Versatility of 7-Substituted Coumarin Molecules as Antimycobacterial Agents, Neuronal Enzyme Inhibitors and Neuroprotective Agents

 ,
,
Abstract
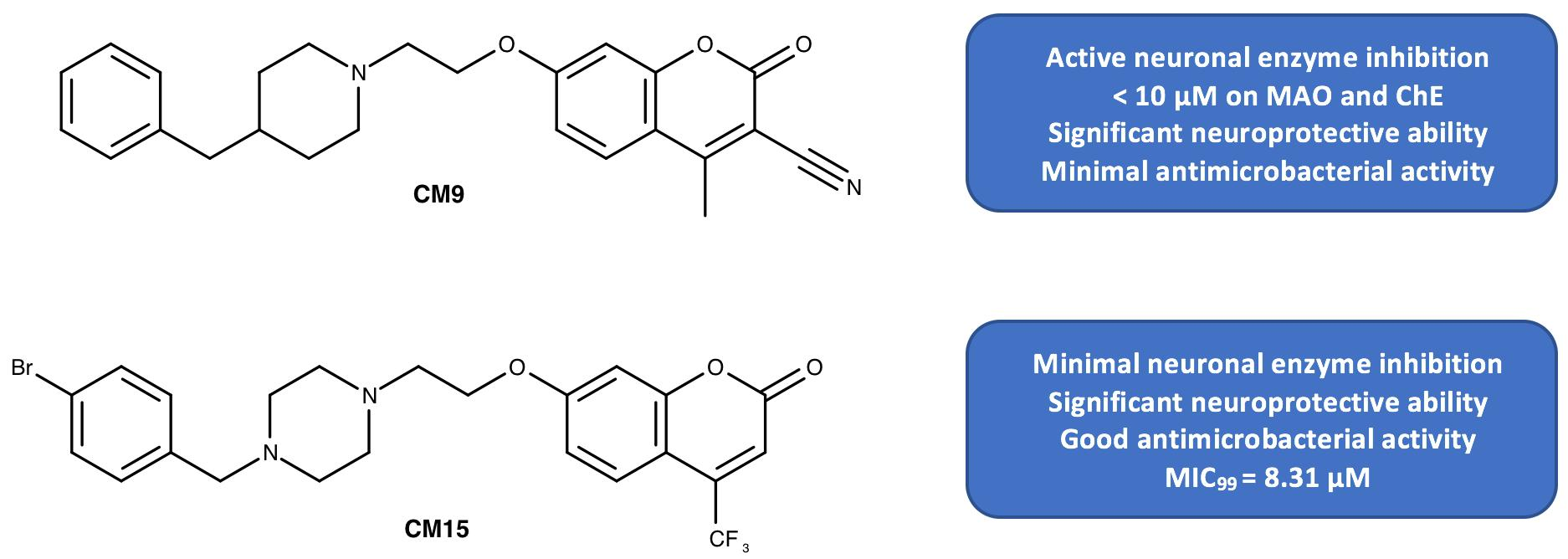
1. Introduction
2. Results and Discussion
2.1. Medium Throughput Screen
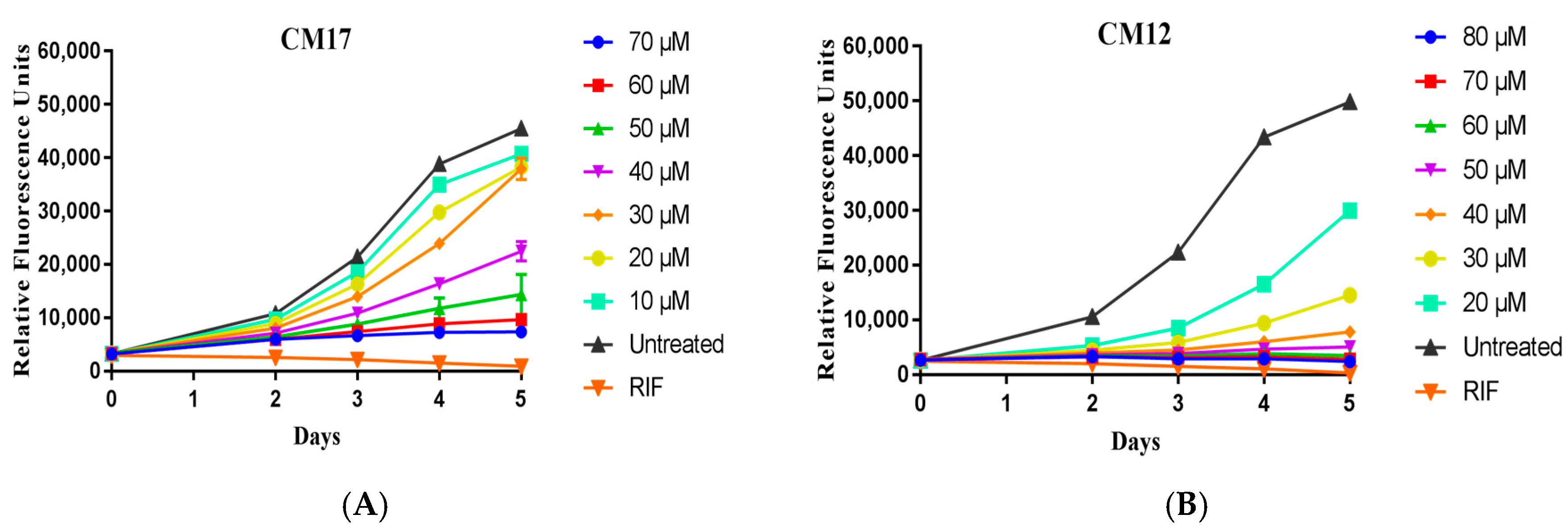
2.2. Evaluation of Compound Activity in Quinolone Resistant Mycobacterium tuberculosis
2.3. Minimum Inhibitory Concentration Determination
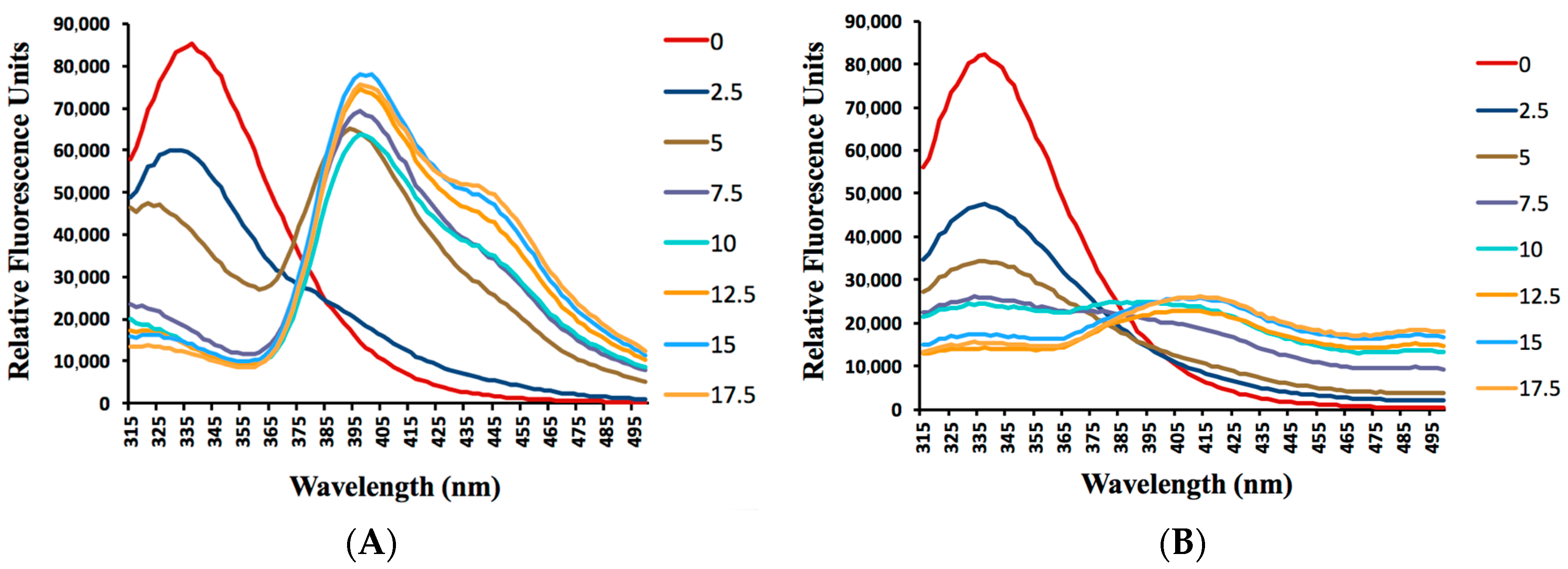
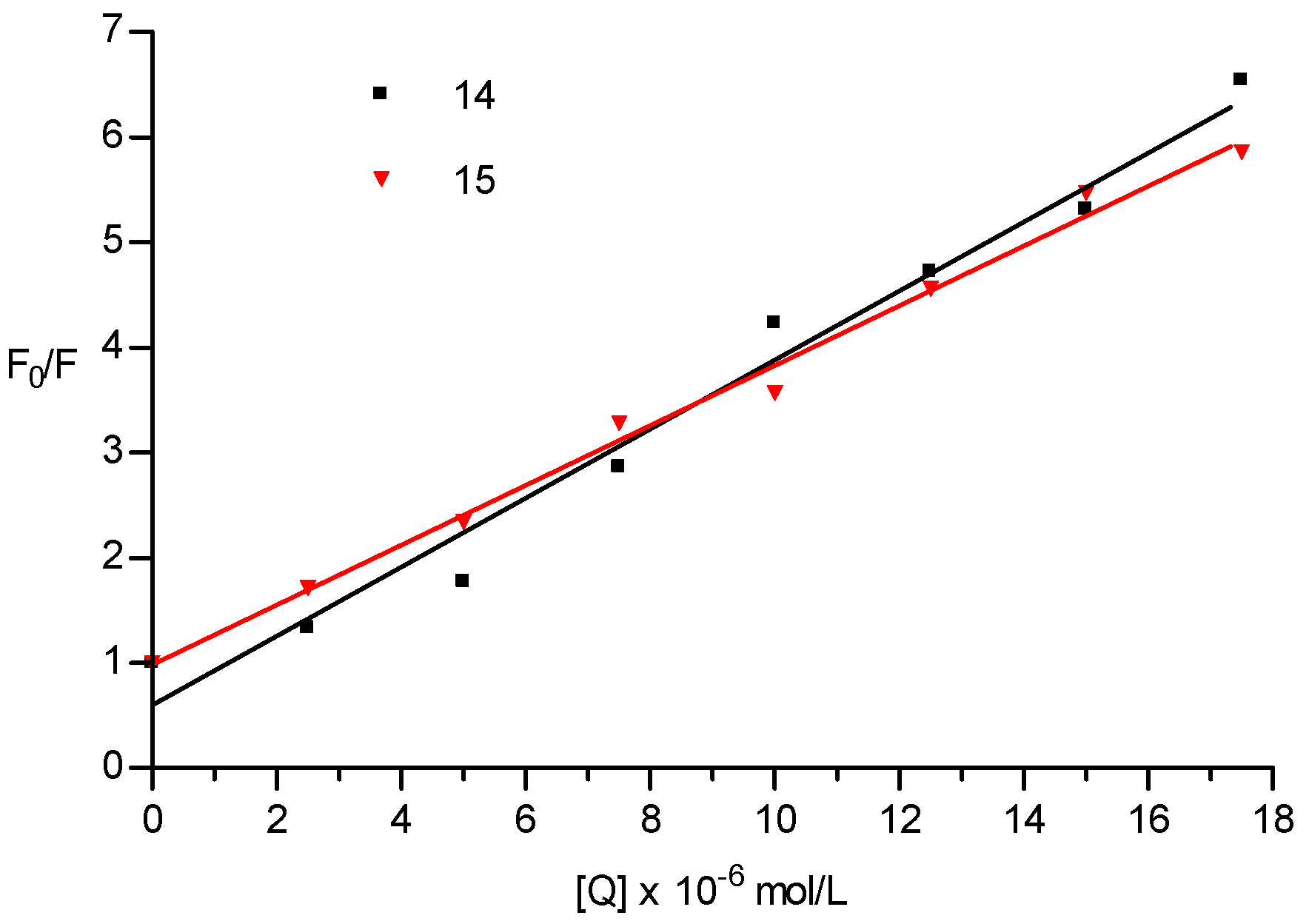
2.4. Albumin Binding Assay
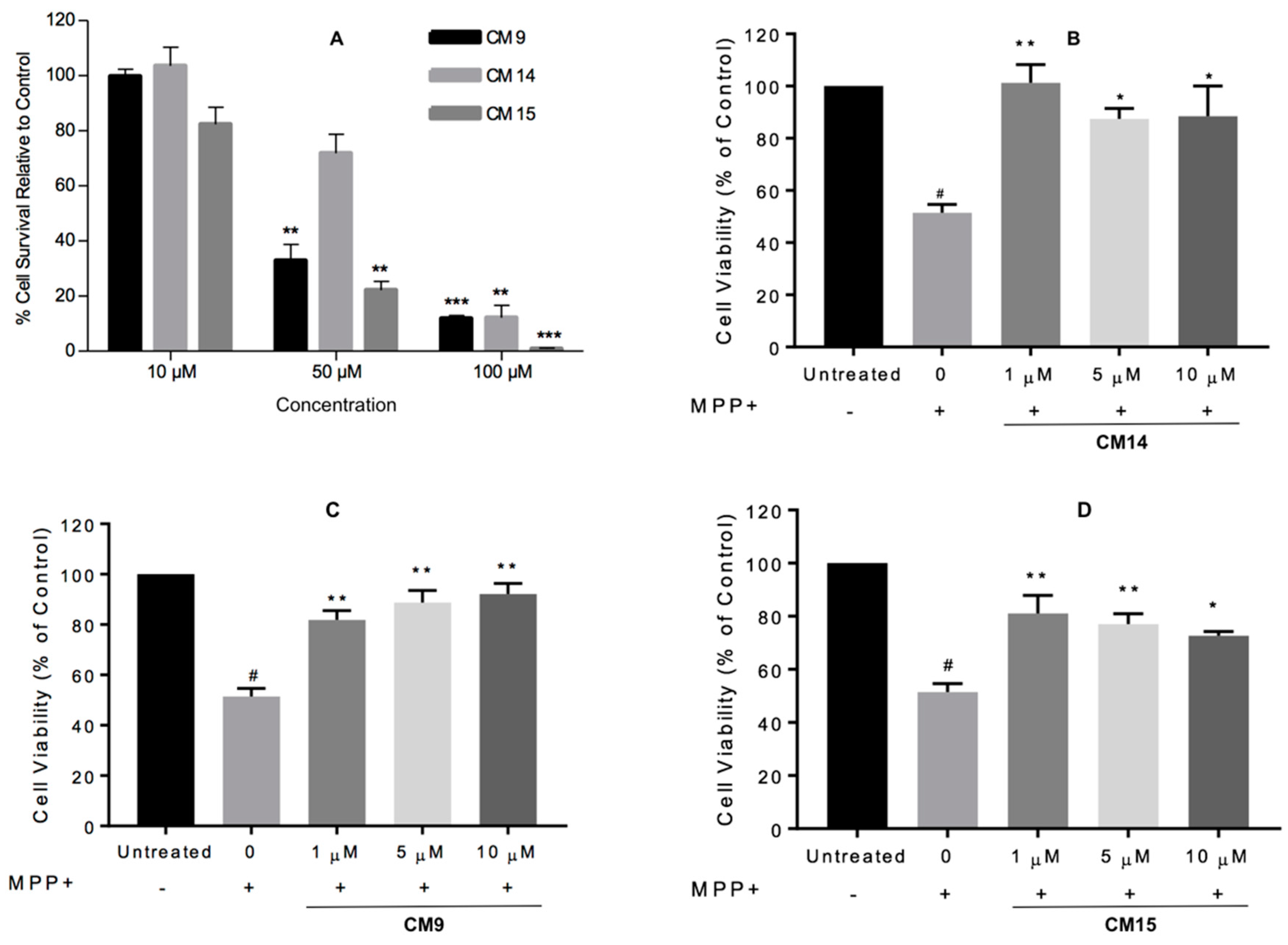
2.5. Cell Viability Assays
3. Materials and Methods
3.1. Compound Synthesis
3.2. Initial Medium Throughput In Vitro Activity Screen
3.3. Activity in Moxifloxacin Resistant M. tuberculosis
3.4. Minimum Inhibitory Concentration Determination
3.4.1. M. tuberculosis pMSP12:GFP in GAST-Fe Media
3.4.2. M. tuberculosis H37Rv:pCHERRY3 in 7H9+OADC
3.5. Bovine Serum Albumin Binding Assay
3.6. Chinese Hamster Ovary Cell Cytotoxicity Assays
3.7. Human Neuroblastoma SH-SY5Y Cell Viability Assays
3.7.1. Cell Line and Culture Conditions
3.7.2. SH-SY5Y Cytotoxicity Assays
3.7.3. MPP+-Induced Cytotoxicity in SH-SY5Y Cells
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, D.G.; Lister, T.; May-Dracka, T.L. New natural products as new leads for antibacterial drug discovery. Bioorg. Med. Chem. Lett. 2014, 24, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Q.; Xu, Z.; Zhang, S.; Wu, X.; Ding, J.W.; Lv, Z.S.; Feng, L.S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 136, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Sasidhar, B.S.; Nagaraja, B.M.; Santos, M.A. Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents. Eur. J. Med. Chem. 2015, 100, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Kawate, T.; Iwase, N.; Shimizu, M.; Clatworthy, A.E.; Kazyanskaya, E.; Sacchettini, J.C.; Ioerger, T.R.; Siddiqi, N.A.; Minami, S.; et al. Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. USA 2013, 110, 11565–11570. [Google Scholar] [CrossRef] [PubMed]
