
Direct Arylation Strategies in the Synthesis of π-Extended Monomers for Organic Polymeric Solar Cells



Abstract
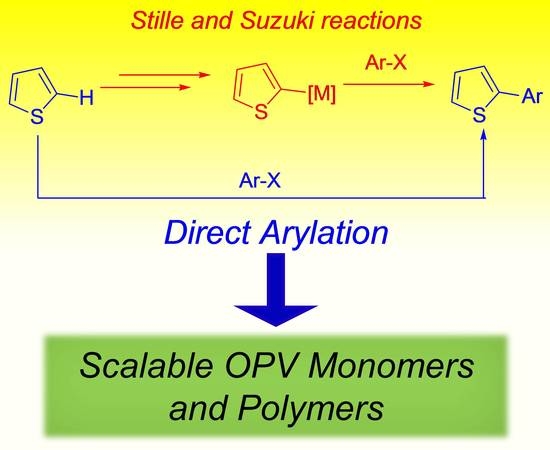
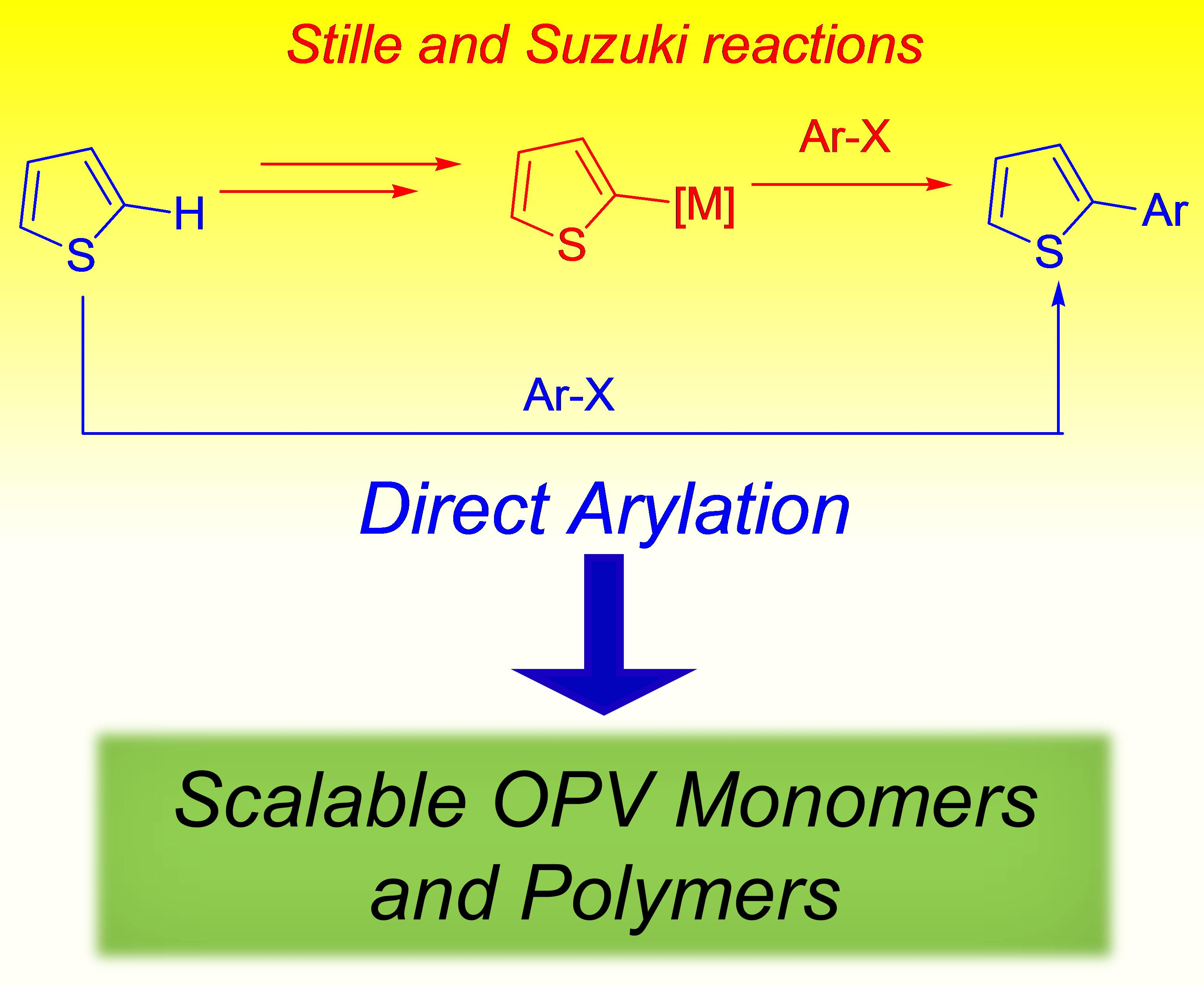
:
{kind=link}
{kind=link}
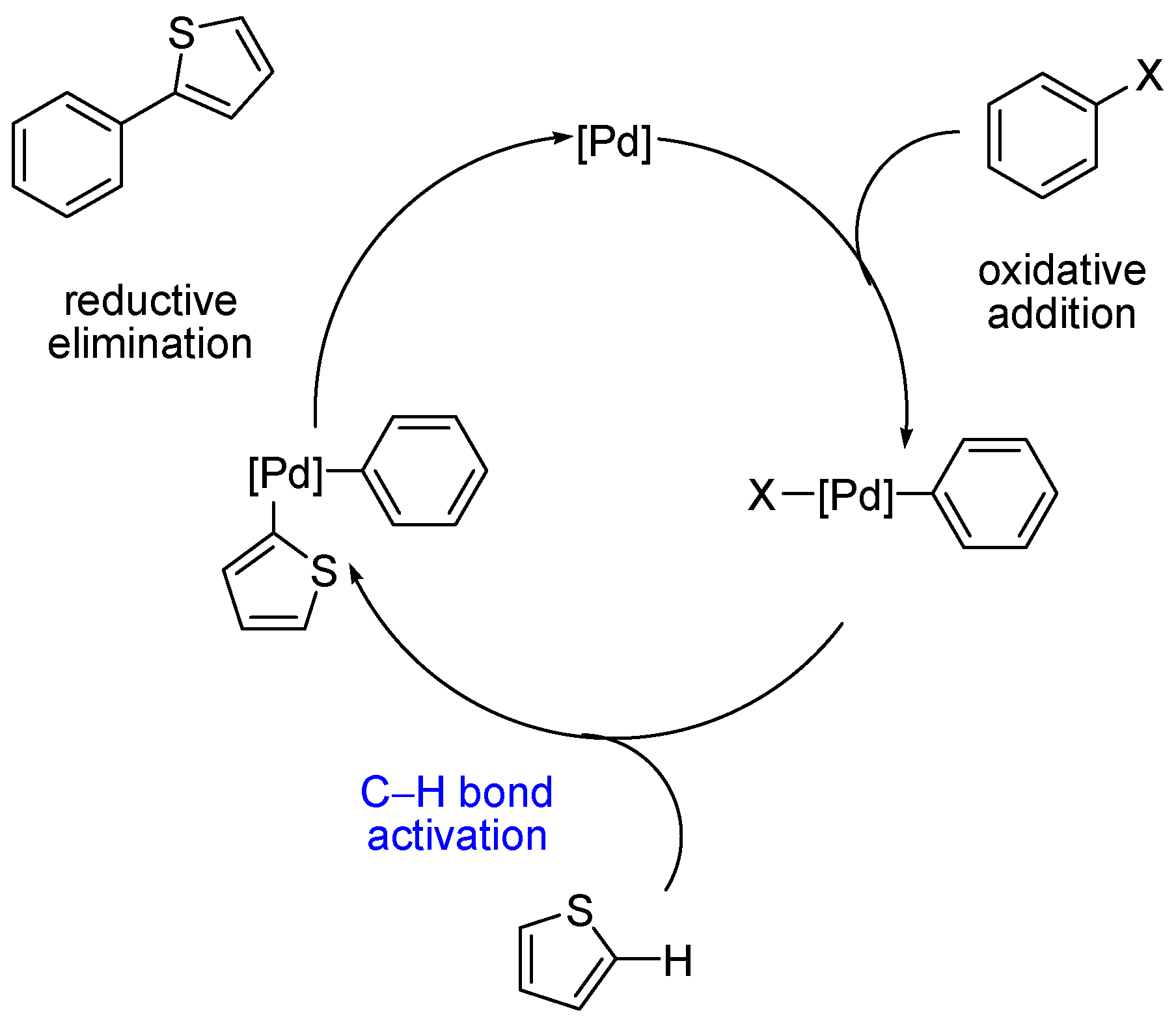{kind=link}
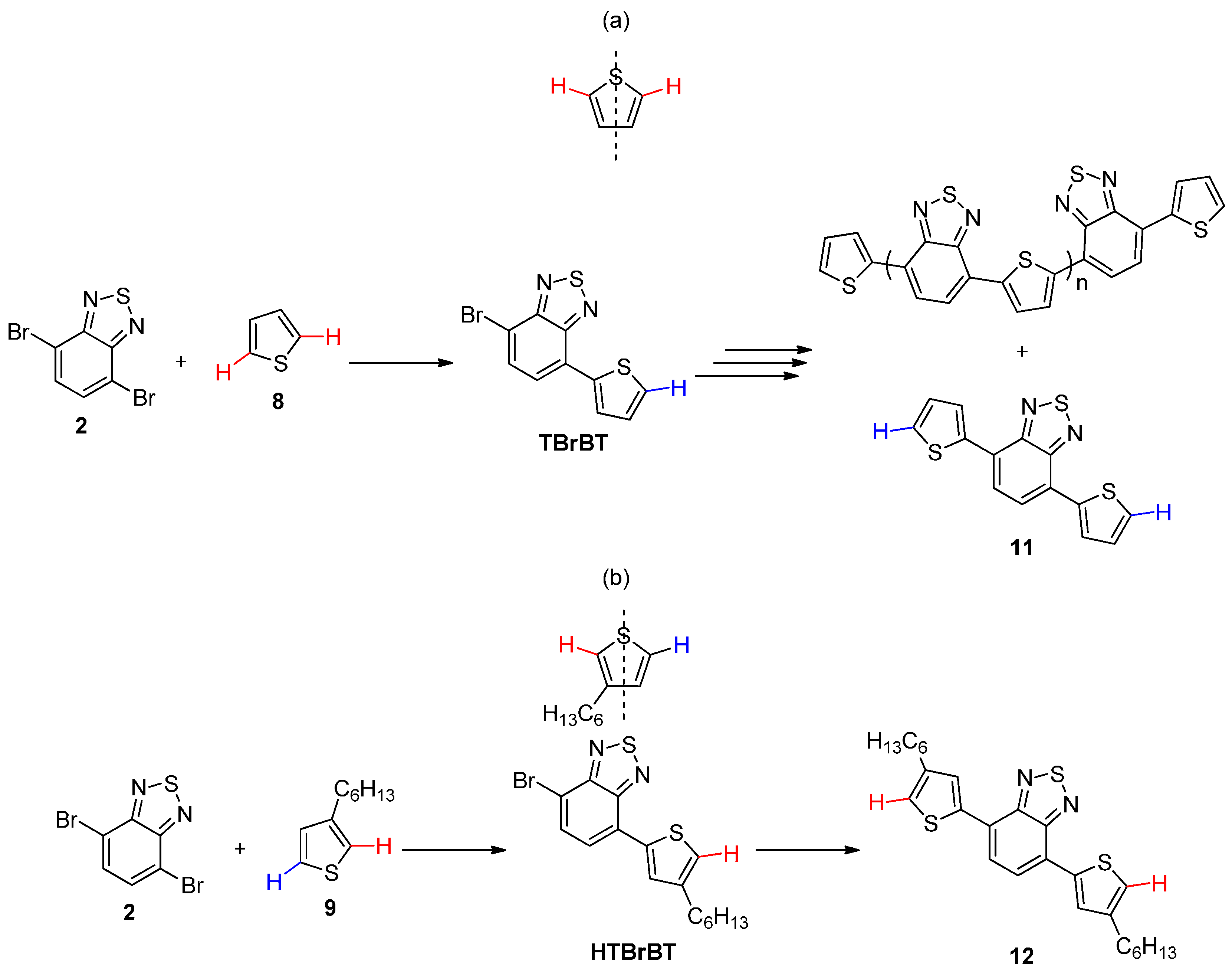{kind=link}
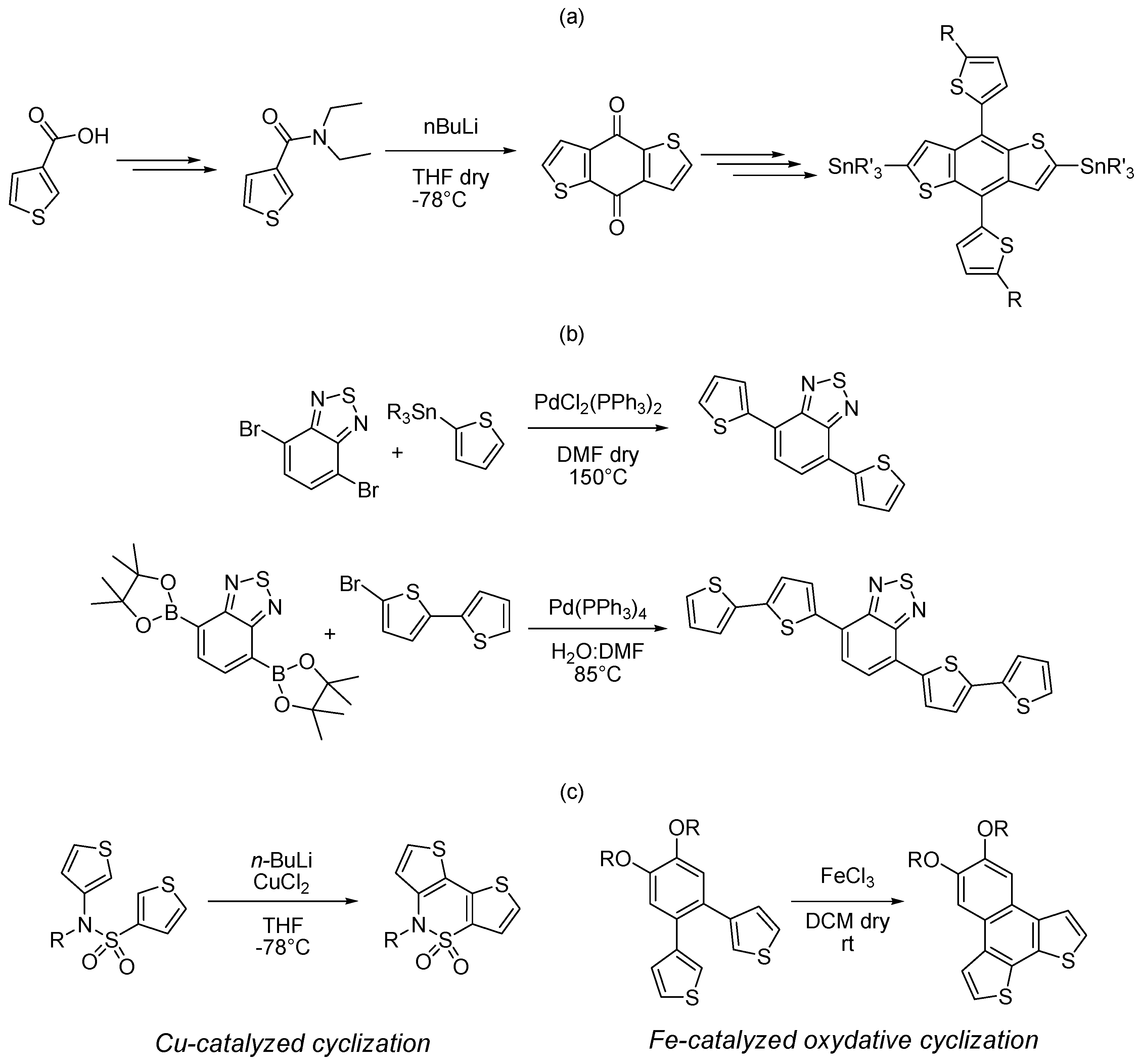{kind=link}
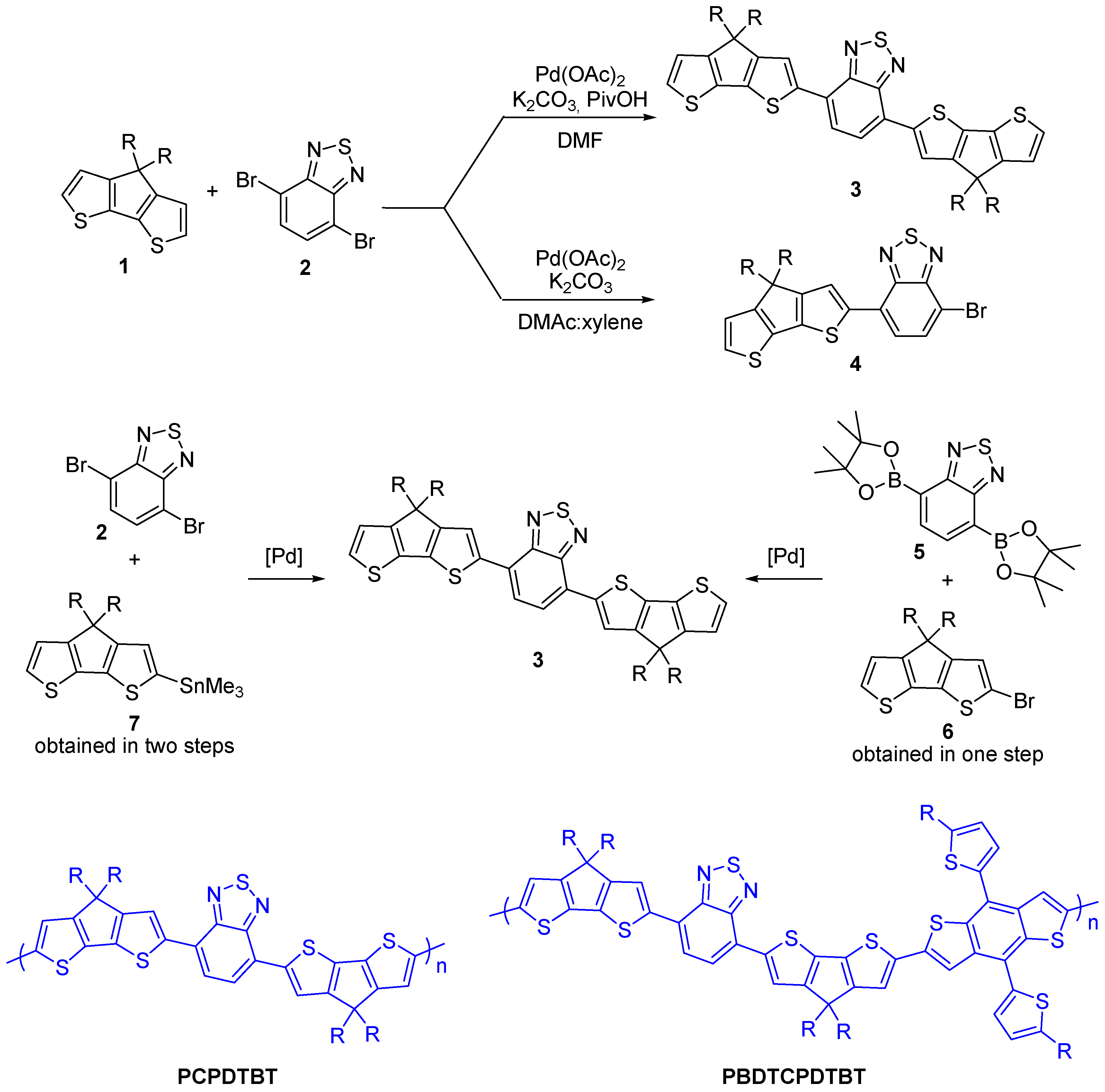{kind=link}
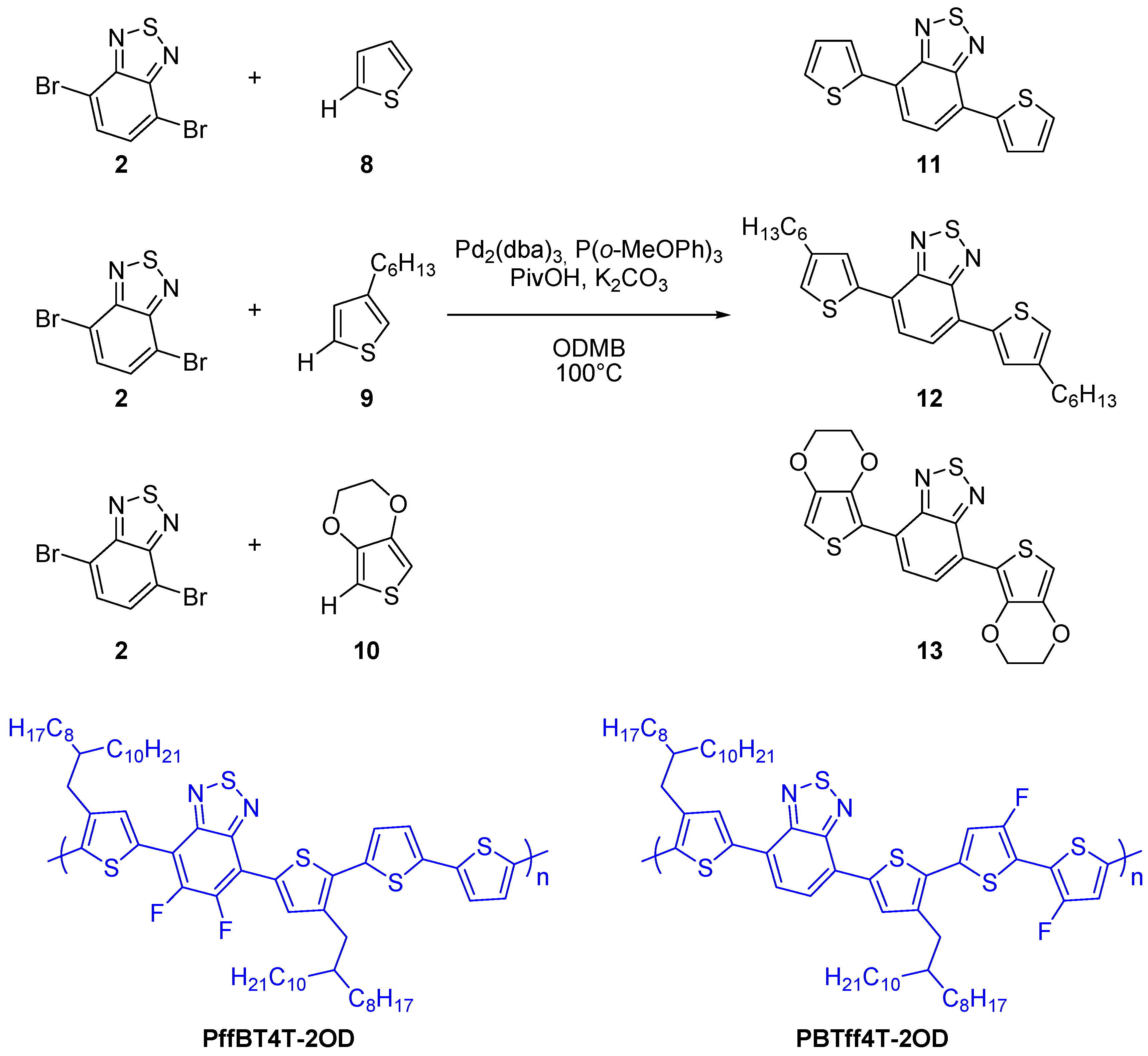{kind=link}
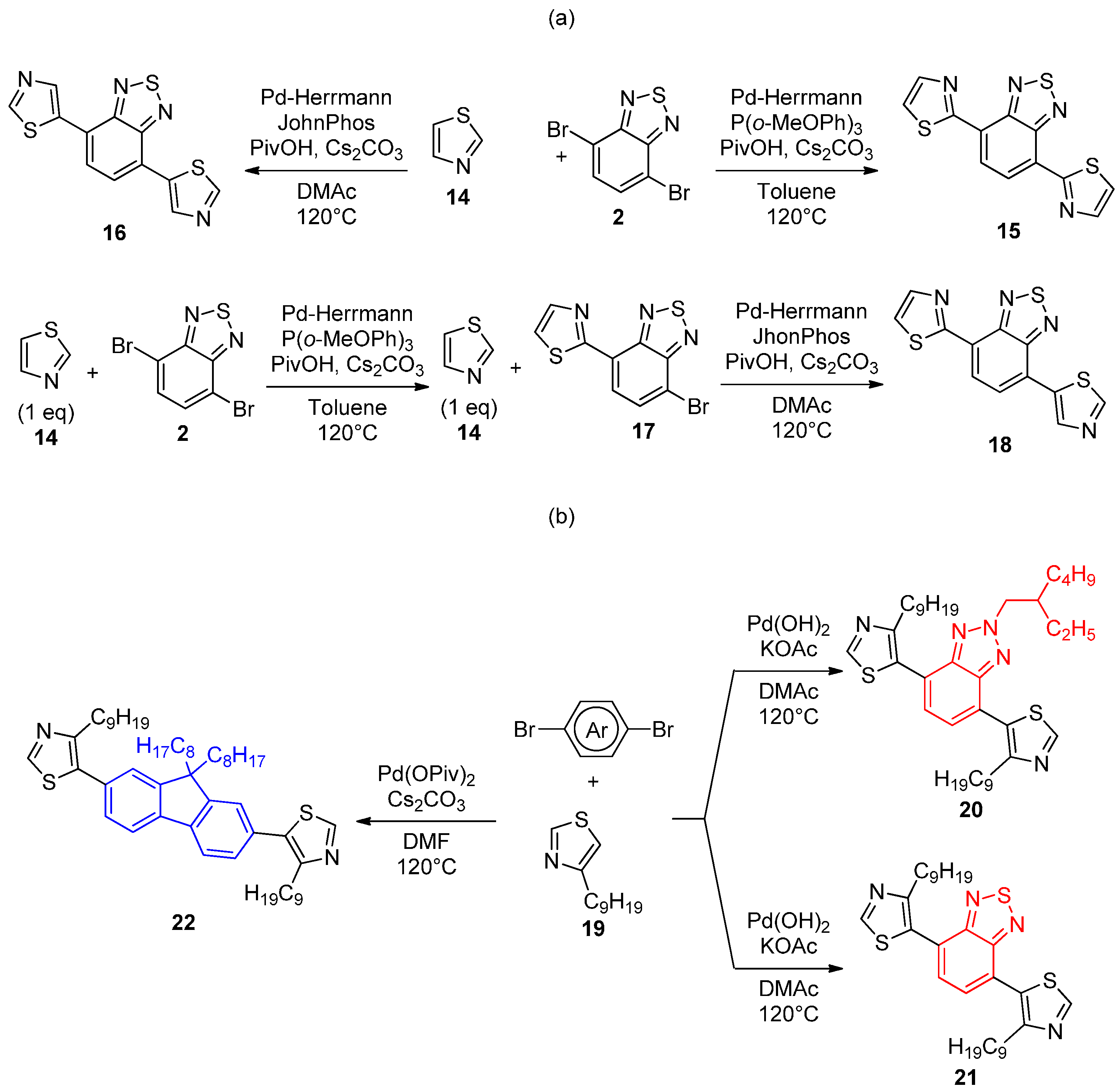{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Discussion
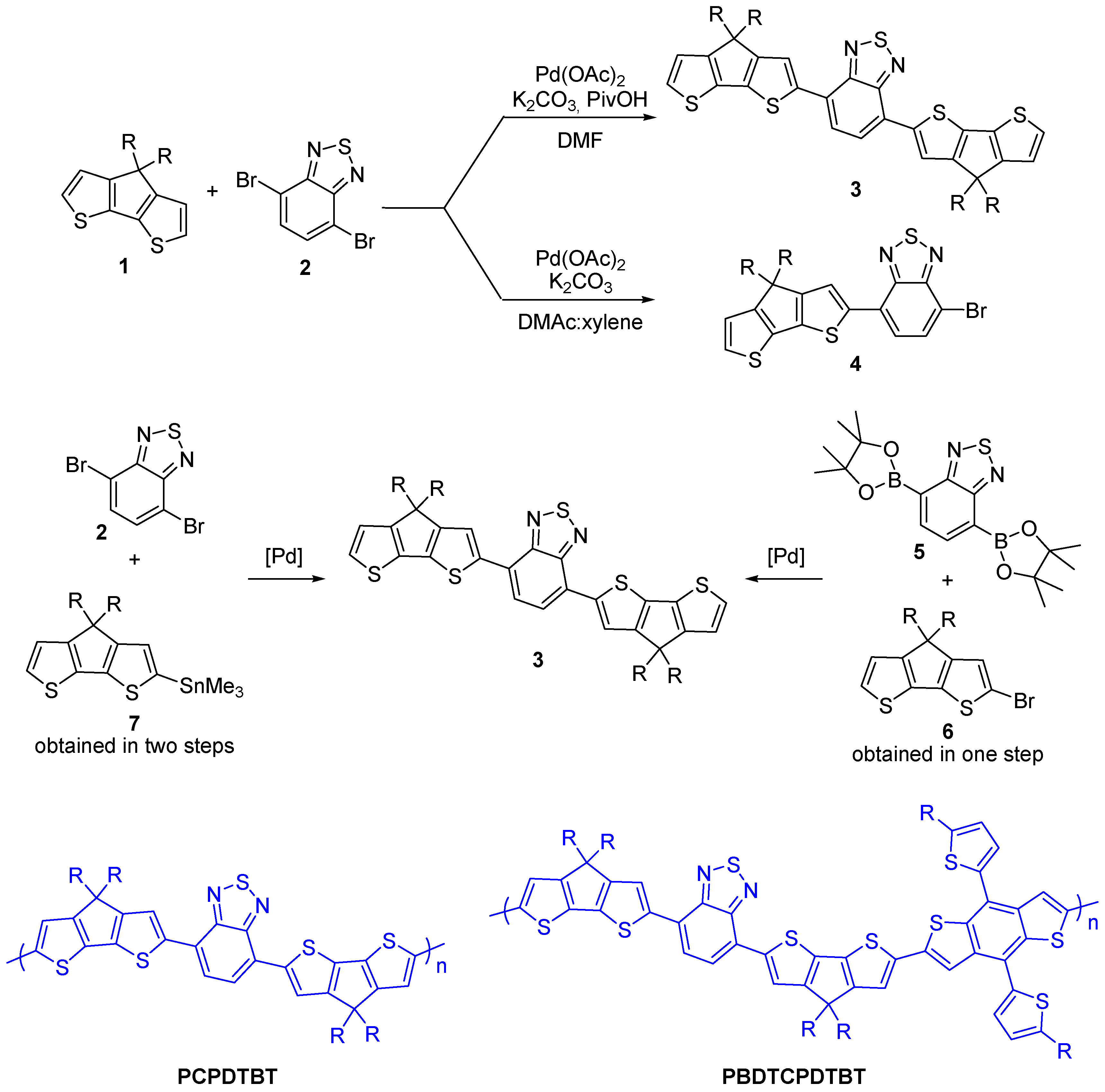
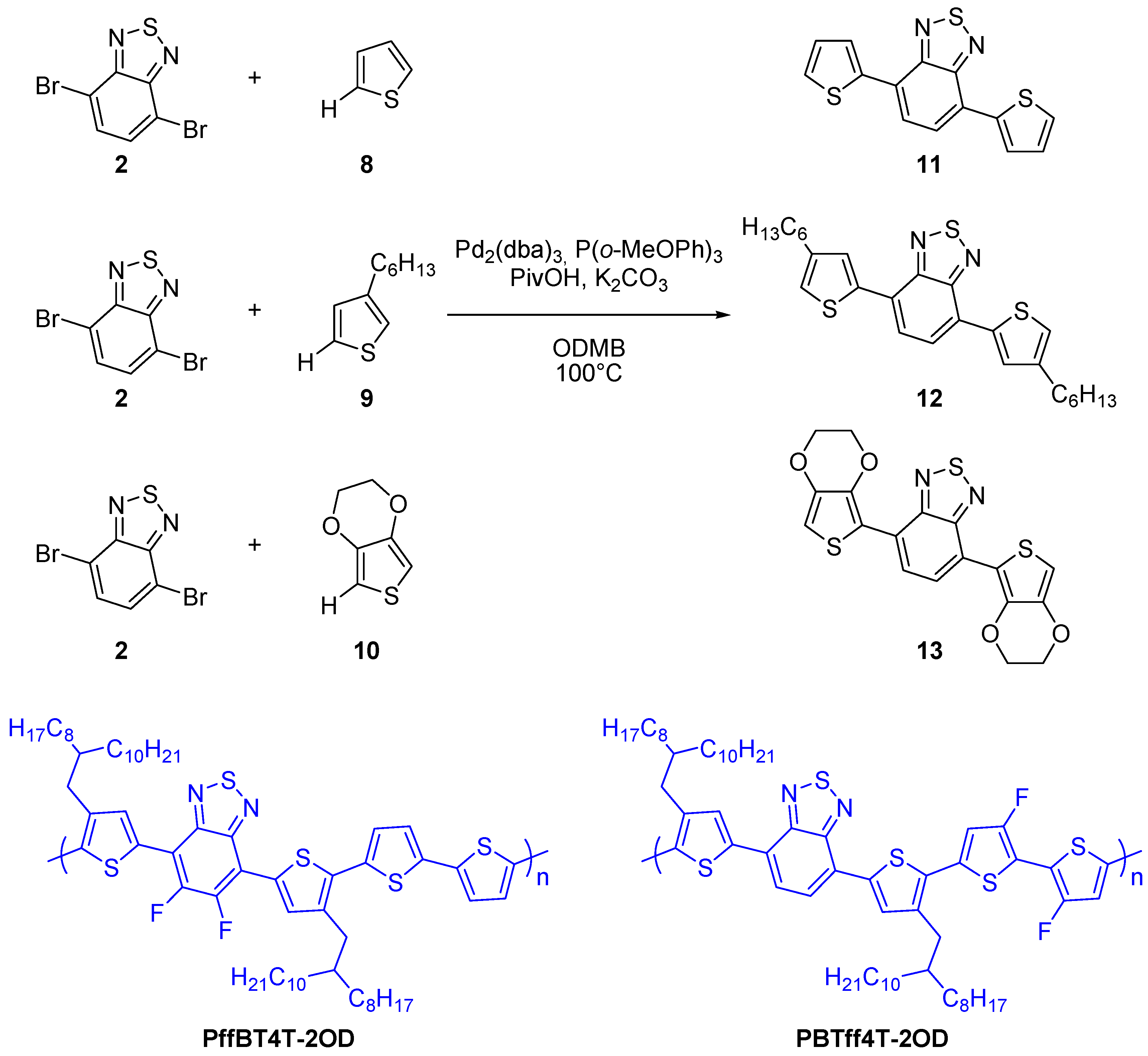
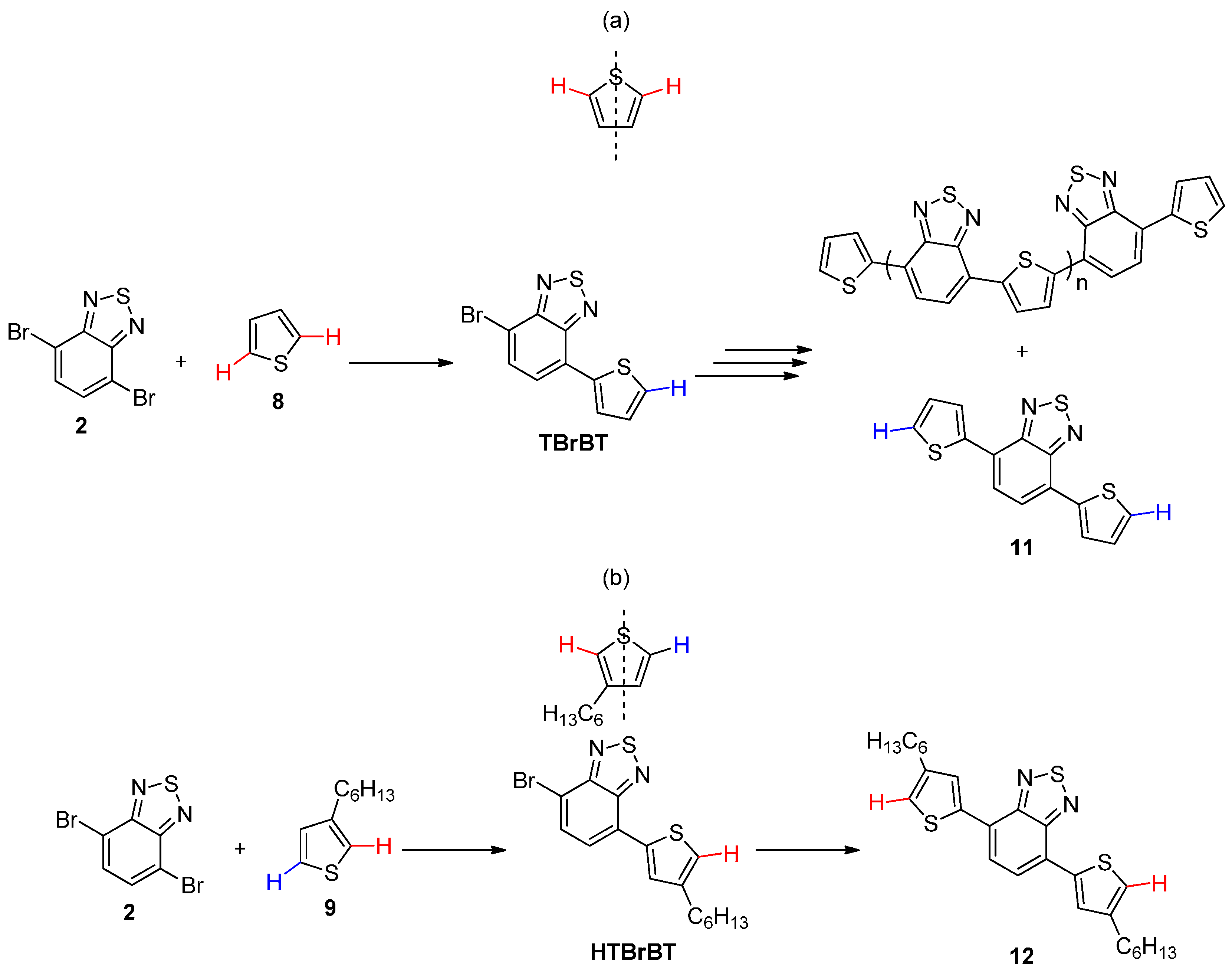
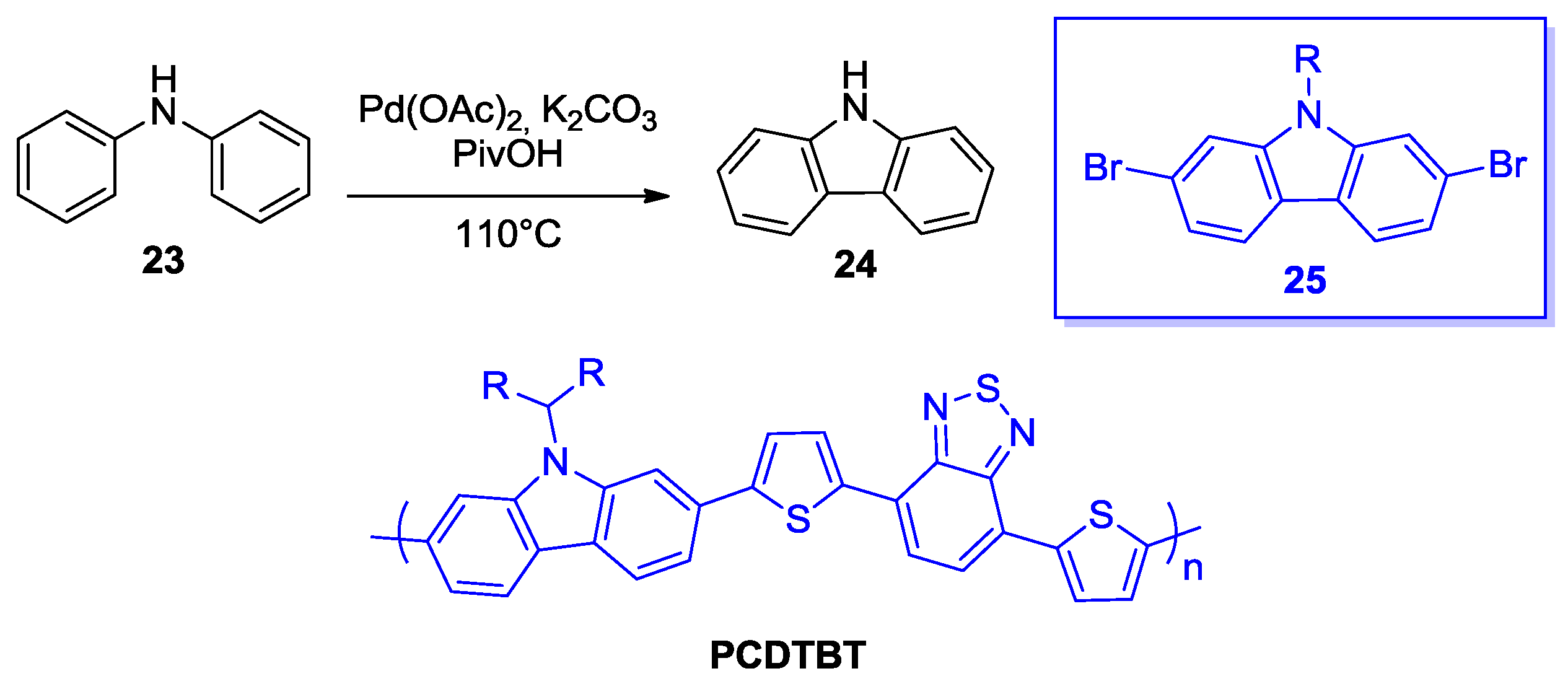
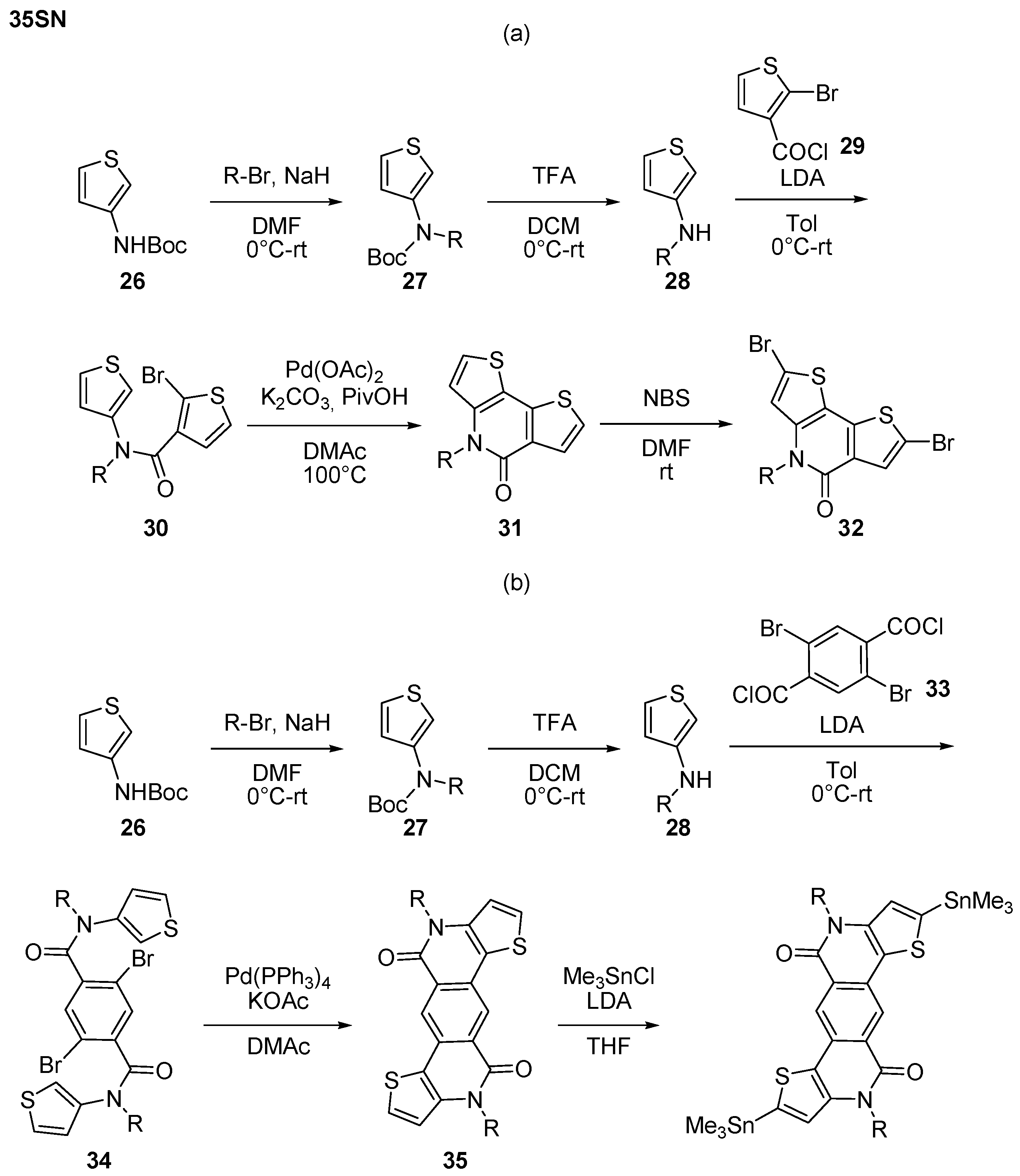
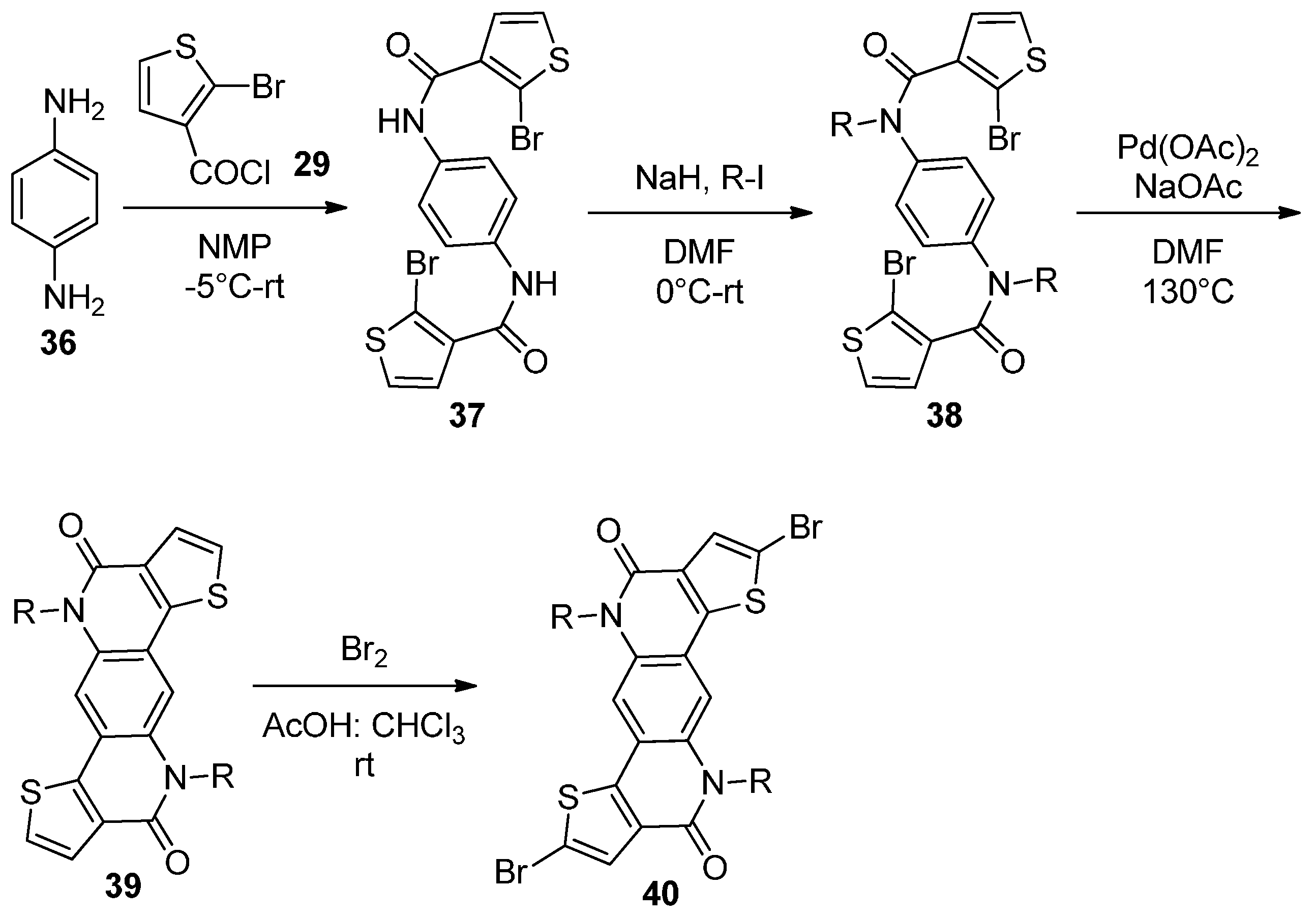
2.1. Intermolecular DHA
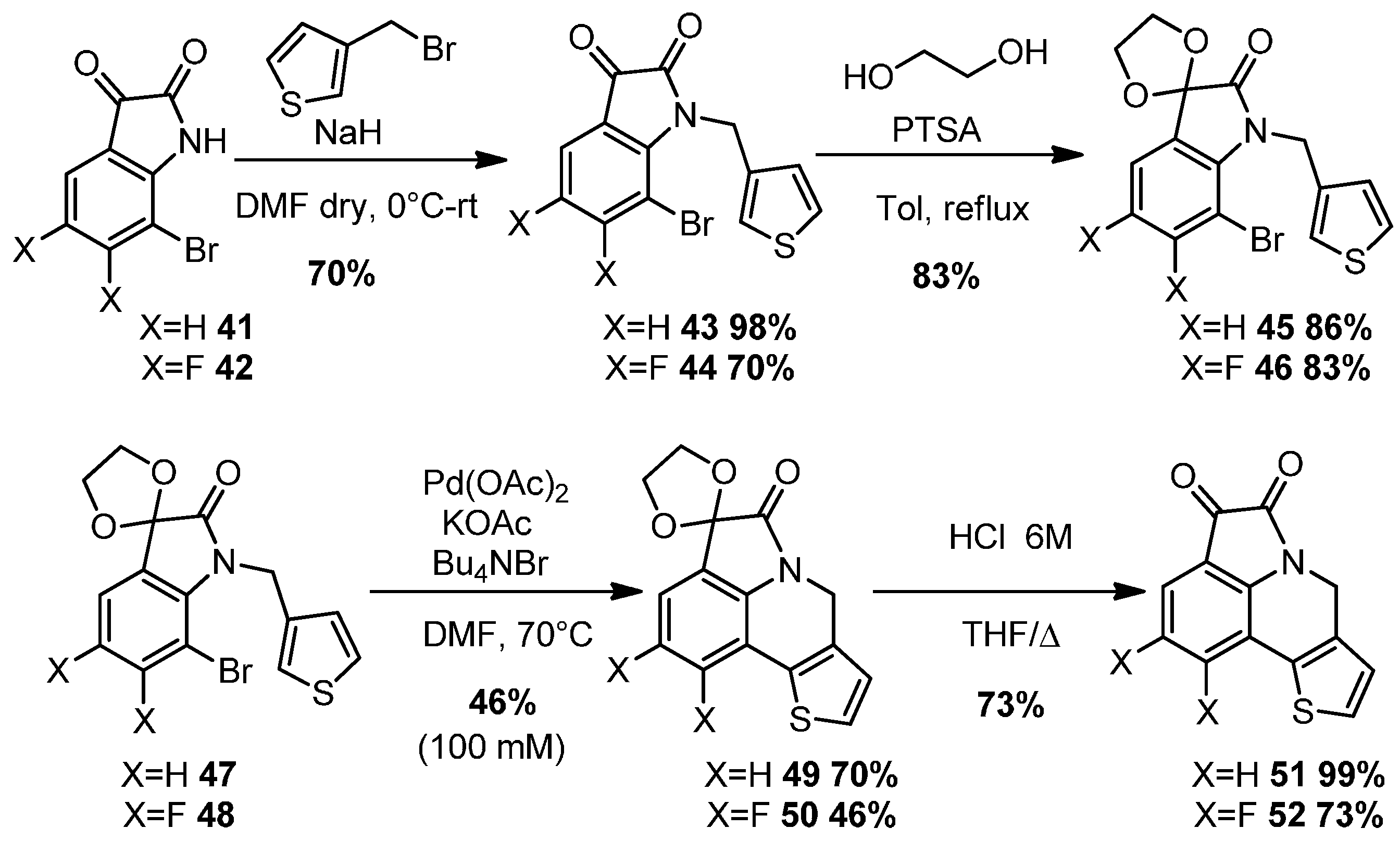
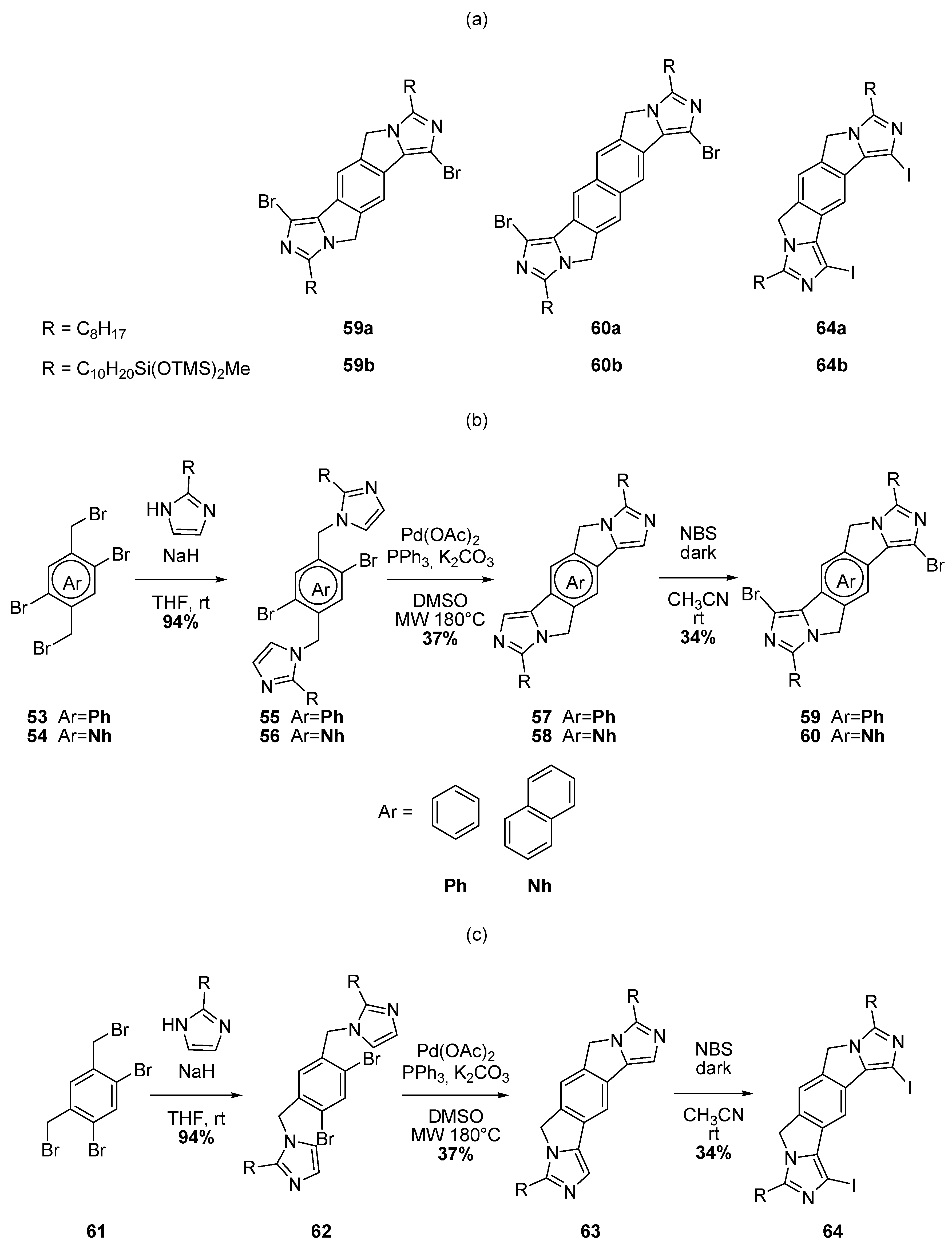
2.2. Intramolecular DHA
3. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| BT | 2,1,3-Benzothiadiazole |
| BTI | dithieno[3,2-c:2′,3′-e]azepine-4,6-dione |
| CPDT | 4H-cyclopenta[1,2-b:5,4-b′]dithiophene |
| Cz | Carbazole |
| DBrBT | 4,7-dibromo-2,1.3-benzothiadiazole |
| DEDOTBT | 4,7-bis(3,4-ethylenedioxythiophene-2-yl)-2,1,3-benzothiadiazole |
| DHTBT | 4,7-bis(4-hexylthiophen-2-yl)-2,1,3-benzothiadiazole |
| DMAc | N,N-dimethylacetamide |
| DMF | N,N-dimethylformamide |
| DTBDT | Dithienobenzodithiophene |
| DTBT | 4,7-di(thiophen-2-yl)-2,1,3-benzothiadiazole |
| EDOT | 3,4-ethylenedioxythiophene |
| F | Fluorene |
| HOMO | Highest occupied molecular orbital |
| HT | 3-hexylthiophene |
