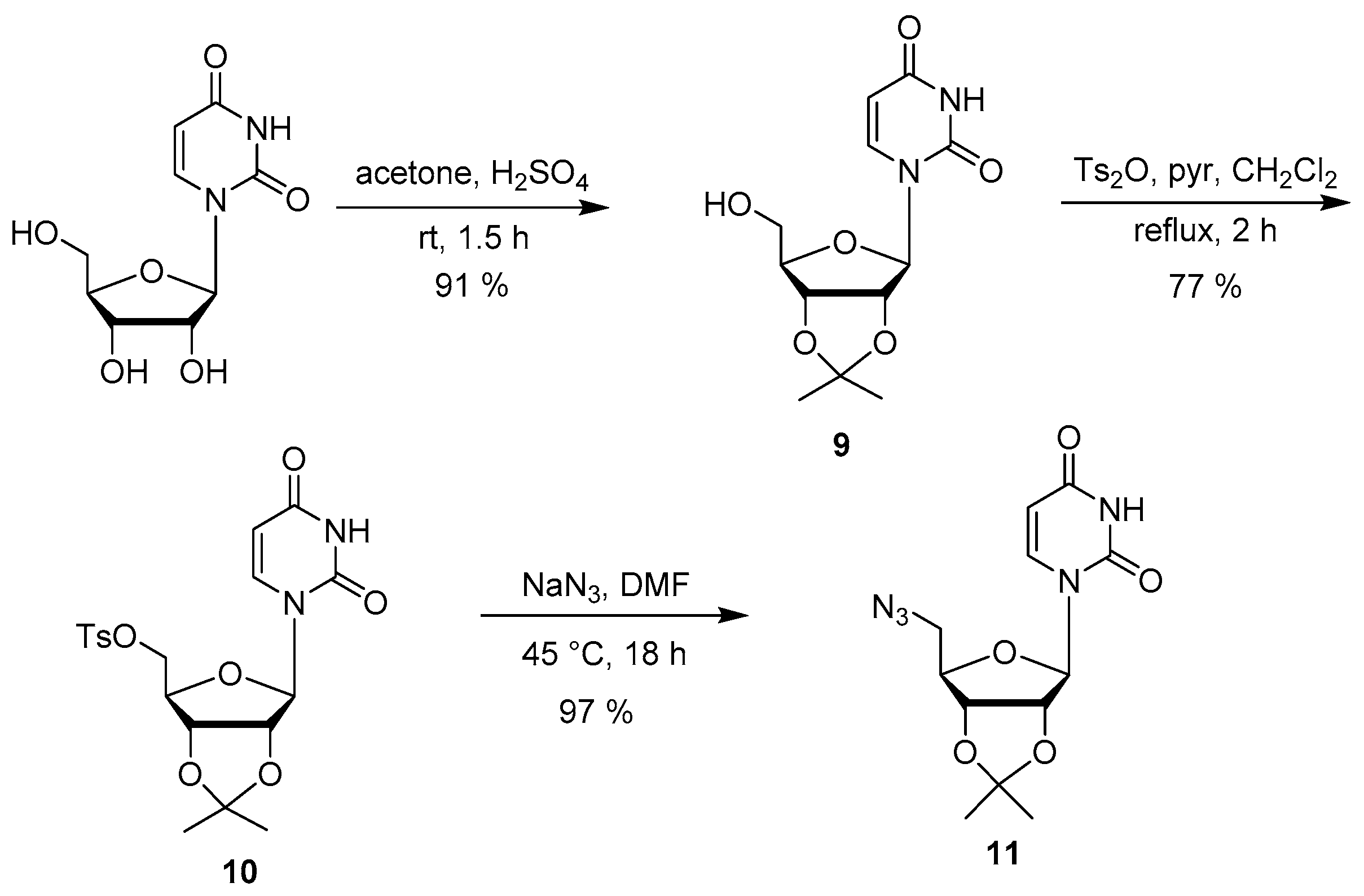
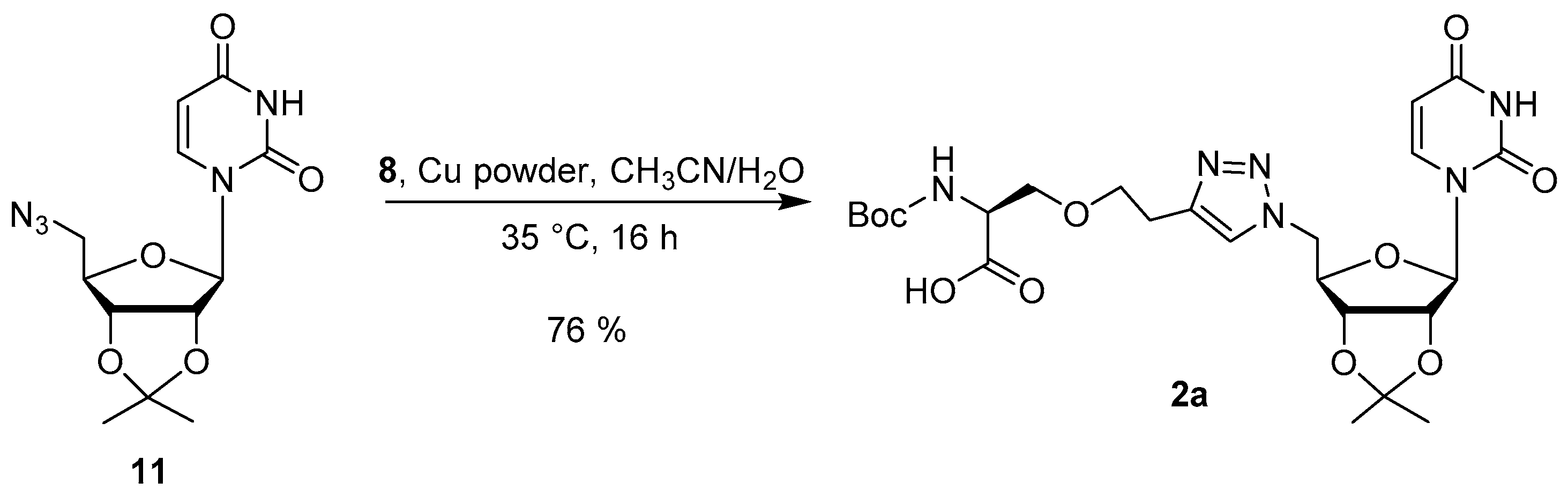
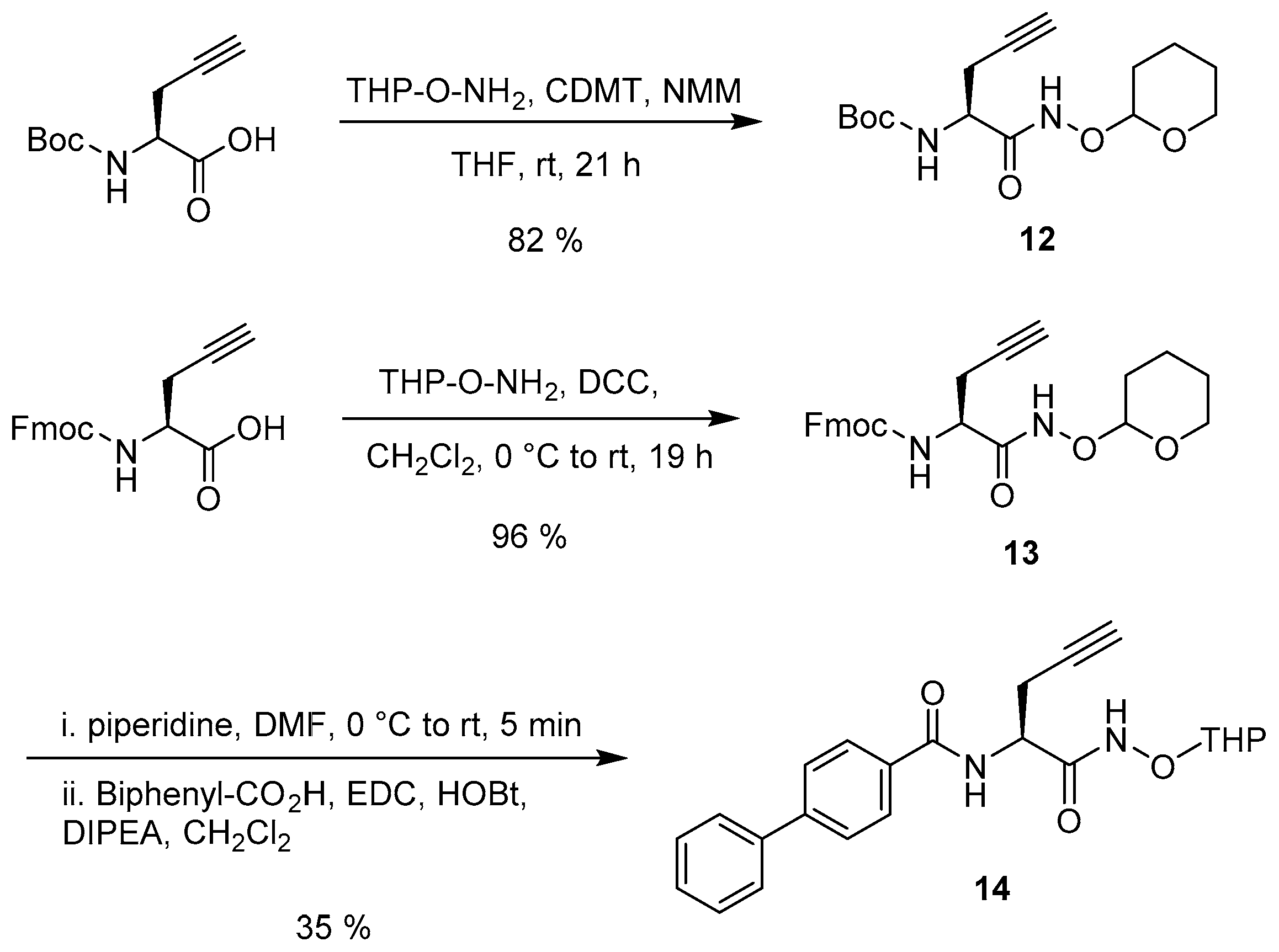
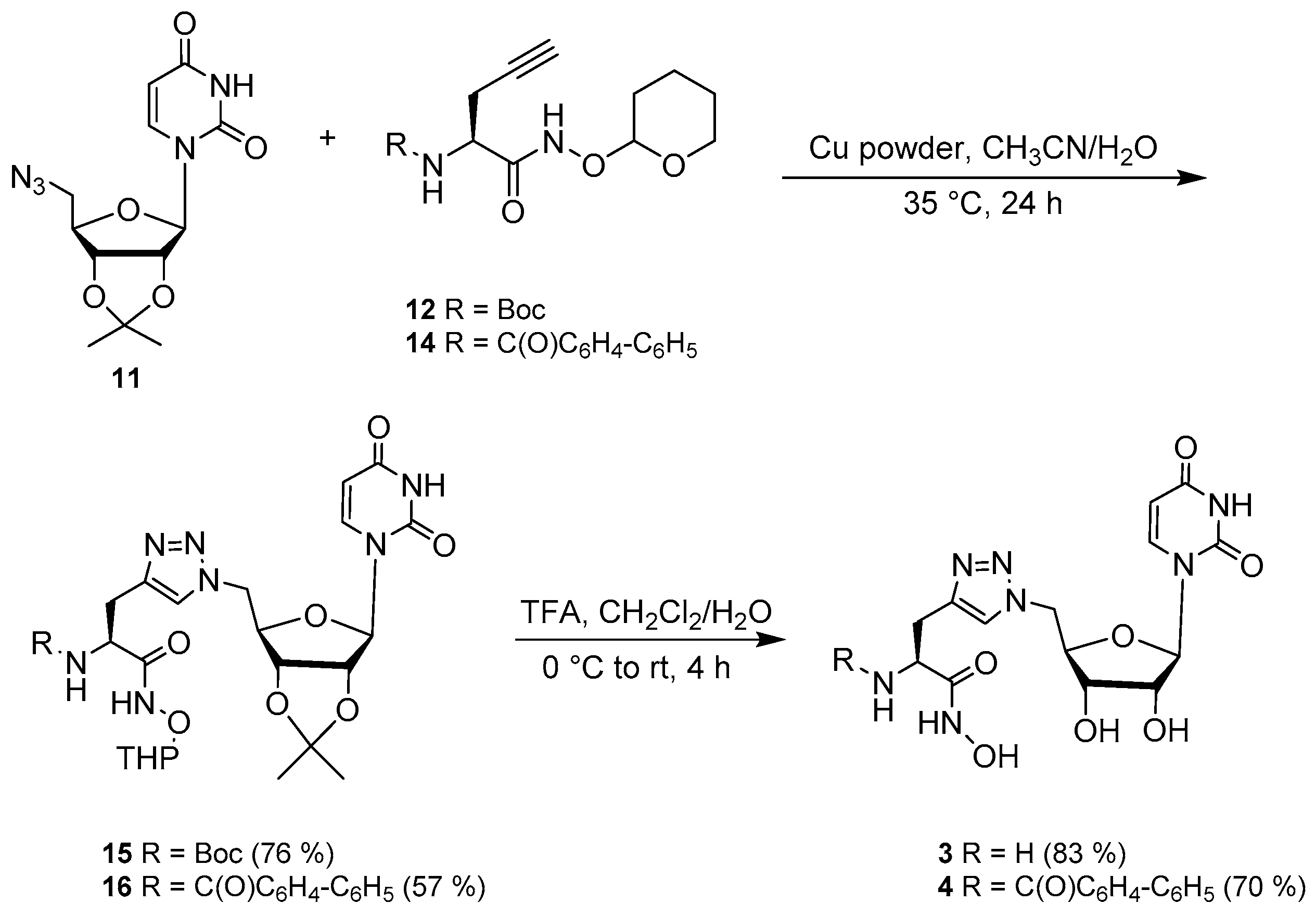
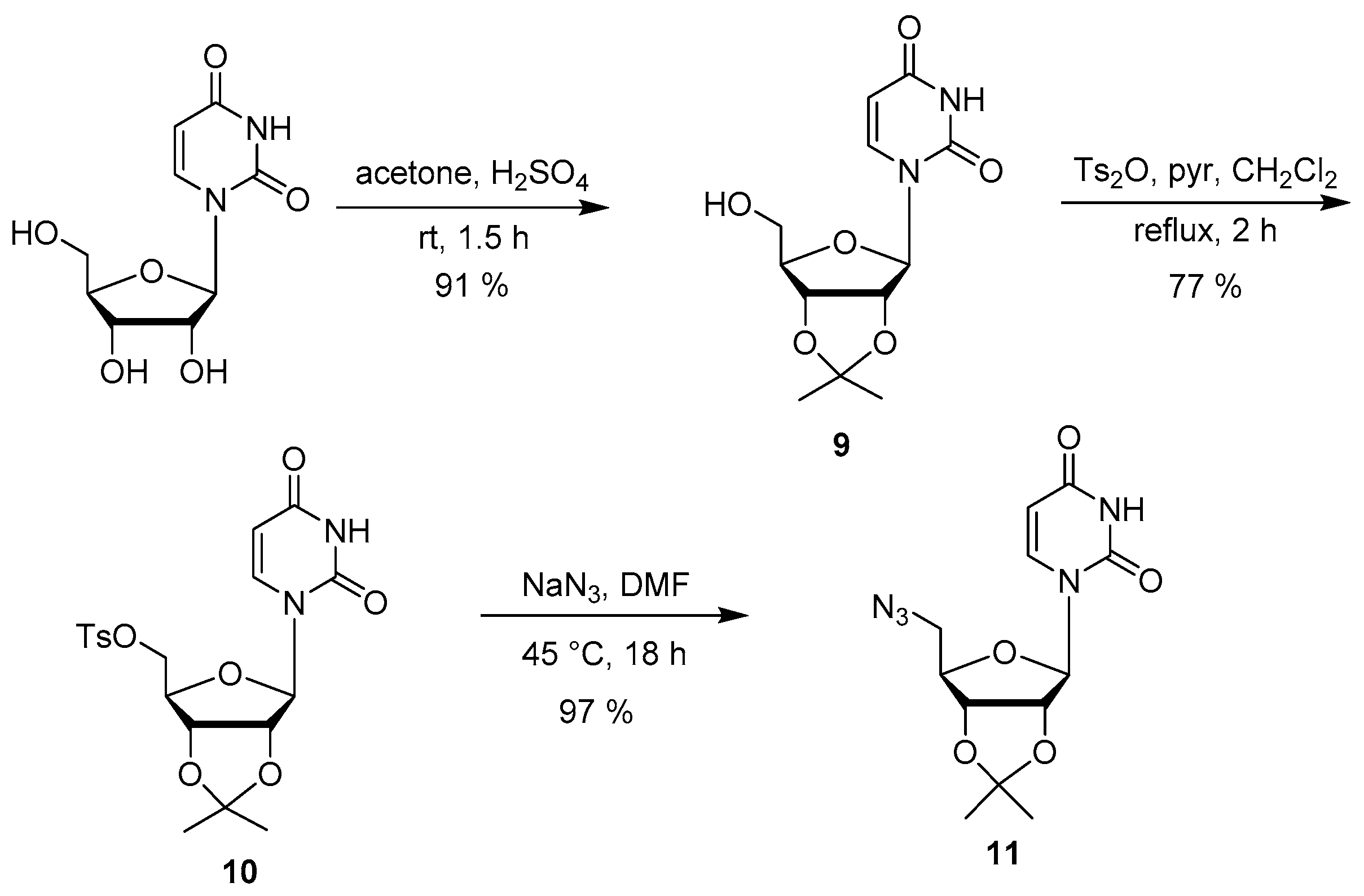
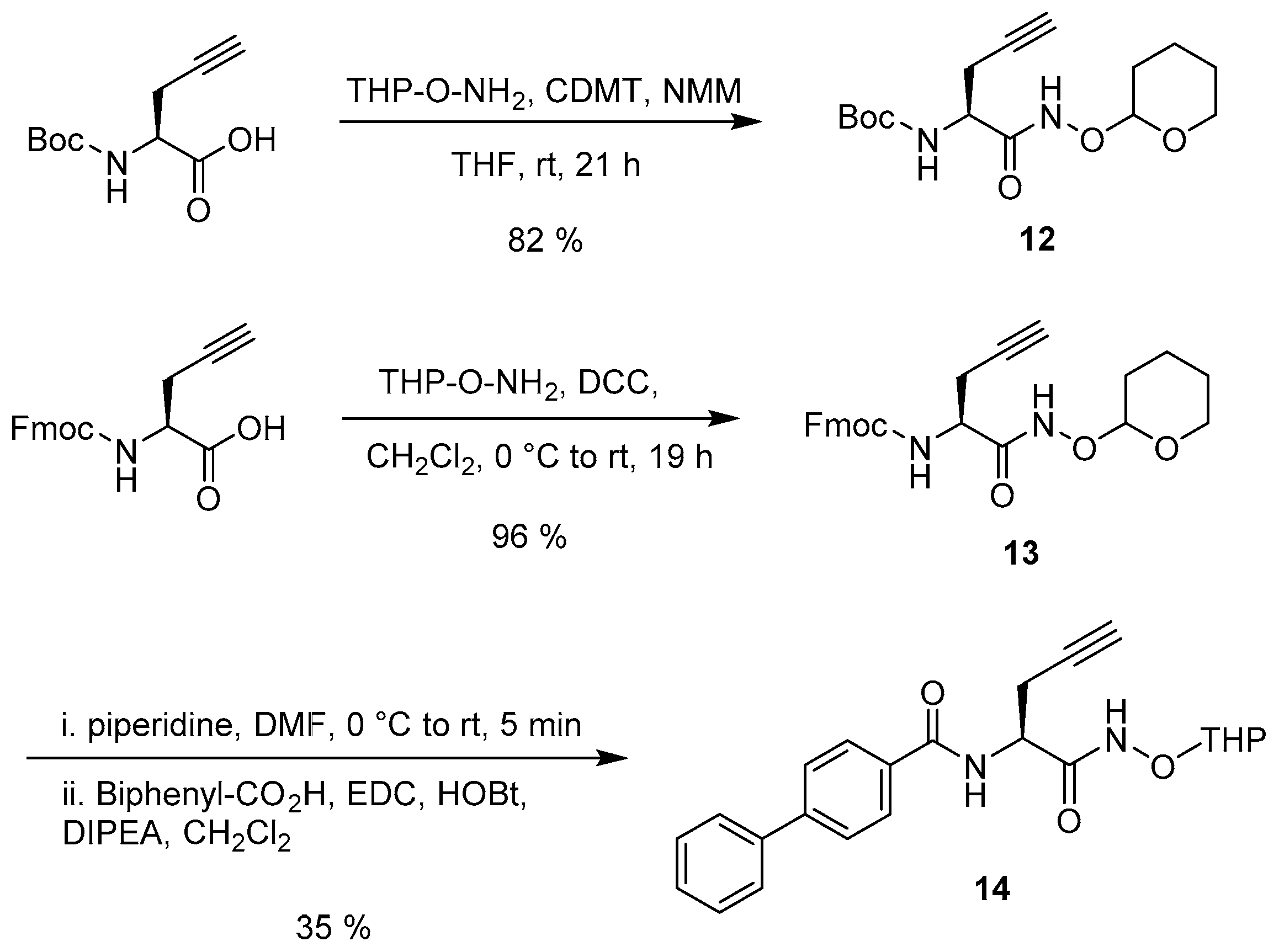
2.2. Synthesis
3. Materials and Methods
3.1. General Methods
3.2. Computational Methods
3.3. Synthesis of Acyclic Nucleoside Conjugate 1
3.4. Synthesis of the Uridine-Amino Acid Conjugates 2–4
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, L.; Zhang, Z. Development and applications of the copper-catalyzed azide-alkyne cycloaddition (cuaac) as a bioorthogonal reaction. Molecules 2016, 21, 1393. [Google Scholar] [CrossRef] [PubMed]
- Bodnár, B.; Mernyák, E.; Wölfling, J.; Schneider, G.; Herman, B.E.; Szécsi, M.; Sinka, I.; Zupkó, I.; Kupihár, Z.; Kovács, L. Synthesis and biological evaluation of triazolyl 13α-estrone–nucleoside bioconjugates. Molecules 2016, 21, 1212. [Google Scholar] [CrossRef] [PubMed]
- Lima-Neto, R.G.; Cavalcante, N.N.M.; Srivastava, R.M.; Mendonca, F.J., Jr.; Wanderley, A.G.; Neves, R.P.; dos Anjos, J.V. Synthesis of 1,2,3-triazole derivatives and in vitro antifungal evaluation on candida strains. Molecules 2012, 17, 5882–5892. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Macaubas, C.; Mellins, E.; Astakhova, K. Synthesis of phospholipid-protein conjugates as new antigens for autoimmune antibodies. Molecules 2015, 20, 10253–10263. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Chavan, S.R.; Shirazi, F.; Razdan, M.; Nimkar, P.; Maybhate, S.P.; Likhite, A.P.; Gonnade, R.; Hazara, B.G.; Deshpande, M.V.; et al. Exploration of click reaction for the synthesis of modified nucleosides as chitin synthase inhibitors. Bioorg. Med. Chem. 2009, 17, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Walvoort, M.T.C.; Lukose, V.; Imperiali, B. A modular approach to phosphoglycosyltransferase inhibitors inspired by nucleoside antibiotics. Chem. Eur. J. 2016, 22, 3856–3864. [Google Scholar] [CrossRef] [PubMed]
- Szabó, J.; Jerkovics, N.; Schneider, G.; Wölfling, J.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and in vitro antiproliferative evaluation of C-13 epimers of triazolyl-d-secoestrone alcohols: The first potent 13-d-secoestrone derivative. Molecules 2016, 21, 611. [Google Scholar] [CrossRef]
- Oldham, E.D.; Nunes, L.M.; Varela-Ramirez, A.; Rankin, S.E.; Knutson, B.L.; Aguilera, R.J.; Lehmler, H.-J. Cytotoxic activity of triazole-containing alkyl β-d-glucopyranosides on a human T-cell leukemia cell line. Chem. Cent. J. 2015, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Pastuch-Gawolek, G.; Plesniak, M.; Komor, R.; Byczek-Wyrostek, A.; Erfurt, K.; Szeja, W. Synthesis