- Heide, L. New aminocoumarin antibiotics as gyrase inhibitors. Int. J. Med. Microbiol. 2014, 304, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Schroder, W.; Goerke, C.; Wolz, C. Opposing effects of aminocoumarins and fluoroquinolones on the SOS response and adaptability in Staphylococcus aureus. J. Antimicrob. Chemother. 2013, 68, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Flatman, R.H.; Howells, A.J.; Heide, L.; Fiedler, H.; Maxwell, A. Simocyclinone D8, an inhibitor of DNA gyrase with a novel mode of action. Antimicrob. Agents Chemother. 2005, 49, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.J.; Williams, M.A.; Maxwell, A.; McKay, A.R. Mass spectrometry reveals that the antibiotic simocyclinone D8 binds to DNA gyrase in a “bent-over” conformation: Evidence of positive cooperativity in binding. Biochemistry 2011, 50, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Hearnshaw, S.J.; Edwards, M.J.; Stevenson, C.E.; Lawson, D.M.; Maxwell, A. A New crystal structure of the bifunctional antibiotic simocyclinone D8 bound to DNA gyrase gives fresh insight into the mechanism of inhibition. J. Mol. Biol. 2014, 426, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Hao, H.; Dai, M.; Liu, Z.; Yuan, Z. Antibacterial action of quinolones: From target to network. Eur. J. Med. Chem. 2013, 66, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Blower, T.R.; Kerns, R.J.; Berger, J.M.; Osheroff, N. Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: Enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. USA 2016, 113, E839–E846. [Google Scholar] [CrossRef] [PubMed]
- Shadrick, W.R.; Ndjomou, J.; Kolli, R.; Mukherjee, S.; Hanson, A.M.; Frick, D.N. Discovering new medicines targeting helicases: Challenges and recent progress. J. Biomol. Screen. 2013, 18, 761–781. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Pai, R.; Di, M.; Aiello, D.; Barnes, M.H.; Butler, M.M.; Tashjian, T.F.; Peet, N.P.; Bowlin, T.L.; Moir, D.T. Coumarin-based inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicase: Chemical optimization, biological evaluation, and antibacterial activities. J. Med. Chem. 2012, 55, 10896–10908. [Google Scholar] [CrossRef] [PubMed]
- Aiello, D.; Barnes, M.H.; Biswas, E.E.; Biswas, S.B.; Gu, S.; Williams, J.D.; Bowlin, T.L.; Moir, D.T. Discovery, characterization and comparison of inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicases. Bioorg. Med. Chem. 2009, 17, 4466–4476. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Matsuyama, K.; Tran, T.; Malerich, J.P.; Wan, B.; Franzblau, S.G.; Lun, S.; Guo, H.; Maiga, M.C.; Bishai, W.R.; et al. Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2011, 67, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Jeankumar, V.U.; Reshma, R.S.; Janupally, R.; Saxena, S.; Sridevi, J.P.; Medapi, B.; Kulkarni, P.; Yogeeswari, P.; Sriram, D. Enabling the (3 + 2) cycloaddition reaction in assembling newer anti-tubercular lead acting through the inhibition of the gyrase ATPase domain: Lead optimization and structure activity profiling. Org. Biomol. Chem. 2015, 13, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Jeyachandran, M.; Ramesh, P.; Sriram, D.; Senthilkumar, P.; Yogeeswari, P. Synthesis and in vitro antitubercular activity of 4-aryl/alkylsulfonylmethylcoumarins as inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2012, 22, 4807–4809. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Kulkarni, M.V.; Joshi, S.D.; Dixit, S.R. One pot Click chemistry: A three component reaction for the synthesis of 2-mercaptobenzimidazole linked coumarinyl triazoles as anti-tubercular agents. Bioorg. Med. Chem. Lett. 2016, 26, 4709–4713. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.S.; Hosamani, K.M.; Devarajegowda, H.C. Design, synthesis of benzocoumarin-pyrimidine hybrids as novel class of antitubercular agents, their DNA cleavage and X-ray studies. Eur. J. Med. Chem. 2015, 101, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Somersan-Karakaya, S.; Lu, S.; Roberts, J.; Pingle, M.; Warrier, T.; Little, D.; Guo, X.; Brickner, S.J.; Nathan, C.F.; et al. Synthetic calanolides with bactericidal activity against replicating and nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2014, 57, 3755–3772. [Google Scholar] [CrossRef] [PubMed]
- Basanagouda, M.; Jambagi, V.B.; Barigidad, N.N.; Laxmeshwar, S.S.; Devaru, V.; Narayanachar. Synthesis, structure-activity relationship of iodinated-4-aryloxymethyl-coumarins as potential anti-cancer and anti-mycobacterial agents. Eur. J. Med. Chem. 2014, 74, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Skalicka-Woźniak, K.; Orhan, I.E.; Cordell, G.A.; Nabavi, S.M.; Budzyńska, B. Implication of coumarins towards central nervous system disorders. Pharmacol. Res. 2016, 103, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; D’Ascenzio, P.; Chimenti, D.; Secci, A.; Bolasco, A. Selective MAO-B inhibitors. Mol. Div. 2016, 254, 219–243. [Google Scholar]