| JHONPHOS | (2-Biphenyl)di-tert-butylphosphine |
| LUMO | Lowest unoccupied molecular orbital |
| NDT | Naphto[2,1-b:3,4-b′]dithiophene |
| NMP | N-methyl-2-pyrrolidone |
| ODMB | o-dimethylbenzene |
| P3HT | Poly(3-hexylthiophene) |
| PC61BM | [6,6]-phenyl-C61-butyric acid methyl esther |
| PC71BM | phenyl-[C71]-butyric acid methyl esther |
| PCE | Power conversion efficiency |
| PDT | 2-pyridone dithiophene |
| PivOH | 2,2-dimethylpropionic acid (Pivalic acid) |
| PSC | Polymer Solar Cell |
| T | Thiophene |
| TDPT | Thieno[2′,3′,:5,6]pyrido[3,4-g]thieno[2,2-c]isoquinoline-5,11-dione |
| TFA | Trifluoroacetic acid |
| TPTBL | Thiophene-phenylene-thiophene fused bilactam |
| TTe | Thieno[3,4-b]thiophene |
| Tz | Thiazole |
References
- Chiang, C.K.; Fincher, C.R.; Park, Y.W.; Heeger, A.J.; Shirakawa, H.; Louis, E.J.; Gau, S.C.; MacDiarmid, A.G. Electrical Conductivity in Doped Polyacetylene. Phys. Rev. Lett. 1977, 39, 1098. [Google Scholar] [CrossRef]
- Shirota, Y. Organic materials for electronic and optoelectronic devices. J. Mater. Chem. 2000, 10, 1–25. [Google Scholar] [CrossRef]
- Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Semiconducting π-Conjugated Systems in field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, H.; Kido, J. Recent Progress in Phosphorescent Organic Light-Emitting Devices. Eur. J. Org. Chem. 2013, 7653–7663. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, T.; Wu, Q.; Schneider, A.M.; Zhao, D.; Yu, L. Recent Advances in Bulk Heterojunction Polymer Solar Cells. Chem. Rev. 2015, 115, 12666–12731. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Lanzani, G. Organic photonics for communications. Nat. Photonics 2010, 4, 438–446. [Google Scholar] [CrossRef]
