
Atom-Economic Synthesis of 4-Pyrones from Diynones and Water

 ,
,
Abstract
:
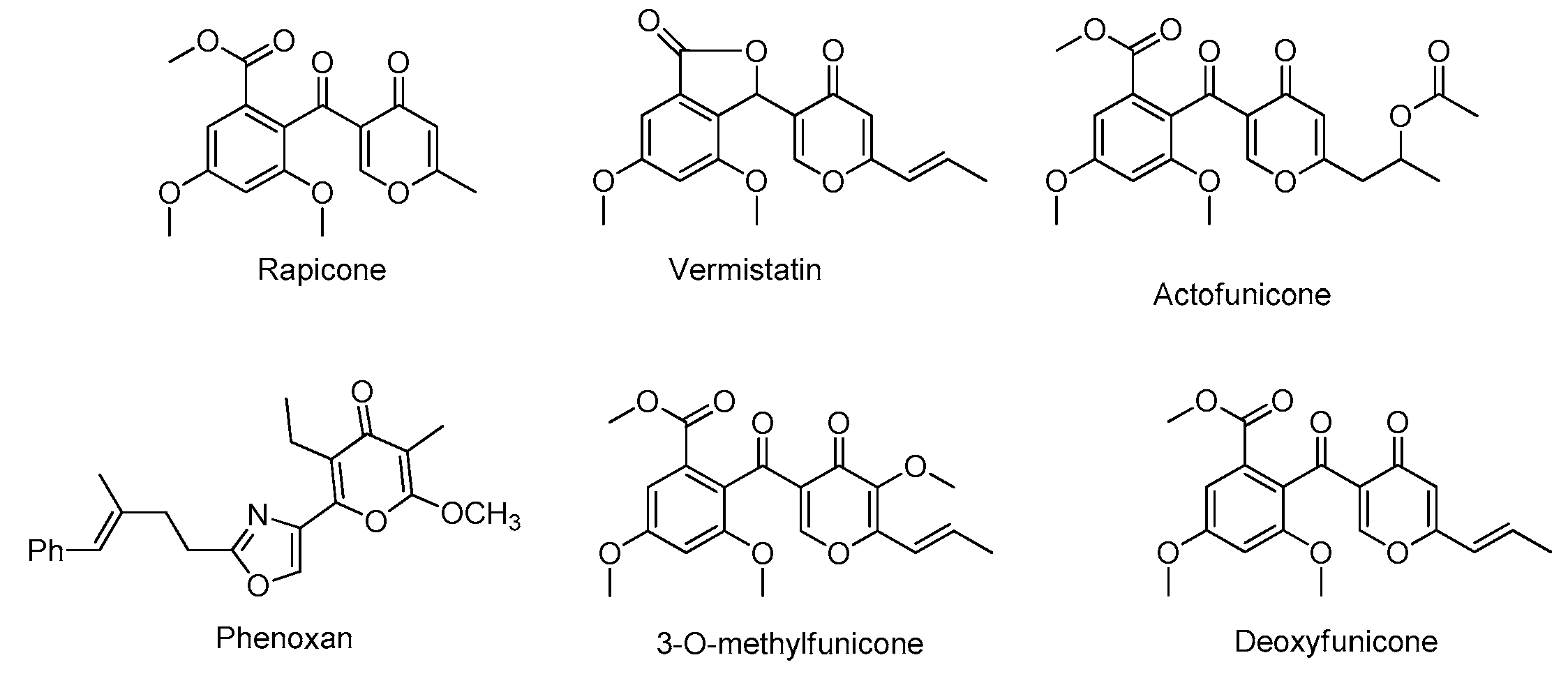
1. Introduction
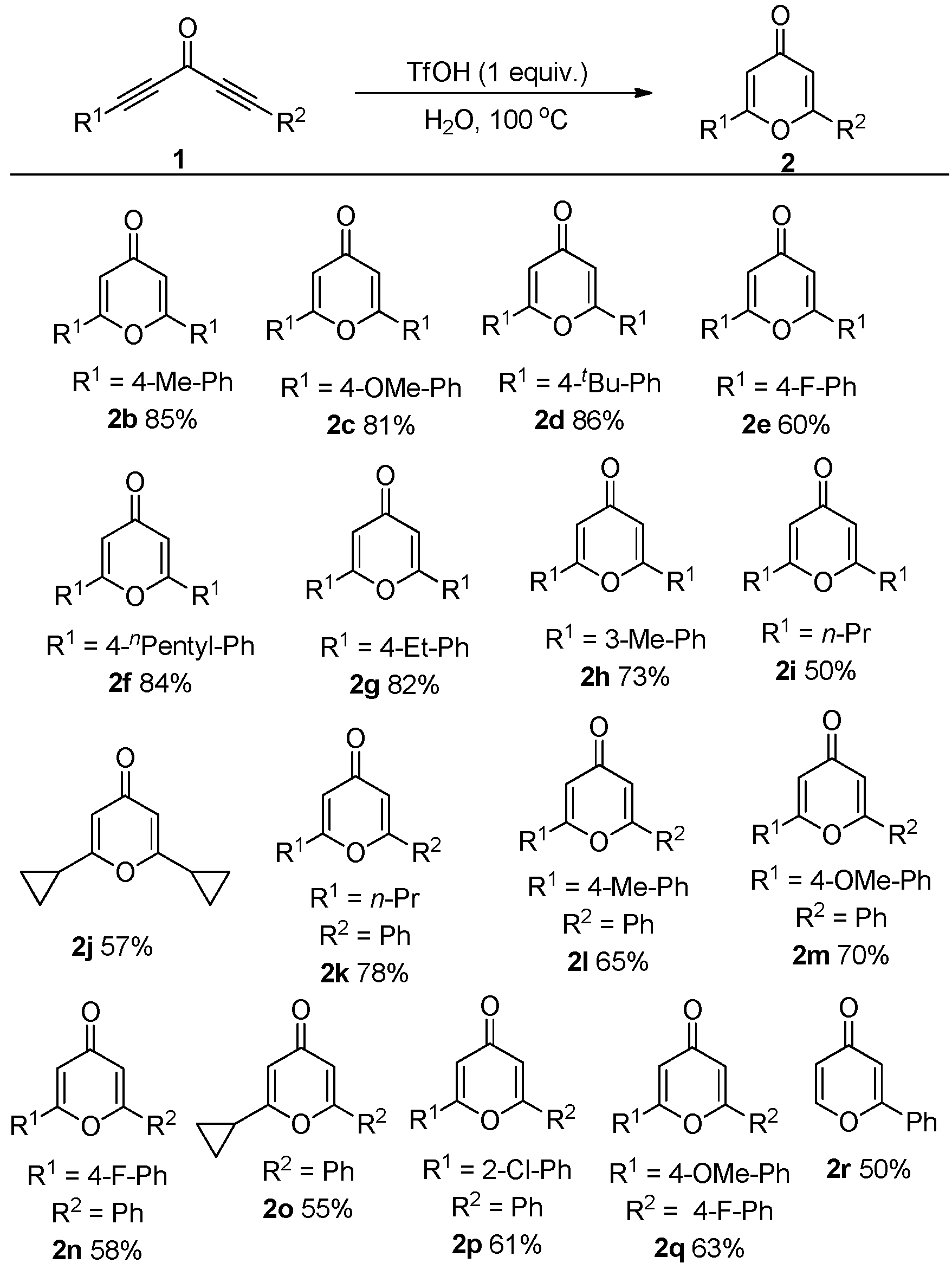
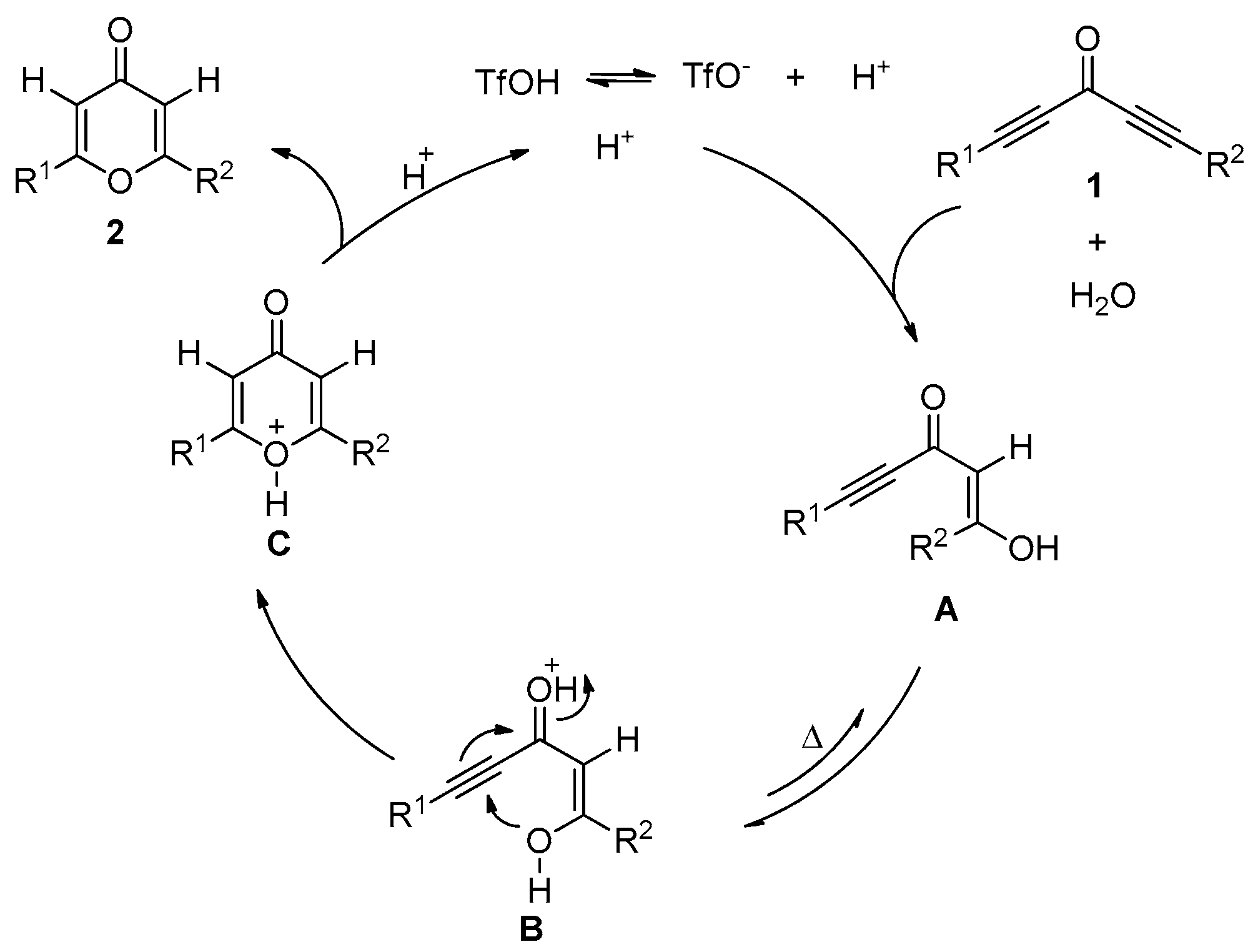
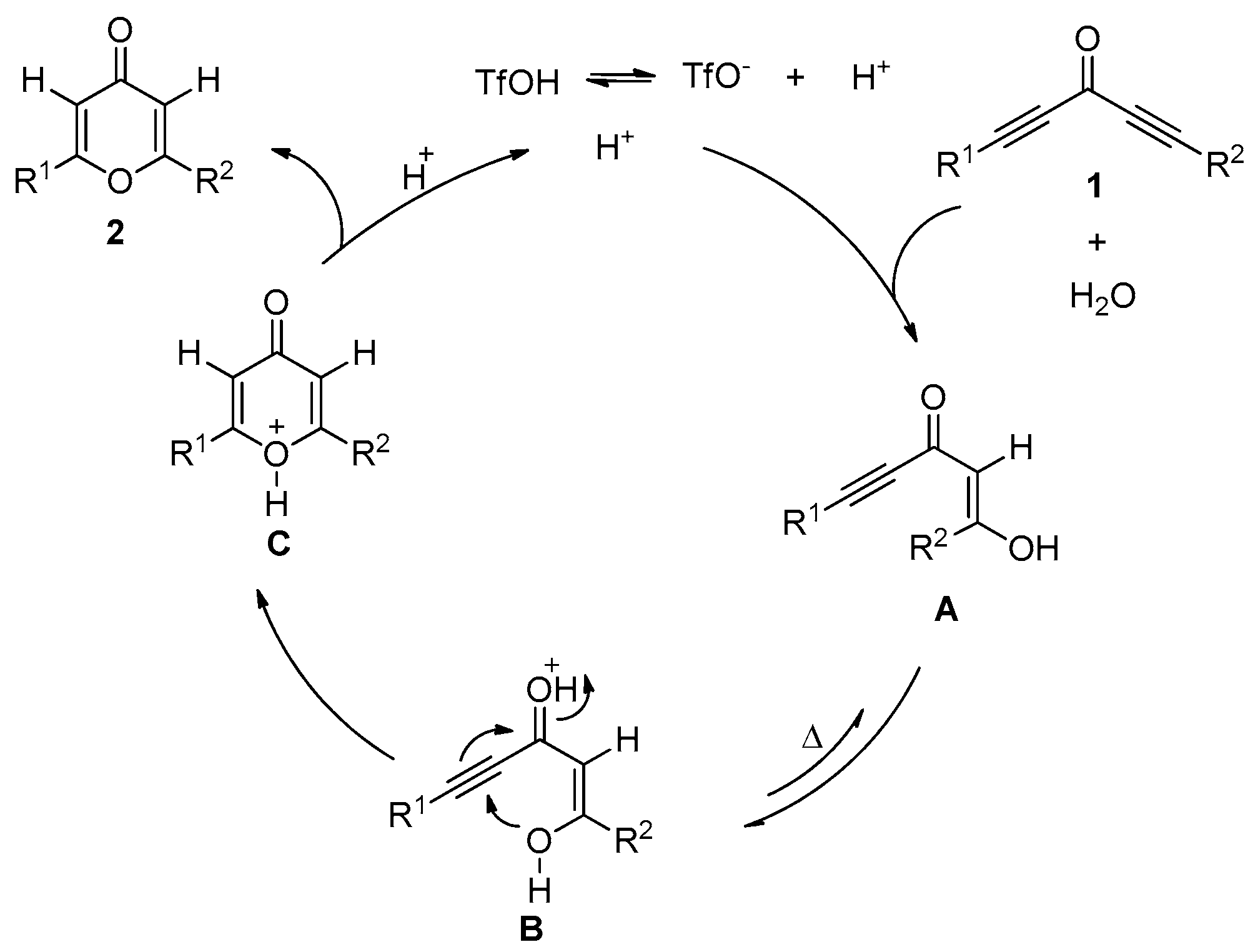
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Compound 2
3.3. Control Experiments
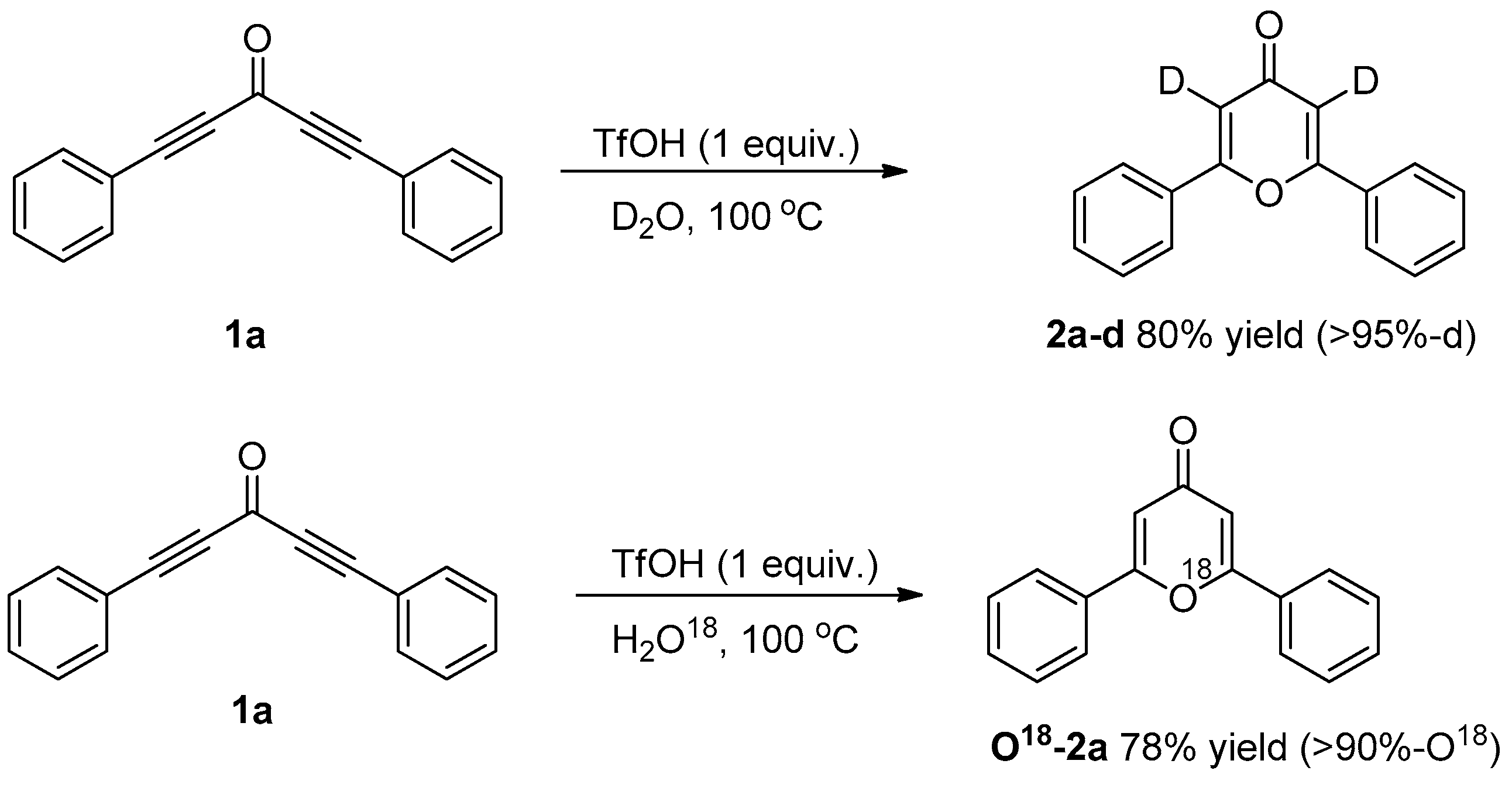
3.3.1. Deuterium Labeling Experiments
3.3.2. O18-Labelling Experiment
3.3.3. Gram-Scale Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TfOH | trifluoromethanesulfonic acid |
| PTSA | 4-methylbenzenesulfonic acid |
| HCl | hydrochloric acid |
| HIV | human immunodeficiency virus |
| HOAc | acetic acid |
| Ph | phenyl |
| Me | methyl |
| OMe | methoxyl |
| Et | ethyl |
| tBu | tertiary butyl |
| nPr | n-propyl |
| NMR | nuclear magnetic resonance |
| HRMS | high-resolution mass |
References
- Padiya, K.J.; Gavade, S.; Kardile, B.; Tiwari, M.; Bajare, S.; Mane, M.; Gaware, V.; Varghese, S.; Harel, D.; Kurhade, S. Unprecedented “In Water” Imidazole Carbonylation: Paradigm Shift for Preparation of Urea and Carbamate. Org. Lett. 2012, 14, 2814–2817. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Muldoon, J.; Finn, M.G.; Fokin, V.V.; Kolb, H.C.; Sharpless, K.B. “On Water”: Unique Reactivity of Organic Compounds in Aqueous Suspension. Angew. Chem. Int. Ed. 2005, 44, 3275–3279. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Sarma, K.D. Multicomponent Reactions Are Accelerated in Water. J. Am. Chem. Soc. 2004, 126, 444–445. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, J.M. Practical Approaches to Green Solvents. Science 2002, 297, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Powner, M.W.; Zheng, S.-L.; Szostak, J.W. Multicomponent Assembly of Proposed DNA Precursors in Water. J. Am. Chem. Soc. 2012, 134, 13889–13895. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, E.; Ghosh, R.; Olofsson, B. Metal-Free Synthesis of Aryl Ethers in Water. Org. Lett. 2013, 15, 6070–6073. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J. Organic Reactions in Aqueous Media-With a Focus on Carbon-Carbon Bond Formation. Chem. Rev. 1999, 93, 2023–2035. [Google Scholar] [CrossRef]
- Huang, Y.-T.; Lu, S.-Y.; Yi, C.-L.; Lee, C.-F. Iron-Catalyzed Synthesis of Thioesters from Thiols and Aldehydes in Water. J. Org. Chem. 2014, 79, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Xu, Z.M.; Lu, H.F.; Dai, X.D.; Yang, T.; Lin, X.C.; Ren, F. Mild and Regioselective N-Alkylation of 2-Pyridones in Water. Org. Lett. 2015, 17, 3382–3385. [Google Scholar] [CrossRef]