- De Souza, L.G.; Rennó, M.N.; Figueroa-Villar, J. Coumarins as cholinesterase inhibitors: A review. Chem. Biol. Interact. 2016, 254, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Foka, G.B.; Repsold, B.P.; Oliver, D.W.; Kapp, E.; Malan, S.F. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 125, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.O.; Bari, S.B.; Firke, S.D.; Deshmukh, P.K.; Donda, S.T.; Patil, D.A. A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 2434–2450. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 1, CD001190. [Google Scholar]
- Kuhn, M.L.; Alexander, E.; Minasov, G.; Page, H.J.; Warwrzak, Z.; Shuvalova, L.; Flores, K.J.; Wilson, D.J.; Shi, C.; Aldrich, C.C.; et al. Structure of the essential Mtb FadD32 Enzyme: A promising drug target for treating tuberculosis. ACS Infect. Dis. 2016, 2, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Maruri, F.; Sterling, T.R.; Kaiga, A.W.; Blackman, A.; van der Heijden, Y.F.; Mayer, C.; Cambau, E.; Aubry, A. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J. Antimicrob. Chemother. 2012, 67, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.; Schreuder, L.J.; Muwanguzi-Karugaba, J.; Wiles, S.; Robertson, B.D.; Ripoll, J.; Ward, T.H.; Bancroft, G.J.; Schaible, U.E.; Parish, T. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS ONE 2010, 5, e9823. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, G.L.; Kumar, A.; Savvi, S.; Hung, A.W.; Wen, S.; Abell, C.; Barry, C.E.; Sherman, D.R.; Boshoff, H.I.; Mizrahi, V. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 2012, 19, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Lounis, N.; Vranckx, L.; Gevers, T.; Kaniga, K.; Andries, K. In vitro culture conditions affecting minimal inhibitory concentration of bedaquiline against M. tuberculosis. Med. Mal. Infect. 2016, 46, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Dockal, M.; Carter, D.C.; Ruker, F. Conformational transitions of the three recombinant domains of human serum albumin depending on pH. J. Biol. Chem. 2000, 275, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Wu, X.; Zheng, J.; Yang, J.; Zhou, H.; Zhang, M.; Tang, Y. Study on the interaction between Florasulam and bovine serum albumin. J. Fluoresc. 