- Brabec, C.J. Organic photovoltaics: Technology and market. Sol. Energy Mater. Sol. Cells 2004, 83, 273–292. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and Morphology Control Enables Multiple cases of High-Efficiency Polymer Solar Cells. Nat. Commun. 2014, 5, 5293. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhu, R.; Yang, Y. Polymer Solar Cells. Nat. Photonics 2012, 6, 153–161. [Google Scholar] [CrossRef]
- Po, R.; Bernardi, A.; Calabrese, A.; Carbonera, C.; Corso, G.; Pellegrino, A. From lab to fab: How must the polymer solar cell materials design change?—An industrial perspective. Energy Environ. Sci. 2014, 7, 925–943. [Google Scholar] [CrossRef]
- Jørgensen, M.; Carlé, J.E.; Søndergaard, R.R.; Lauritzen, M.; Dagnæs-Hansen, N.A.; Byskov, S.L.; Andersen, T.R.; Larsen-Olsen, T.T.; Böttiger, A.P.L.; Andreasen, B.; et al. The state of organic solar cells—A meta analysis. Sol. Energy Mater. Sol. Cells 2013, 119, 84–93. [Google Scholar] [CrossRef]
- Maluenda, I.; Navarro, O. Recent Developments in the Suzuki-Miyaura Reaction: 2010–2014. Molecules 2015, 20, 7528–7557. [Google Scholar] [CrossRef] [PubMed]
- Cordovilla, C.; Bartolome, C.; Martínez-Ilarduya, J.M.; Espinet, P. The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053. [Google Scholar] [CrossRef]
- Morin, P.-O.; Bura, T.; Leclerc, M. Realizing the full potential of conjugated polymers: Innovation in polymer synthesis. Mater. Horiz. 2016, 3, 11–20. [Google Scholar] [CrossRef]