and preliminary biological assay of uridine glycoconjugate derivatives containing amide and/or 1,2,3-triazole linkers. Bioorg. Chem. 2017, 72, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-N.; Zhang, Z.-F.; Liu, N.-N.; Xiang, Y.-H.; Zhang, Z.-Y.; Andrei, G.; Snoeck, R.; Schols, D.; Zhang, Q.-S.; Wu, Q.-P. Synthesis and bioactivity of novel trisubstituted triazole nucleosides. Nucleosides Nucleotides Nucleic Acids 2016, 35, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Krajczyk, A.; Kulinska, K.; Kulinski, T.; Hurst, B.L.; Day, C.W.; Smee, D.F.; Ostrowski, T.; Januszczyk, P.; Zeidler, J. Antivirally active ribavirin analogues—4,5-disubstituted 1,2,3-triazole nucleosides: Biological evaluation against certain respiratory viruses and computational modelling. Antiviral Chem. Chemother. 2013, 23, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Chandra Babu Tirumalasetty, M.; Wudayagiri, R.; Donthabakthuni, S.; Maroju, R.; Baburao, K.; Parasa, L.S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Alaoui, S.; Dufies, M.; Driowya, M.; Demange, L.; Bougrin, K.; Robert, G.; Auberger, P.; Pagès, G.; Benhida, R. Synthesis and anti-cancer activities of new sulfonamides 4-substituted-triazolyl nucleosides. Bioorg. Med. Chem. Lett. 2017, 27, 1989–1992. [Google Scholar] [CrossRef] [PubMed]