- Sberegaeva, A.V.; Zavalij, P.Y.; Vedernikov, A.N. Oxidation of a Monomethylpalladium(II) Complex with O2 in Water: Tuning Reaction Selectivity to Form Ethane, Methanol, or Methylhydroperoxide. J. Am. Chem. Soc. 2016, 138, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Katsuki, T. Fe(salan)-Catalyzed Asymmetric Oxidation of Sulfides with Hydrogen Peroxide in Water. J. Am. Chem. Soc. 2007, 129, 8940–8941. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.M.; Emmanuvel, A.L.; Sudalai, A. NaIO4-Mediated Selective Oxidation of Alkylarenes and Benzylic Bromides/Alcohols to Carbonyl Derivatives Using Water as Solvent. J. Org. Chem. 2006, 71, 5043–5046. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wang, Z.T.; Cheng, X.M.; Li, C.Z. Silver-Catalyzed Decarboxylative Alkynylation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc. 2012, 134, 14330–14333. [Google Scholar] [CrossRef] [PubMed]
- Barker, T.J.; Boger, D.L. Fe(III)/NaBH4-Mediated Free Radical Hydrofluorination of Unactivated Alkenes. J. Am. Chem. Soc. 2012, 134, 13588–13591. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.D.; Song, L.Y.; Li, C.Z. Silver-Catalyzed Radical Aminofluorination of Unactivated Alkenes in Aqueous Media. J. Am. Chem. Soc. 2013, 135, 4640–4643. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Nagano, T.; Schneider, U.; Hamada, T.; Ogawa, C.; Kobayashi, S. Zn-Catalyzed Asymmetric Allylation for the Synthesis of Optically Active Allylglycine Derivatives. Regio- and Stereoselective Formal α-Addition of Allylboronates to Hydrazono Esters. J. Am. Chem. Soc. 2008, 130, 2914–2915. [Google Scholar] [CrossRef] [PubMed]
- Gordillo, A.; Ortuño, M.A.; Mardomingo, C.L.; Lledós, A.; Ujaque, G.; Jesús, E. Mechanistic Studies on the Pd-Catalyzed Vinylation of Aryl Halides with Vinylalkoxysilanes in Water: The Effect of the Solvent and NaOH Promoter. J. Am. Chem. Soc. 2013, 135, 13749–13763. [Google Scholar] [CrossRef] [PubMed]
- Felpin, F.-X.; Landais, Y. Practical Pd/C-Mediated Allylic Substitution in Water. J. Org. Chem. 2005, 70, 6441–6446. [Google Scholar] [CrossRef] [PubMed]
- Candeias, N.R.; Gois, P.M.P.; Afonso, C.A.M. Rh(II)-Catalyzed Intramolecular C–H Insertion of Diazo Substrates in Water: Scope and Limitations. J. Org. Chem. 2006, 71, 5489–5497. [Google Scholar] [CrossRef] [PubMed]
- Sotto, N.; Billamboz, M.; Carole, C.-V.; Len, C. Selective Pinacol Coupling on Regeneratable Supported Acids in Sole Water. J. Org. Chem. 2015, 80, 6375–6380. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Dong, Y.; Pang, B.; Ma, J.H. [Bmim]PF6-Promoted Ligandless Suzuki–Miyaura Coupling Reaction of Potassium Aryltrifluoroborates in Water. J. Org. Chem. 2014, 79, 7193–7198. [Google Scholar] [CrossRef] [PubMed]
- Botella, L.; Nájera, C. Mono- and β,β-Double-Heck Reactions of α,β-Unsaturated Carbonyl Compounds in Aqueous Media. J. Org. Chem. 2005, 70, 4360–4369. [Google Scholar] [CrossRef] [PubMed]