- Po, R.; Bianchi, G.; Carbonera, C.; Pellegrino, A. “All That Glisters Is Not Gold”: An Analysis of the Synthetic Complexity of Efficient Polymer Donors for Polymer Solar Cells. Macromolecules 2015, 48, 453–461. [Google Scholar] [CrossRef]
- Okamoto, K.; Zhang, J.; Housekeeper, J.B.; Marder, S.R.; Luscombe, C.K. C–H Arylation Reaction: Atom Efficient and Greener Syntheses of π-Conjugated Small Molecules and Macromolecules for Organic Electronic Materials. Macromolecules 2013, 46, 8059–8078. [Google Scholar] [CrossRef]
- Schipper, D.J.; Fagnou, K. Direct Arylation as a Synthetic Tool for the Synthesis of Thiophene-Based Organic Electronic Materials. Chem. Mater. 2011, 23, 1594–1600. [Google Scholar] [CrossRef]
- Kudrjasova, J.; Kesters, J.; Verstappen, P.; Brebels, J.; Vangerven, T.; Cardinaletti, I.; Drijkoningen, J.; Penxten, H.; Manca, J.; Lutsen, L.; et al. A direct arylation approach towards efficient small molecule organic solar cells. J. Mater. Chem. A 2016, 4, 791–795. [Google Scholar] [CrossRef]
- Lin, P.-H.; Lu, T.-J.; Cai, D.-J.; Lee, K.-M.; Liu, C.-Y. Connecting Direct C–H Arylation Reactions with Dye-Sensitized Solar Cells: A Shortcut to D–A–π–A Organic Dyes. ChemSusChem 2015, 8, 3222–3227. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Park, M.-H.; Zhang, S.; Yao, Y.; Chen, L.-M.; Li, J.-H.; Yang, Y. Bandgap and Molecular Energy Level Control of Conjugated Polymer Photovoltaic Materials Based on Benzo[1,2-b:4,5-b′]dithiophene. Macromolecules 2008, 41, 6012–6018. [Google Scholar] [CrossRef]