2007, 17, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, J.; Zhang, J.; Hu, Z.; Chen, X. Fluorescence studies on the interactions of barbaloin with bovine serum albumin. Chem. Pharm. Bull. 2003, 51, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Seetharamappa, J.; Kamat, B.P. Spectroscopic studies on the mode of interaction of an anticancer drug with bovine serum albumin. Chem. Pharm. Bull. (Tokyo) 2004, 52, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Sulkowska, A.; Rownicka, J.; Bojko, B.; Sulkowski, W. Interaction of anticancer drugs with human and bovine serum albumin. J. Mol. Struct. 2003, 651, 133–140. [Google Scholar] [CrossRef]
- Kun, R.; Kis, L.; Dekany, I. Hydrophobization of bovine serum albumin with cationic surfactants with different hydrophobic chain length. Colloids Surf. B Biointerfaces 2010, 79, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Guo, Y.; Wang, J.; Xu, R.; Wang, X.; Wang, D.; Zhang, L.; Xu, Y. Spectroscopic studies on the interaction and sonodynamic damage of neutral red (NR) to bovine serum albumin (BSA). J. Lumin. 2010, 130, 1036–1043. [Google Scholar] [CrossRef]
- Xiang, G.; Tong, C.; Lin, H. Nitroaniline isomers interaction with bovine serum albumin and toxicological implications. J. Fluoresc. 2007, 17, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, Y.; Zhang, L.; Zhao, R.; Qu, S. Studies of interaction between colchicine and bovine serum albumin by fluorescence quenching method. J. Mol. Struct. 2005, 750, 174–178. [Google Scholar] [CrossRef]
- Gök, E.; Öztürk, C.; Akbay, N. Interaction of Thyroxine with 7 Hydroxycoumarin: A fluorescence quenching study. J. Fluoresc. 2008, 18, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gao, S.; Lv, J.; Liu, Q.; Zuo, Y.; Wang, X. Spectroscopic investigations on the mechanism of interaction of crystal violet with bovine serum albumin. J. Mol. Struct. 2009, 919, 334–338. [Google Scholar] [CrossRef]
- Shaikh, S.M.T.; Seetharamappa, J.; Kandagal, P.B.; Manjunatha, D.H.; Ashoka, S. Spectroscopic investigations on the mechanism of interaction of bioactive dye with bovine serum albumin. Dyes Pigment. 2007, 74, 665–671. [Google Scholar] [CrossRef]