- Fer, M.J.; Bouhss, A.; Patrao, M.; Le Corre, L.; Pietrancosta, N.; Amoroso, A.; Joris, B.; Mengin-Lecreulx, D.; Calvet-Vitale, S.; Gravier-Pelletier, C. 5′-methylene-triazole-substituted-aminoribosyl uridines as mray inhibitors: Synthesis, biological evaluation and molecular modeling. Org. Biomol. Chem. 2015, 13, 7193–7222. [Google Scholar] [CrossRef] [PubMed]
- Barb, A.W.; Jiang, L.; Raetz, C.R.H.; Zhou, P. Structure of the deacetylase lpxc bound to the antibiotic chir-090: Time-dependent inhibition and specificity in ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 18433–18438. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.B. Antibiotic resistance—The problem intensifies. Adv. Drug Deliv. Rev. 2005, 57, 1446–1450. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, U.F.; Reddy, P.A. Hydantoin Derivatives Useful as Antibacterial Agents. U.S. Patent 7,998,961, 29 August 2007. [Google Scholar]
- Barb, A.W.; Zhou, P. Mechanism and inhibition of lpxc: An essential zinc-dependent deacetylase of bacterial lipid a synthesis. Curr. Pharm. Biotechnol. 2008, 9, 9–15. [Google Scholar] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Gennadios, H.A.; Christianson, D.W. Binding of uridine 5′-diphosphate in the “basic patch” of the zinc deacetylase lpxc and implications for substrate binding. Biochemistry 2006, 45, 15216–15223. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Sauer, R.; El-Tayeb, A.; Kaulich, M.; Mueller, C.E. Synthesis of uracil nucleotide analogs with a modified, acyclic ribose moiety as P2Y2 receptor antagonists. Bioorg. Med. Chem. 2009, 17, 5071–5079. [Google Scholar] [CrossRef] [PubMed]
- Winans, K.A.; Bertozzi, C.R. An inhibitor of the human udp-glcnac 4-epimerase identified from a uridine-based library. A strategy to inhibit O-linked glycosylation. Chem. Biol. 2002, 9, 113–129. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, X.; Yan, H.; Chen, H. Synthesis, spectroscopic characterization, axial base coordination equilibrium and photolytic kinetics studies of a new coenzyme B12 analogue-3′-deoxy-2′,3′-anhydrothymidylcobalamin. Dalton Trans. 2007, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Prusoff, W.H. A novel synthesis and biological activity of several 5-halo-5′-amino analogues of deoxyribopyrimidine nucleosides. J. Med. Chem. 1978, 21, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Okabe, T.; Okudaira, S.; Nishimasu, H.; Ishitani, R.; Kojima, H.; Nureki, O.; Aoki, J.; Nagano, T. Screening and X-ray crystal structure-based optimization of autotaxin (enpp2) inhibitors, using a newly developed fluorescence probe. ACS Chem. Biol. 2013, 8, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Pontikis, R.; Benhida, R.; Aubertin, A.