- Livi, F.; Zawacka, N.K.; Angmo, D.; Joergensen, M.; Krebs, F.C.; Bundgaard, E. Influence of Side Chain Position on the Electrical Properties of Organic Solar Cells Based on Dithienylbenzothiadiazole-alt-phenylene Conjugated Polymers. Macromolecules 2015, 48, 3481–3492. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Q.; Chen, T.; Wang, Y.; Yuan, J.; Zheng, C.; Chen, R.; Huang, W. Molecular rearrangement at charged states: Intrinsic effects upon photo and electroluminescence. Dyes Pigment. 2015, 113, 529–535. [Google Scholar] [CrossRef]
- Melkonyan, F.S.; Zhao, W.; Dress, M.; Eastham, N.D.; Leonardi, M.J.; Butler, M.R.; Chen, Z.; Yu, X.; Chang, R.P.H.; Ratmer, M.A.; et al. Bithiophenesulfonamide Building Block for π-Conjugated Donor–Acceptor Semiconductors. J. Org. Chem. Soc. 2016, 138, 6944–6947. [Google Scholar] [CrossRef] [PubMed]
- Tovar, J.D.; Rose, A.; Swager, T.M. Functionalizable Polycyclic Aromatics through Oxidative Cyclization of Pendant Thiophenes. J. Am. Chem. Soc. 2002, 124, 7762–7769. [Google Scholar] [CrossRef] [PubMed]
- Alberico, D.; Scot, M.E.; Lautens, M. Aryl–Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef] [PubMed]
- Mercier, L.G.; Leclerc, M. Direct(Hetero)Arylation: A New Tool for Polymer Chemists. Acc. Chem. Res. 2013, 46, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-W.; Waters, H.; Kettle, J.; Horie, M. Cyclopentadithiophene–benzothiadiazole oligomers: Synthesis via direct arylation, X-ray crystallography, optical properties, solution casted field-effect transistor and photovoltaic characteristics. Org. Electron. 2012, 13, 2967–2974. [Google Scholar] [CrossRef]
- Chang, S.-W.; Waters, H.; Kettle, J.; Kuo, Z.-R.; Li, C.-H.; Yu, C.-Y.; Horie, M. Pd-catalysed direct arylation polymerisation for synthesis of low-bandgap conjugated polymers and photovoltaic performance. Macromol. Rapid Commun. 