- Papadopoulou, A.; Green, R.J.; Frazier, R.A. Interaction of flavonoids with bovine serum albumin: A fluorescence quenching study. J. Agric. Food Chem. 2005, 53, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, X.; Gui, M.; Zhou, H.; Yang, R.; Zhang, H.; Jin, Y. Studies on interaction between flavonoids and bovine serum albumin by spectral methods. J. Lumin. 2010, 130, 637–644. [Google Scholar] [CrossRef]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Z.; Li, W.; Hu, S.; Mak, S.; Zhang, H.; Han, R.; Yuan, S.; Li, S.; Sa, F.; et al. The anti-cancer agent SU4312 unexpectedly protects against MPP(+)-induced neurotoxicity via selective and direct inhibition of neuronal NOS. Br. J. Pharmacol. 2013, 168, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Greda, A.; Golda, S.; Korostynski, M.; Grygier, B.; Roman, A.; Pilc, A.; Lason, W. Neuroprotective effects of metabotropic glutamate receptor group II and III activators against MPP(+)-induced cell death in human neuroblastoma SH-SY5Y cells: The impact of cell differentiation state. Neuropharmacology 2014, 83, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Pyszko, J.; Strosznajder, J.B. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, J.; Miao, Y.; Cui, Q.; Zhao, W.; Zhang, J.; Wang, H. Pinocembrin protects SH-SY5Y cells against MPP+-induced neurotoxicity through the mitochondrial apoptotic pathway. J. Mol. Neurosci. 2014, 53, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Nakaso, K.; Tajima, N.; Horikoshi, Y.; Nakasone, M.; Hanaki, T.; Kamizaki, K.; Matsura, T. The estrogen receptor beta-PI3K/Akt pathway mediates the cytoprotective effects of tocotrienol in a cellular Parkinson’s disease model. Biochim. Biophys. Acta 2014, 1842, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, Y.; Omura, T.; Yamada, T.; Ohkubo, T.; Suno, M.; Iida, S.; Sakaguchi, T.; Asari, M.; Shimizu, K.; Matsubara, K. Meloxicam protects cell damage from 1-methyl-4-phenyl pyridinium toxicity via the phosphatidylinositol 3-kinase/Akt pathway in human dopaminergic neuroblastoma SH-SY5Y cells. Brain Res. 2010, 1344, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, X.; Wu, S.; Li, Q. Involvement of activation of PI3K/Akt pathway in the protective effects of puerarin against MPP+-induced human neuroblastoma SH-SY5Y cell death. Neurochem. Int. 2012, 60, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Ioerger, T.R.; Feng, Y.; Ganesula, K.; Chen, X.; Dobos, K.M.; Fortune, S.; Jacobs, W.R., Jr.; Mizrahi, V.; Parish, T.; Rubin, E.; et al. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 2010, 192, 3645–3653. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, R.M.; Goonesekera, S.D.; Bloom, B.R.; Sampson, S.L. The transcriptional regulator Rv0485 modulates the expression of a pe and ppe gene pair and is required for Mycobacterium tuberculosis virulence. Infect. Immun. 2009, 77, 4654–4667. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, J.; Bailey, M.A.; Moraski, G.C.; Casey, A.; Florio, S.; Alling, T.; Miller, M.J.; Parish, T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS ONE 2013, 8, e60531. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds CM1–CM19 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | ||||||
|---|---|---|---|---|---|---|---|
| Series 1 | Series 2 | ||||||
| % M. tuberculosis Growth, Day 5 | |||||||
| Number | X | R1 | R2 | R3 | 100 µM | 50 µM | 1 µM |
| Series 1 | |||||||
| CM12 ** | N | Br | CH3 | H | 3.93 | 4.35 | 95.23 |
| CM14 ** | N | Br | CH3 | CN | 4.44 | 4.40 | 99.53 |
| CM8 ** | CH | H | CH3 | Cl | 4.62 | 5.69 | 93.59 |
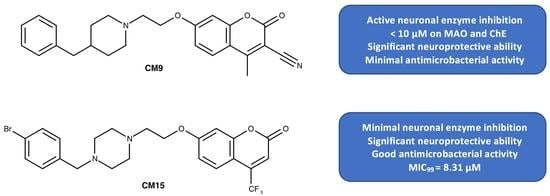
| CM15 ** | N | Br | CF3 | H | 4.88 | 6.68 | 106.71 |
| CM11 * | N | Br | H | H | 4.46 | 7.05 | 95.18 |
| CM9 * | CH | H | CH3 | CN | 6.19 | 7.75 | 92.82 |
| CM7 * | CH | H | CH3 | H | 8.18 | 29.50 | 95.10 |
| CM6 | CH | H | H | H | 15.43 | 52.71 | 98.19 |
| CM13 * | N | Br | CH3 | Cl | 32.06 | 42.47 | 97.03 |
| Series 2 | |||||||
| CM17 * | H | CH3 | H | 19.81 | 28.65 | 97.59 | |
| CM2 * | Br | CH3 | H | 33.16 | 40.86 | 89.98 | |
| CM4 * | Br | CH3 | CN | 33.52 | 44.60 | 95.27 | |
| CM5 | Br | CF3 | H | 56.09 | 65.04 | 106.87 | |
| CM19 | H | CH3 | CN | 55.44 | 66.92 | 98.28 | |
| CM16 | H | H | H | 50.66 | 72.70 | 97.53 | |
| CM1 | Br | H | H | 63.63 | 79.77 | 96.28 | |
| CM18 | H | CH3 | Cl | 75.52 | 87.91 | 97.30 | |
| CM3 | Br | CH3 | Cl | 92.86 | 96.11 | 97.82 | |
| Compound | GFP GAST-Fe | mCHERRY—7H9+OADC | ||||
|---|---|---|---|---|---|---|
| MIC90 (µM) | MIC99 (µM) | MIC90 (µM) | St. Dev. | MIC99 (µM) | St. Dev. | |
| CM8 | 19.6 | 25.2 | 40.55 | 1.16 | 44.15 | 1.37 |
| CM12 | 14 | 29.7 | 40.89 | 0.06 | 50.40 | 8.96 |
| CM14 | 16.7 | 28 | 45.22 | 3.04 | 54.46 | 9.58 |
| CM15 | 3.2 | 8.31 | 43.78 | 1.46 | 57.17 | 10.05 |