- Erb, W.; Albini, M.; Rouden, J.; Blanchet, J. Sequential One-Pot Access to Molecular Diversity through Aniline Aqueous Borylation. J. Org. Chem. 2014, 79, 10568–10580. [Google Scholar] [CrossRef] [PubMed]
- Kalutharage, N.; Yi, C.S. Chemoselective Formation of Unsymmetrically Substituted Ethers from Catalytic Reductive Coupling of Aldehydes and Ketones with Alcohols in Aqueous Solution. Org. Lett. 2015, 17, 1778–1781. [Google Scholar] [CrossRef] [PubMed]
- Hikawa, H.; Suzuki, H.; Azumaya, I. Au(III)/TPPMS-Catalyzed Benzylation of Indoles with Benzylic Alcohols in Water. J. Org. Chem. 2013, 78, 12128–12135. [Google Scholar] [CrossRef] [PubMed]
- Handa, S.; Slack, E.D.; Lipshutz, B.H. Nanonickel-Catalyzed Suzuki–Miyaura Cross-Couplings in Water. Angew. Chem. Int. Ed. 2015, 54, 11994–11998. [Google Scholar] [CrossRef] [PubMed]
- Nishikata, T.; Lipshutz, B.H. AllylicEthersas Educts for Suzuki–Miyaura Couplings in Water at Room Temperature. J. Am. Chem. Soc. 2009, 131, 12103–12105. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, Y.; Guo, R. Facile Synthesis of Highly Stable Gold Nanoparticles and Their Unexpected Excellent Catalytic Activity for Suzuki–Miyaura Cross-Coupling Reaction in Water. J. Am. Chem. Soc. 2009, 131, 2060–2061. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-H.; Mannathan, S.; Cheng, C.-H. Enantioselective Synthesis of β-Substituted Cyclic Ketones via Cobalt-Catalyzed Asymmetric Reductive Coupling of Alkynes with Alkenes. J. Am. Chem. Soc. 2011, 133, 6942–6944. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-T.; Jayanth, T.T.; Wang, C.-C.; Cheng, C.-H. Cobalt-Catalyzed Reductive Coupling of Activated Alkenes with Alkynes. J. Am. Chem. Soc. 2007, 129, 12032–12041. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, Z.-Z.; He, Y.-T.; Li, L.-H.; Ma, J.-W.; Qiu, Y.-F.; Zhou, P.-X.; Liu, X.-Y.; Xu, P.-F.; Liang, Y.-M. Iodine-Promoted Radical Cyclization in Water: A Selective Reaction of 1,6-Enynes with Sulfonyl Hydrazides. J. Org. Chem. 2016, 81, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.-F.; Zhu, S.-L.; Chen, C.; Wang, H.J.; Liang, Y.-M. Silver-Catalyzed Decarboxylative Addition/Cyclization of Activated Alkenes with Aliphatic Carboxylic Acids. J. Org. Chem. 2016, 81, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-L.; Shen, S.-L.; Yang, P.; Qu, J. A Catalyst-Free One-Pot Construction of Skeletons of 5-Methoxyseselin and Alloxanthoxyletin in Water. Org. Lett. 2013, 15, 3856–3859. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.S.; Bulger, P.G.; Maloney, K.M. A High-Yielding Method for the Preparation of Isoxazolopyridin-3-amine Derivatives. Green Chem. 2016, 18, 4941–4946. [Google Scholar] [CrossRef]
- Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F., III. Organocatalytic Direct Asymmetric Aldol Reactions in Water. J. Am. Chem. Soc. 2006, 128, 734–735. [Google Scholar] [CrossRef] [PubMed]