-M.; Grierson, D.S.; Monneret, C. Synthesis and anti-hiv activity of novel N-1 side chain-modified analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT). J. Med. Chem. 1997, 40, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.; Brinks, M.K.; Hirtz, M.; Schaefer, A.; Chi, L.-F.; Studer, A. Chemical surface modification of self-assembled monolayers by radical nitroxide exchange reactions. Chem. Eur. J. 2011, 17, 9107–9112. [Google Scholar] [CrossRef] [PubMed]
- Margalith, I.; Suter, C.; Ballmer, B.; Schwarz, P.; Tiberi, C.; Sonati, T.; Falsig, J.; Nystroem, S.; Hammarstroem, P.; Aslund, A.; et al. Polythiophenes inhibit prion propagation by stabilizing prion protein (prp) aggregates. J. Biol. Chem. 2012, 287, 18872–18887. [Google Scholar] [CrossRef] [PubMed]
- Jawalekar, A.M.; Meeuwenoord, N.; Cremers, J.G.O.; Overkleeft, H.S.; van der Marel, G.A.; Rutjes, F.P.J.T.; van Delft, F.L. Conjugation of nucleosides and oligonucleotides by [3+2] cycloaddition. J. Org. Chem. 2008, 73, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.M.; Poduch, E.; Fujihashi, M.; Amani, M.; Li, Y.; Crandall, I.; Hui, R.; Lee, P.I.; Kain, K.C.; Pai, E.F.; et al. A potent, covalent inhibitor of orotidine 5′-monophosphate decarboxylase with antimalarial activity. J. Med. Chem. 2007, 50, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Steensma, D.H.; Takaoka, Y.; Yun, J.W.; Kajimoto, T.; Wong, C.