2012, 33, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ford, M.; Phan, H.; Coughlin, J.; Nguyena, T.-Q.; Bazan, G.C. Fluorine substitution influence on benzo[2,1,3]thiadiazole based polymers for field-effect transistor applications. Chem. Commun. 2016, 52, 3207–3210. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Sarothia, Y.; Singh, R.; Kan, Z.; Keivanidis, P.E.; Jacob, J. Synthesis and Characterization of Light-Absorbing Cyclopentadithiophene-Based Donor–Acceptor Copolymers. Polym. Int. 2016, 65, 57–65. [Google Scholar] [CrossRef]
- Wang, X.; Wang, K.; Wang, M. Synthesis of Conjugated Polymers via an Exclusive Direct-Arylation Coupling Reaction: A Facile and Straightforward Way to Synthesize Thiophene-Flanked Benzothiadiazole Derivatives and their Copolymers. Polym. Chem. 2015, 6, 1846–1855. [Google Scholar] [CrossRef]
- Hou, Q.; Xu, Y.S.; Yang, W.; Yuan, M.; Peng, J.B.; Cao, Y. Novel Red-Emitting Fluorene-Based Copolymers. J. Mater. Chem. 2002, 12, 2887–2892. [Google Scholar] [CrossRef]
- Liu, B.; Najari, A.; Pan, C.Y.; Leclerc, M.; Xiao, D.Q.; Zou, Y.P. New Low Bandgap Dithienylbenzothiadiazole Vinylene Based Copolymers: Synthesis and Photovoltaic Properties. Macromol. Rapid Commun. 2010, 31, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Chàvez, P.; Ngov, C.; Frémont, P.; Lévêque, P.; Leclerc, N. Synthesis by Direct Arylation of Thiazole–Derivatives: Regioisomer Configurations–Optical Properties Relationship Investigation. J. Org. Chem. 2014, 79, 10179–10188. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jiang, R.; Wu, D.; You, J. Rapid Access to 2,2′-Bithiazole-Based Copolymers via Sequential Palladium-Catalyzed C–H/C–X and C–H/C–H Coupling Reactions. Macromol. Rapid Commun. 