- Lerebours, R.; Wolf, C. Palladium(II)-Catalyzed Conjugate Addition of Arylsiloxanes in Water. Org. Lett. 2007, 9, 2737–2740. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-L.; Wang, G.-W. Aminochlorination in Water: First Brønsted Acid-Promoted Synthesis of Vicinal Chloramines. J. Org. Chem. 2007, 72, 9398–9401. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.L.; Perkins, B.L.; Ni, B. Diarylprolinol Silyl Ether Salts as New, Efficient, Water-Soluble, and Recyclable Organocatalysts for the Asymmetric Michael Addition on Water. J. Am. Chem. Soc. 2010, 132, 50–51. [Google Scholar] [CrossRef] [PubMed]
- He, R.J.; Shirakawa, S.; Maruoka, K. Enantioselective Base-Free Phase-Transfer Reaction in Water-Rich Solvent. J. Am. Chem. Soc. 2009, 131, 16620–16621. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Nishijima, Y.; Fujita, M. Cage-Catalyzed Knoevenagel Condensation under Neutral Conditions in Water. J. Am. Chem. Soc. 2012, 134, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, C.B. Neighboring Heteroatom Effect Unique to Aqueous Aldol Reactions of Water-Insoluble Substrates. J. Org. Chem. 2014, 79, 2242–2254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Tian, H.; Shi, Y. Catalytic Asymmetric Bromination of UnfunctionalizedOlefins with H2 Oasa Nucleophile. Chem. Eur. J. 2015, 21, 11658–11663. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.-K.; Wang, L.-J.; Liu, Q.-J.; Li, J.-F.; Tang, Y. Asymmetric H2O-Nucleophilic Ring Opening of D−A Cyclopropanes: Catalyst Serves as a Source of Water. J. Am. Chem. Soc. 2015, 137, 14594–14597. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.C.; Rao, S.N.; Adimurthy, S. Synthesis of Imidazo[1,2-a]pyridines: “Water-Mediated” Hydroamination and Silver-Catalyzed Aminooxygenation. J. Org. Chem. 2013, 78, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Yehoshoa, B.-D.; Milstein, D. General Synthesis of Amino Acid Salts from Amino Alcohols and Basic Water Liberating H2. J. Am. Chem. Soc. 2016, 138, 6143–6146. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Biyajima, T.; Fujii, T.; Murakami, M. Synthesis of α-Amino Ketones from Terminal Alkynes via Rhodium Catalyzed Denitrogenative Hydration of N-Sulfonyl-1,2,3-triazoles. J. Am. Chem. Soc. 2012, 134, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Buyck, T.; Wang, Q.; Zhu, J.P. Triple Role of Phenylselenonyl Group Enabled a One-Pot Synthesis of 1,3-Oxazinan-2-ones Fromα-Isocyanoacetates, Phenyl Vinyl Selenones, and Water. J. Am. Chem. Soc. 2014, 136, 11524–11528. [Google Scholar] [CrossRef] [PubMed]
- Colemana, M.T.D.; Garson, M.J. Marine polypropionates. Nat. Prod. Rep. 1998, 15, 477–493. [Google Scholar] [CrossRef]