-H. A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases. Bioorg. Med. Chem. 1997, 5, 661–672. [Google Scholar] [CrossRef]
- Jackman, J.E.; Fierke, C.A.; Tumey, L.N.; Pirrung, M.; Uchiyama, T.; Tahir, S.H.; Hindsgaul, O.; Raetz, C.R.H. Antibacterial agents that target lipid a biosynthesis in gram-negative bacteria: Inhibition of diverse udp-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylases by substrate analogs containing zinc binding motifs. J. Biol. Chem. 2000, 275, 11002–11009. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Lee, C.-J.; Chen, X.; Chung, H.S.; Zeng, D.; Raetz, C.R.H.; Li, Y.; Zhou, P.; Toone, E.J. Syntheses, structures and antibiotic activities of lpxc inhibitors based on the diacetylene scaffold. Biorg. Med. Chem. 2011, 19, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2009.
- Abrams, H.M.; Ho, L.; Chu, S.H. Synthesis of pyrimidine acyclonucleosides. J. Heterocycl. Chem. 1981, 18, 947–951. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Vacuum Model | Solvated Model | ||
|---|---|---|---|---|
| Zn2+ | Total | Zn2+ | Total | |
| 1b | −296 | −544 | nd | nd |
| 2b | −293 | −529 | nd | nd |
| 3 | −496 | −613 | −491 | −710 |