2016, 37, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Liégault, B.; Lee, D.; Huestis, M.P.; Stuart, D.R.; Fagnou, K. Intramolecular Pd(II)-Catalyzed Oxidative Biaryl Synthesis Under Air: Reaction Development and Scope. J. Org. Chem. 2008, 73, 5022–5028. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.M.; Lu, L.; Manley, E.F.; Zheng, T.; Sharapov, V.; Xu, T.; Marks, T.J.; Chen, L.X.; Yu, L. Wide bandgap OPV polymers based on pyridinonedithiophene unit with efficiency >5%. Chem. Sci. 2015, 6, 4860–4866. [Google Scholar] [CrossRef]
- Jung, I.H.; Lo, W.-Y.; Jang, J.; Chen, W.; Zhao, D.; Landry, E.S.; Lu, L.; Talapin, D.V.; Yu, L. Synthesis and Search for Design Principles of New Electron Accepting Polymers for All-Polymer Solar Cells. Chem. Mater. 2014, 26, 3450–3459. [Google Scholar] [CrossRef]
- Poduval, M.K.; Burrezo, P.M.; Cadaso, J.; López Navarrete, J.T.; Ortiz, R.P.; Kim, T.-H. Novel Thiophene–Phenylene–Thiophene Fused Bislactam-Based Donor–Acceptor Type Conjugate Polymers: Synthesis by Direct Arylation and Properties. Macromolecules 2013, 46, 9220–9230. [Google Scholar] [CrossRef]
- Nitti, A.; Signorile, M.; Boiocchi, M.; Bianchi, G.; Po, R.; Pasini, D. Conjugated Thiophene-Fused Isatin Dyes through Intramolecular Direct Arylation. J. Org. Chem. 2016, 81, 11035–11042. [Google Scholar] [CrossRef] [PubMed]
- Gsänger, M.; Bialas, D.; Huang, L.; Stolte, M.; Würthner, F. Organic Semiconductors based on Dyes and Color Pigments. Adv. Mater. 2016, 28, 3615–3645. [Google Scholar] [CrossRef] [PubMed]