- Sakakura, A.; Watanabe, H.; Ishihara, K. Rate-Accelerating Effect by the Neighboring-Group Participation of Protecting Groups in the Dehydrative Cyclization of 1,3,5-Triketones. Org. Lett. 2008, 10, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Boukouvalas, J.; Wang, J.-X. Structure Revision and Synthesis of a Novel Labdane Diterpenoid from Zingiber ottensii. Org. Lett. 2008, 10, 3397–3399. [Google Scholar] [CrossRef] [PubMed]
- Molenda, J.J.; Jones, M.M.; Johnston, D.S.; Walker, E.M.; Cannon, D.J. Mobilization of Iron by Chiral and Achiral Anionic 3-Hydroxypyrid-4-one. J. Med. Chem. 1994, 37, 4363–4370. [Google Scholar] [CrossRef] [PubMed]
- Stossel, D.; Chan, T.H. A 5C + 5C Bicycloaromatization Reaction via an Aldol Condensation Acyclic Precursors Cascade: A Regioselective Synthesis of Functionalized Naphthalenes from Acyclic Precursors. J. Org. Chem. 1988, 53, 4901–4908. [Google Scholar] [CrossRef]
- Reddy, D.S.; Velde, D.V.; Aube, J. Synthesis and Conformational Studies of Dipeptides Constrained by Disubstituted 3-(Aminoethoxy)propionic Acid Linkers. J. Org. Chem. 2004, 69, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.M.; Luo, W.; Quinn, P.J.; Liu, Z.D.; Hider, R.C. Design, Synthesis, Physicochemical Properties, and Evaluation of Novel Iron Chelators with Fluorescent Sensors. J. Med. Chem. 2004, 47, 6349–6362. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.Z.; Mi, X.L.; Xu, H.; Wang, P.G.; Cheng, J.-P. Efficient Baylis-Hillman Reactions of Cyclic Enones in Methanol As Catalyzed by Methoxide Anion. J. Org. Chem. 2004, 69, 8413–8422. [Google Scholar] [CrossRef] [PubMed]
- Yeates, C.L.; Batchelor, J.F.; Capon, E.C.; Cheesman, N.J.; Fry, M.; Hudson, A.T.; Pudney, M.; Trimming, H.; Woolven, J.; Bueno, J.M.; et al. Synthesis and Structure–Activity Relationships of 4-Pyridones as Potential Antimalarials. J. Med. Chem. 2008, 51, 2845–2852. [Google Scholar] [CrossRef] [PubMed]
- Fakih, S.; Podinovskaia, M.; Kong, X.L.; Collins, H.L.; Schaible, U.E.; Hider, R.C. Targeting the Lysosome: Fluorescent Iron(III) Chelators To Selectively Monitor Endosomal/Lysosomal Labile Iron Pools. J. Med. Chem. 2008, 51, 4539–4552. [Google Scholar] [CrossRef] [PubMed]
- Fabiola, B.-J.; Ward, D.E. On the Origin of Siphonariid Polypropionates: Total Synthesis of Caloundrin B and Its Isomerization to Siphonarin B. Org. Lett. 2012, 14, 1648–1651. [Google Scholar]
- Li, D.-F.; Hu, P.-P.; Liu, M.-S.; Kong, X.-L.; Zhang, J.-C.; Hider, R.C.; Zhou, T. Design and Synthesis of Hydroxypyridinone-l-phenylalanine Conjugates as Potential Tyrosinase Inhibitors. J. Agric. Food Chem. 2013, 61, 6597–6603. [Google Scholar] [CrossRef] [PubMed]