| Compound | T (°C) | λEx (nm) | λEm (nm) | ksv (mol L−1) | kq (mol L−1 S−1) | R2 |
|---|---|---|---|---|---|---|
| CM14 | 22 | 350 a 350 b | 395 a 400 b | 3.80 × 105 | 2.63 × 1014 | 0.9780 |
| CM15 | 22 | 335 a 335 b | 415 a 415 b | 4.88 × 105 | 2.00 × 1014 | 0.9927 |
| CM | CC50 µM CHO | SI GFP a | SI mCHERRY b | CC 50% SH-SY5Y c | MAO-A IC50 µM d | MAO-B IC50 µM d | AChE IC50 µM d | BuChE IC50 µM d |
|---|---|---|---|---|---|---|---|---|
| CM8 | 33.0 | 1.31 | 0.75 | ND | n.a. | 0.29 | 31.30 | 1.27 |
| CM9 | ND | ND | ND | 10–50 | n.a. | 0.30 | 9.10 | 5.90 |
| CM12 | 15.5 | 0.52 | 0.31 | ND | n.a. | 3.60 | 12.8 | 9.70 |
| CM14 | 41.2 | 1.47 | 0.76 | 50–100 | n.a. | 1.41 | 38.5 | 13.3 |
| CM15 | 26.3 | 3.16 | 0.46 | 10–50 | n.a. | 5.64 | >100 | >100 |
| Emetine | 0.06 | ND | ND | ND | ND | ND | ND | ND |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapp, E.; Visser, H.; Sampson, S.L.; Malan, S.F.; Streicher, E.M.; Foka, G.B.; Warner, D.F.; Omoruyi, S.I.; Enogieru, A.B.; Ekpo, O.E.; et al. Versatility of 7-Substituted Coumarin Molecules as Antimycobacterial Agents, Neuronal Enzyme Inhibitors and Neuroprotective Agents. Molecules 2017, 22, 1644. https://doi.org/10.3390/molecules22101644
Kapp E, Visser H, Sampson SL, Malan SF, Streicher EM, Foka GB, Warner DF, Omoruyi SI, Enogieru AB, Ekpo OE, et al. Versatility of 7-Substituted Coumarin Molecules as Antimycobacterial Agents, Neuronal Enzyme Inhibitors and Neuroprotective Agents. Molecules. 2017; 22(10):1644. https://doi.org/10.3390/molecules22101644
Chicago/Turabian StyleKapp, Erika, Hanri Visser, Samantha L. Sampson, Sarel F. Malan, Elizabeth M. Streicher, Germaine B. Foka, Digby F. Warner, Sylvester I. Omoruyi, Adaze B. Enogieru, Okobi E. Ekpo, and et al. 2017. "Versatility of 7-Substituted Coumarin Molecules as Antimycobacterial Agents, Neuronal Enzyme Inhibitors and Neuroprotective Agents" Molecules 22, no. 10: 1644. https://doi.org/10.3390/molecules22101644
APA StyleKapp, E., Visser, H., Sampson, S. L., Malan, S. F., Streicher, E. M., Foka, G. B., Warner, D. F., Omoruyi, S. I., Enogieru, A. B., Ekpo, O. E., Zindo, F. T., & Joubert, J. (2017). Versatility of 7-Substituted Coumarin Molecules as Antimycobacterial Agents, Neuronal Enzyme Inhibitors and Neuroprotective Agents. Molecules, 22(10), 1644. https://doi.org/10.3390/molecules22101644