| 4 | nd | nd | −470 | −602 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malkowski, S.N.; Dishuck, C.F.; Lamanilao, G.G.; Embry, C.P.; Grubb, C.S.; Cafiero, M.; Peterson, L.W. Design, Modeling and Synthesis of 1,2,3-Triazole-Linked Nucleoside-Amino Acid Conjugates as Potential Antibacterial Agents. Molecules 2017, 22, 1682. https://doi.org/10.3390/molecules22101682
Malkowski SN, Dishuck CF, Lamanilao GG, Embry CP, Grubb CS, Cafiero M, Peterson LW. Design, Modeling and Synthesis of 1,2,3-Triazole-Linked Nucleoside-Amino Acid Conjugates as Potential Antibacterial Agents. Molecules. 2017; 22(10):1682. https://doi.org/10.3390/molecules22101682
Chicago/Turabian StyleMalkowski, Sarah N., Carolyn F. Dishuck, Gene G. Lamanilao, Carter P. Embry, Christopher S. Grubb, Mauricio Cafiero, and Larryn W. Peterson. 2017. "Design, Modeling and Synthesis of 1,2,3-Triazole-Linked Nucleoside-Amino Acid Conjugates as Potential Antibacterial Agents" Molecules 22, no. 10: 1682. https://doi.org/10.3390/molecules22101682
APA StyleMalkowski, S. N., Dishuck, C. F., Lamanilao, G. G., Embry, C. P., Grubb, C. S., Cafiero, M., & Peterson, L. W. (2017). Design, Modeling and Synthesis of 1,2,3-Triazole-Linked Nucleoside-Amino Acid Conjugates as Potential Antibacterial Agents. Molecules, 22(10), 1682. https://doi.org/10.3390/molecules22101682