- Garey, D.; Ramirez, M.-L.; Gonzales, S.; Wertsching, A.; Tith, S.; Keefe, K.; Peña, M.R. An Approach to Substituted 4-Hydroxypyran-2-ones: The Total Synthesis of Phenoxan. J. Org. Chem. 1996, 61, 4853–4856. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Ohba, S.; Nishiyama, S.; Yamamura, S. Total Synthesis of Phenoxan and a Related Pyrone Derivative. Tetrahedron Lett. 1996, 37, 2997–3000. [Google Scholar] [CrossRef]
- Ehrlich, M.; Carell, T. Total Syntheses and Biological Evaluation of 3-O-Methylfunicone and Its Derivatives Prepared by TMPZnCl·LiCl-Mediated Halogenation and Carbonylative Stille Cross-Coupling. Eur. J. Org. Chem. 2013, 1, 77–83. [Google Scholar] [CrossRef]
- Light, R.J.; Hauser, C.R. Aroylations of p-Diketones at the Terminal Methyl Group to Form 1,3,5-Triketones. Cyclizations to 4-Pyrones and 4-Pyridones. J. Org. Chem. 1960, 25, 538–546. [Google Scholar] [CrossRef]
- Morris, J.; Luke, G.P.; Wishka, D.G. Reaction of Phosgeniminium Salts with Enolates Derived from Lewis Acid Complexes of 2′-Hydroxypropiophenones and Related β-Diketones. J. Org. Chem. 1996, 61, 3218–3220. [Google Scholar] [CrossRef] [PubMed]
- Bunescu, A.; Reimann, S.; Lubbe, M.; Spannenberg, A.; Langer, P. Synthesis of Trifluoromethyl-Substituted Arenes, Cyclohexenones and Pyran-4-ones by Cyclocondensation of 1,3-Bis(silyloxy)-1,3-butadienes with 4,4-Dimethoxy-1,1,1-trifluorobut-3-en-2-one: Influence of the Lewis Acid on the Product Distribution. J. Org. Chem. 2009, 74, 5002–5010. [Google Scholar] [CrossRef] [PubMed]
- Malamasa, M.S.; Barnes, K.; Johnson, M.; Hui, Y.; Zhou, P.; Turner, J.; Hu, Y.; Wagner, E.; Fan, K.; Chopra, R.; et al. Di-substituted pyridinyl aminohydantoins as potent and highly selective human β-secretase (BACE1) inhibitors. Bioorg. Med. Chem. 2010, 18, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Brückner, R. Total Syntheses of the Dihydrofuranonecarboxylate Natural Products Gregatin B and E: Gram-Scale Synthesis of (+)-Gregatin B and Unambiguous Assignment of the Stereostructure of (+)-Gregatin E. Org. Lett. 2014, 16, 6428–6431. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-S.; Lacasse, E. Synthesis of Pyran-4-ones from Isoxazoles. Tetrahedron Lett. 2002, 43, 3565–3568. [Google Scholar] [CrossRef]
- Jo, Y.-J.; Cho, I.H.; Song, C.K.; Shin, H.W.; Kim, Y.-S. Comparison of Fermented Soybean Paste (Doenjang) Prepared by Different Methods Based on Profiling of Volatile Compounds. J. Food Sci. 2011, 76, C368–C379. [Google Scholar] [CrossRef] [PubMed]
- Teng, Q.-H.; Xu, Y.-L.; Liang, Y.; Wang, H.-S.; Wang, Y.-C.; Pan, Y.-M. Transition Metal-Free Synthesis of 3-Alkynylpyrrole-2-carboxylates via Michael Addition/Intramolecular Cyclodehydration. Adv. Synth. Catal. 2016, 358, 1897–1902. [Google Scholar] [CrossRef]