- Takagi, K.; Miwa, T.; Masu, H. Synthesis and Optical Properties of π-Conjugated Polymers Containing Fused Imidazole Skeleton. Macromolecules 2016, in press. [Google Scholar] [CrossRef]
- Tennyson, A.G.; Kamplain, J.W.; Bielawski, C.W. Oxidation of Poly(enetetramine)s: A New Strategy for the Synthesis of Conjugated Polyelectrolytes. Chem. Commun. 2009, 2124–2126. [Google Scholar] [CrossRef] [PubMed]
- Tennyson, A.G.; Norris, B.; Bielawski, C.W. Structurally Dynamic Conjugated Polymers. Macromolecules 2010, 43, 6923–6935. [Google Scholar] [CrossRef]
- Neilson, B.M.; Tennyson, A.G.; Bielawski, C.W. Advances in Bis(N-heterocyclic carbene) Chemistry: New Classes of Structurally Dynamic Materials. J. Phys. Org. Chem. 2012, 25, 531–543. [Google Scholar] [CrossRef]



© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nitti, A.; Po, R.; Bianchi, G.; Pasini, D. Direct Arylation Strategies in the Synthesis of π-Extended Monomers for Organic Polymeric Solar Cells. Molecules 2017, 22, 21. https://doi.org/10.3390/molecules22010021
Nitti A, Po R, Bianchi G, Pasini D. Direct Arylation Strategies in the Synthesis of π-Extended Monomers for Organic Polymeric Solar Cells. Molecules. 2017; 22(1):21. https://doi.org/10.3390/molecules22010021
Chicago/Turabian StyleNitti, Andrea, Riccardo Po, Gabriele Bianchi, and Dario Pasini. 2017. "Direct Arylation Strategies in the Synthesis of π-Extended Monomers for Organic Polymeric Solar Cells" Molecules 22, no. 1: 21. https://doi.org/10.3390/molecules22010021
APA StyleNitti, A., Po, R., Bianchi, G., & Pasini, D. (2017). Direct Arylation Strategies in the Synthesis of π-Extended Monomers for Organic Polymeric Solar Cells. Molecules, 22(1), 21. https://doi.org/10.3390/molecules22010021