- Tan, X.-C.; Liang, Y.; Bao, F.-P.; Wang, H.-S.; Pan, Y.-M. Silver-Mediated C–H Bond Functionalization: One-Pot to Construct Substituted Indolizines from 2-Alkylazaarenes with Alkynes. Tetrahedron 2014, 70, 6717–6722. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.-Y.; Pan, Y.-M.; Wang, H.-S.; Liang, H.; Chen, Z.-F.; Qin, X.-H. Samarium(III)-Catalyzed C (sp3)–H Bond Activation: Synthesis of Indolizines via C–C and C–N Coupling between 2-Alkylazaarenes and Propargylic Alcohols. Org. Lett. 2014, 16, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Pan, Y.-M.; Xu, Y.-L.; Wang, H.-S. PTSA-Catalyzed Mannich-Type–Cyclization–Oxidation Tandem Reactions: One-Pot Synthesis of 1, 3, 5-Substituted Pyrazoles from Aldehydes, Hydrazines and Alkynes. Org. Biomol. Chem. 2012, 10, 4696–4698. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, Y.-M.; Huang, X.-C.; Mao, Z.-Y.; Wang, H.-S. A novel methodology for synthesis of dihydropyrazole derivatives as potential anticancer agents. Org. Biomol. Chem. 2014, 12, 2028–2032. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-M.; Zheng, F.-J.; Lin, H.-X.; Zhan, Z.-P. Brønsted Acid-Catalyzed Propargylation/Cycloisomerization Tandem Reaction: One-Pot Synthesis of Substituted Oxazoles from Propargylic Alcohols and Amides. J. Org. Chem. 2009, 74, 3148–3151. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.-Z.; Gao, Q.; Liang, Y.; Wang, H.-S.; Pan, Y.-M. Palladium-Catalyzed Synthesis of Benzoxazoles by the Cleavage Reaction of Carbon–Carbon Triple Bonds with o-Aminophenol. Green Chem. 2014, 16, 2132–2135. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Wang, H.-S.; Huang, G.-B.; Huang, F.-P.; Hu, K.; Pan, Y.-M. A One-Pot Approach to 4,5-Dihydropyrazoles from Ketones, Arylacetylenes, and Hydrazines. Tetrahedron 2014, 70, 1621–1628. [Google Scholar] [CrossRef]
- Qiu, Y.-F.; Yang, F.; Qiu, Z.-H.; Zhong, M.-J.; Wang, L.-J.; Ye, Y.-Y.; Song, B.; Liang, Y.-M. Brønsted Acid Catalyzed and NIS-Promoted Cyclization of Diynones: Selective Synthesis of 4-Pyrone, 4-Pyridone, and 3-Pyrrolone Derivatives. J. Org. Chem. 2013, 78, 12018–12028. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Qiu, Y.-F.; Ji, K.-G.; Niu, Y.-N.; Ali, S.; Liang, Y.-M. Divergent Synthesis of Benzene Derivatives: Brønsted Acid Catalyzed and Iodine-Promoted Tandem Cyclization of 5,2-Enyn-1-ones. J. Org. Chem. 2012, 77, 9029–9037. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Ji, K.-G.; Zhao, S.-C.; Ali, S.; Ye, Y.-Y.; Liu, X.-Y.; Liang, Y.-M. Brønsted Acid Catalyzed Cycloisomerizations of 5,2-Enyne-1-ones: Highly Regioselective Synthesis of 2,3-Dihydro-4H-pyran-4-ones. Chem. Eur. J. 2012, 18, 6470–6474. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, Y.; Luu, T.; Tykwinski, R.R. A One-Pot Synthesis and Functionalization of Polyynes. Org. Lett. 2006, 8, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.; Metz, C.R.; Beam, C.F.; Pennington, W.T.; Derveer, D.G.V. New Strong Base Synthesis of Symmetrical 1,5-Diaryl-1,3,5-pentanetriones from Acetone and Benzoate Esters. Synth. Commun. 2008, 38, 2465–2482. [Google Scholar] [CrossRef]
- Aziz, S.; Mahnaz, S. Synthesis of Pyrone Carbaldehydes, Pyrone Sulfonium Ylides and Related Epoxides. J. Heterocycl. Chem. 2009, 46, 268–272. [Google Scholar]
- Jobour, A.; Nazar, H.; Shandala, M.Y. Synthesis and Spectral Data of Some Heterocyclic Compounds. The Reaction of Arylpropiolic Esters with Tetralones and Acetylcyclopropane. J. Heterocycl. Chem. 1980, 17, 941–944. [Google Scholar] [CrossRef]
- Toshiaki, S.; Taichi, O.; Kiyomi, S.; Yosihiro, N.; Mitsuru, H.; Yoshie, H.; Jun, T.; Takehiro, S. Thermal Addition Reaction of Aroylketene with 1-Aryl-1-trimethylsilyloxyethylenes: Aromatic Substituent Effects of Aroylketene and Aryltrimethylsilyloxyethylene on Their Reactivity. Chem. Pharm. Bull. 1996, 44, 956–966. [Google Scholar]
- Groundwater, P.W.; Hibbs, D.E.; Hursthousea, M.B.; Nyerges, M. Synthesis and Reactions of Reduced Flavones. J. Chem. Soc. Perkin Trans. 1 1997, 163–170. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2a–2r, 2a-d and 2a-o18 are available from the authors.
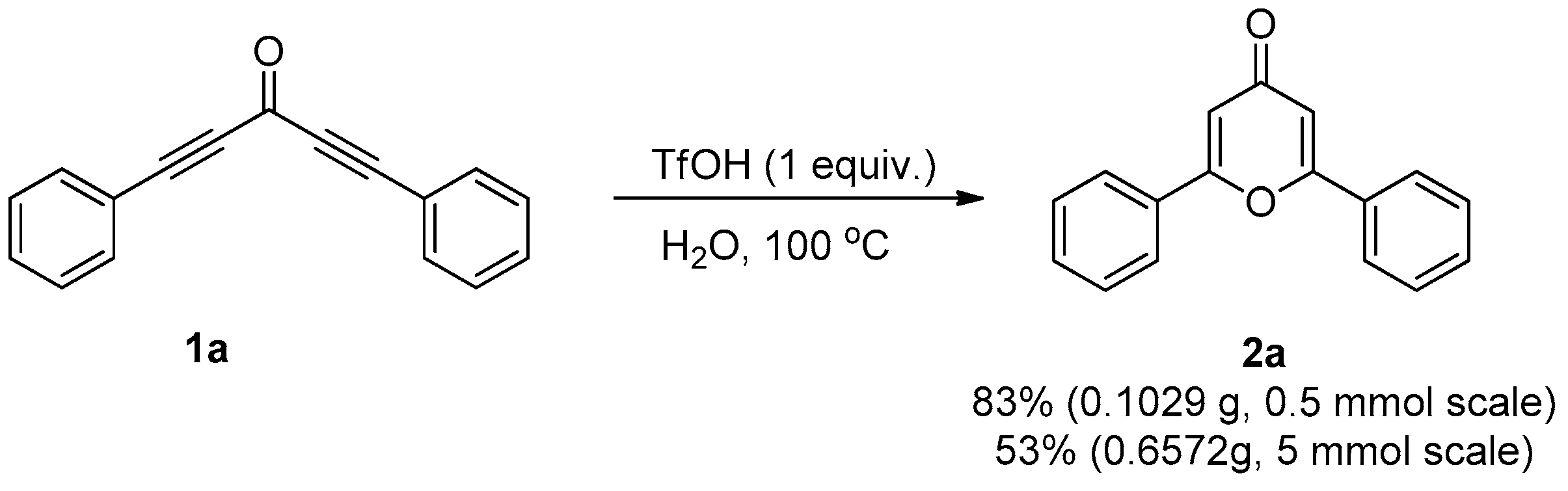
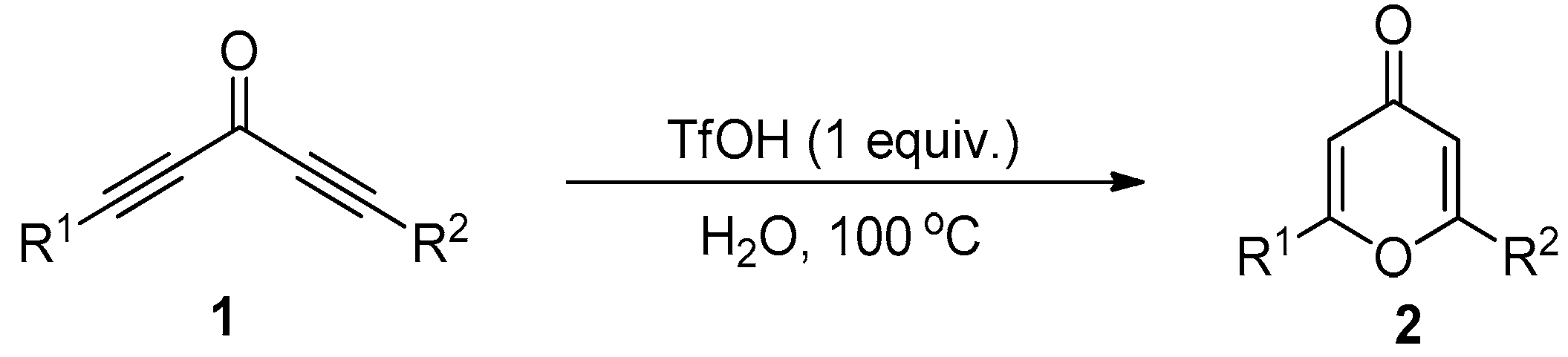
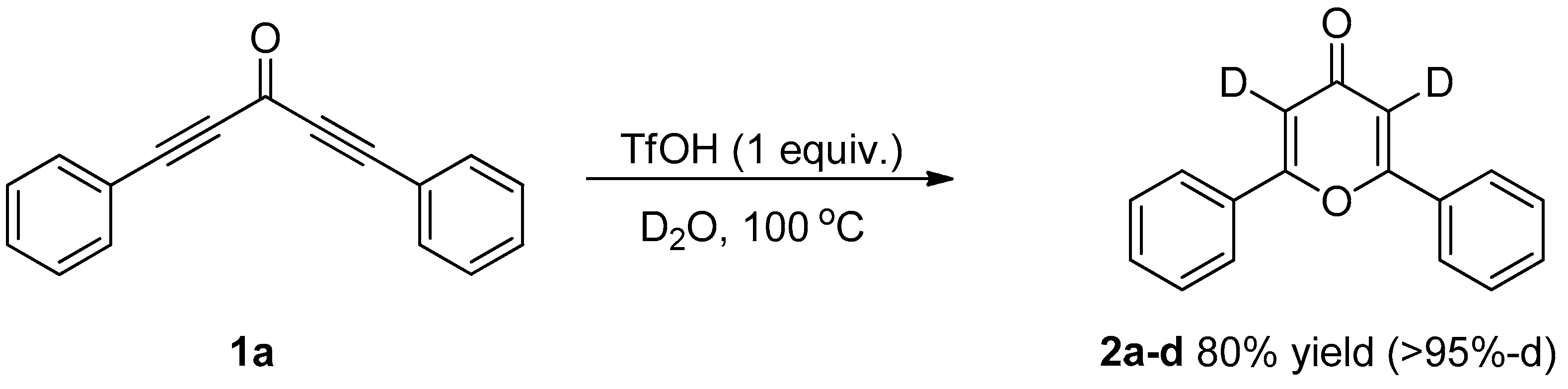
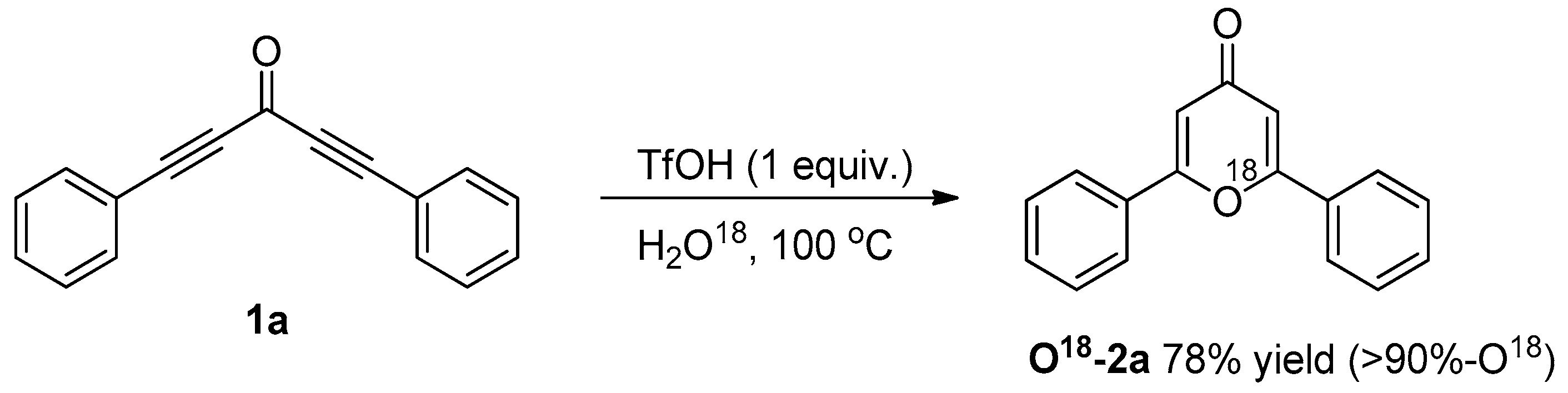
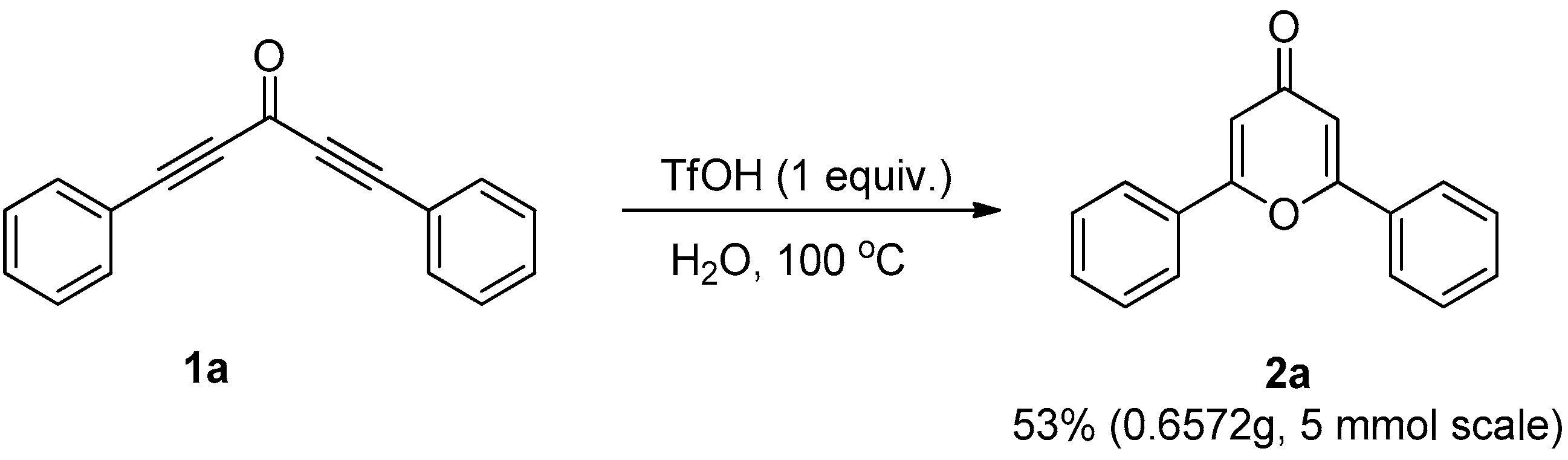

| Entry | Catalyst | Time (h) | Yield (%) b |
|---|---|---|---|
| 1 | TfOH | 24 | 70 |
| 2 | CH3COOH | 24 | 0 |
| 3 | PTSA | 24 | 50 |
| 4 | HCl | 24 | 0 |
| 5 | H3PO4 | 24 | 0 |
| 6 | PhCOOH | 24 | 10 |
| 7 c | TfOH | 24 | 10 |
| 8 d | TfOH | 24 | 50 |
| 9 e | TfOH | 24 | 80 |
| 10 f | TfOH | 24 | 20 |
| 11 g | TfOH | 24 | 75 |
| 12 | TfOH | 36 | 83 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
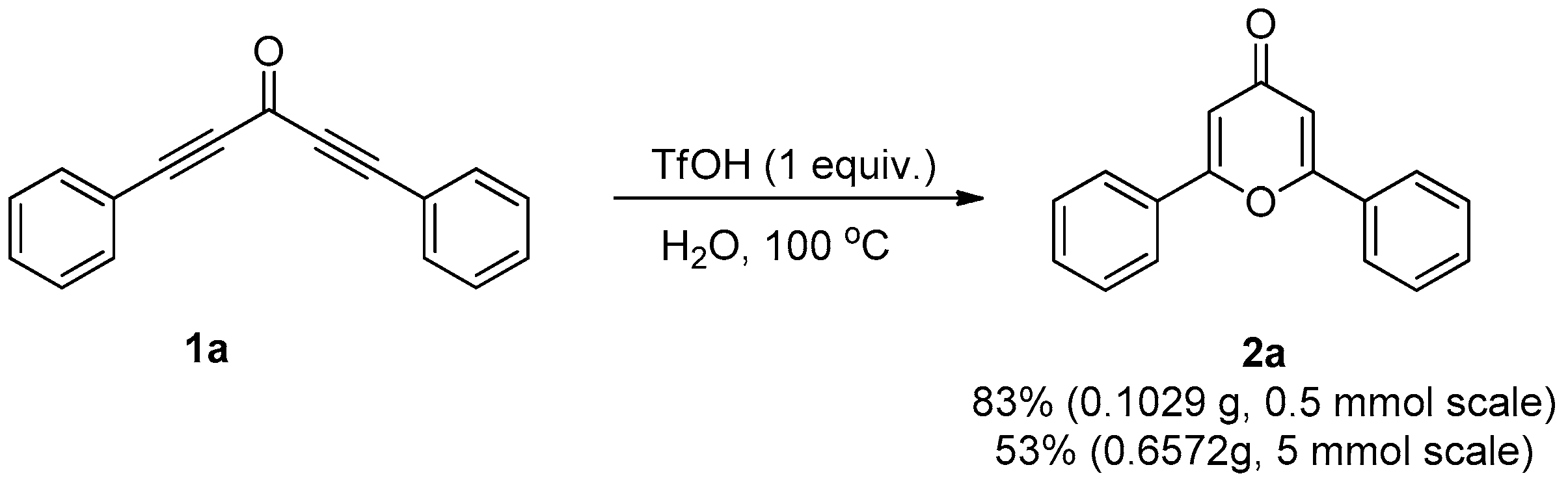{kind=link}
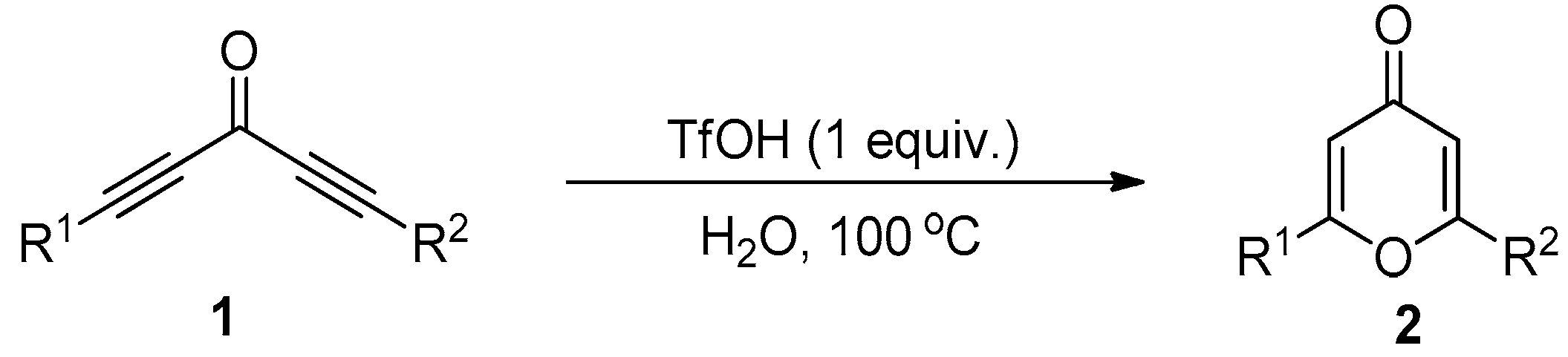{kind=link}
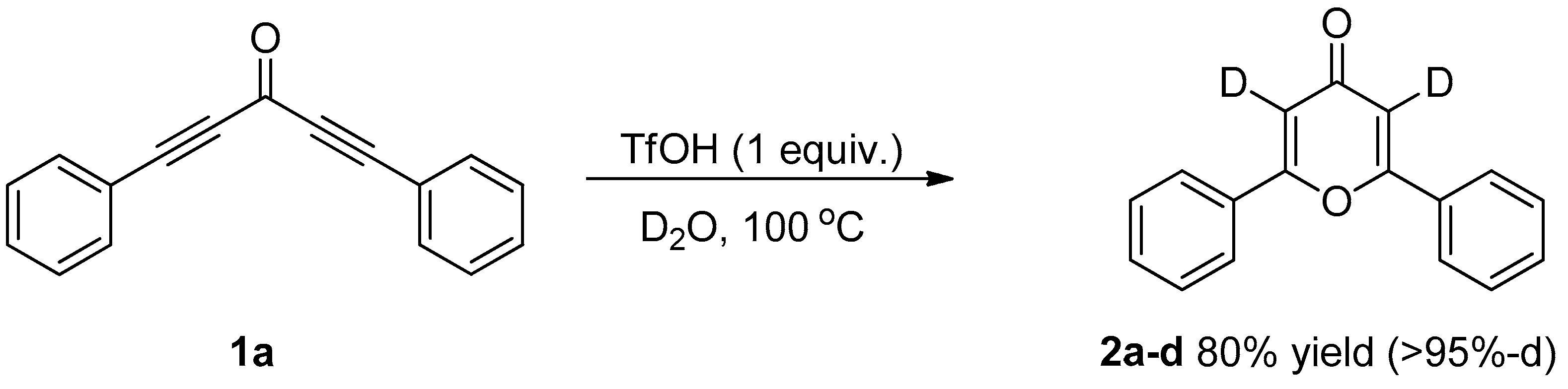{kind=link}
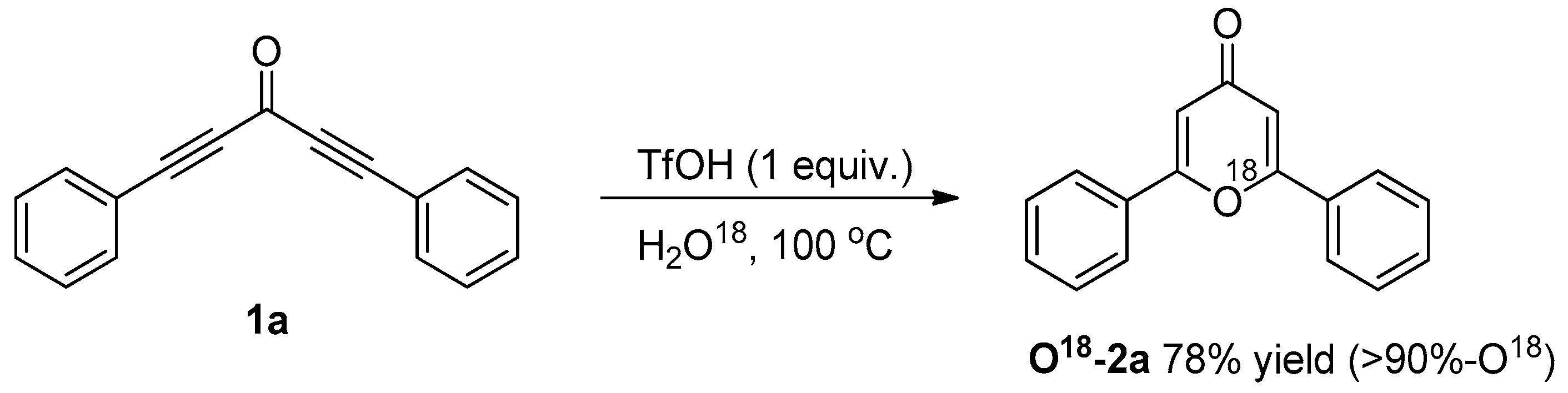{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.-L.; Teng, Q.-H.; Tong, W.; Wang, H.-S.; Pan, Y.-M.; Ma, X.-L. Atom-Economic Synthesis of 4-Pyrones from Diynones and Water. Molecules 2017, 22, 109. https://doi.org/10.3390/molecules22010109
Xu Y-L, Teng Q-H, Tong W, Wang H-S, Pan Y-M, Ma X-L. Atom-Economic Synthesis of 4-Pyrones from Diynones and Water. Molecules. 2017; 22(1):109. https://doi.org/10.3390/molecules22010109
Chicago/Turabian StyleXu, Yan-Li, Qing-Hu Teng, Wei Tong, Heng-Shan Wang, Ying-Ming Pan, and Xian-Li Ma. 2017. "Atom-Economic Synthesis of 4-Pyrones from Diynones and Water" Molecules 22, no. 1: 109. https://doi.org/10.3390/molecules22010109
APA StyleXu, Y.-L., Teng, Q.-H., Tong, W., Wang, H.-S., Pan, Y.-M., & Ma, X.-L. (2017). Atom-Economic Synthesis of 4-Pyrones from Diynones and Water. Molecules, 22(1), 109. https://doi.org/10.3390/molecules22010109