
Self-Supported N-Heterocyclic Carbenes and Their Use as Organocatalysts


Abstract
:1. Introduction
2. Results and Discussion
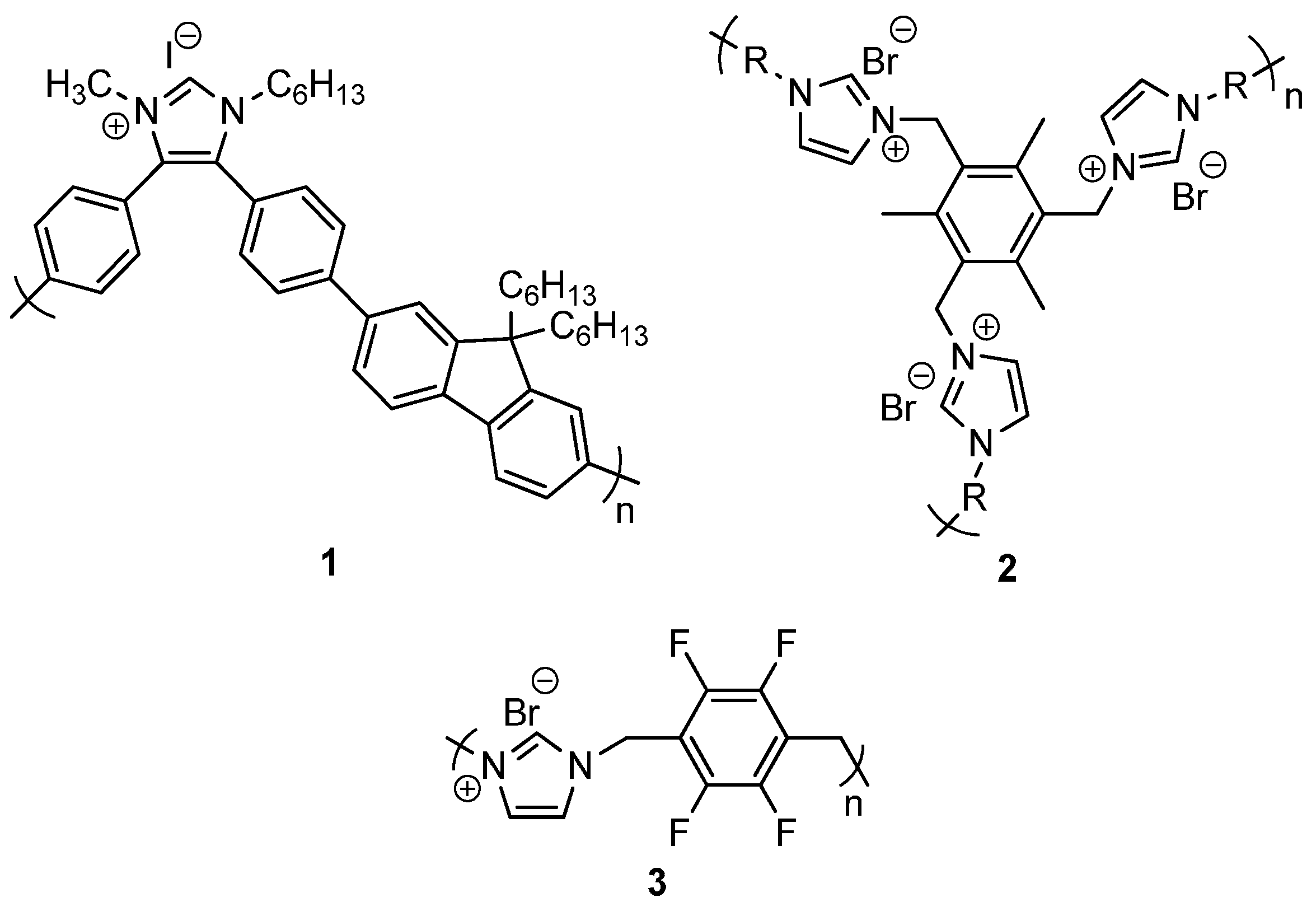
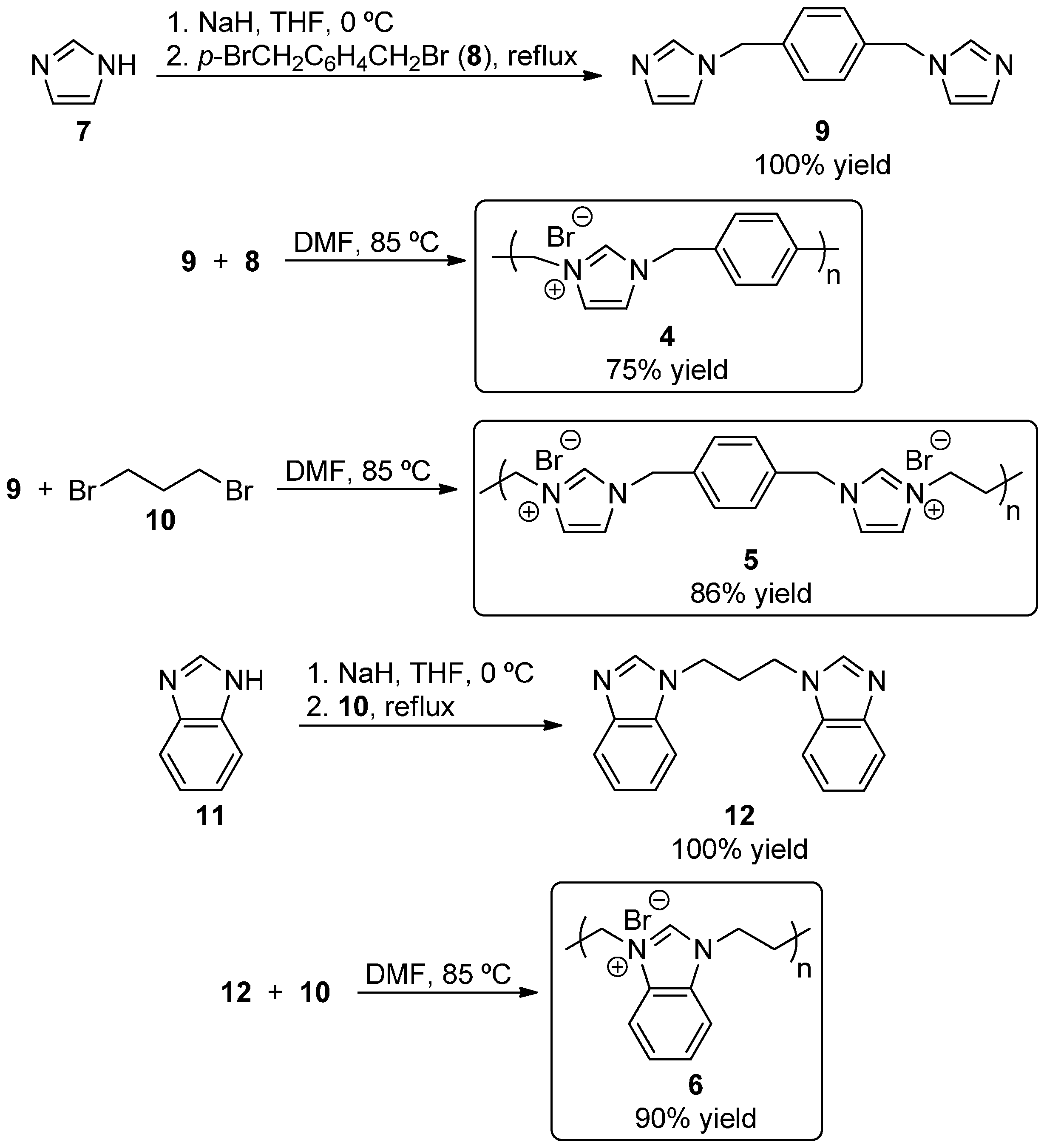
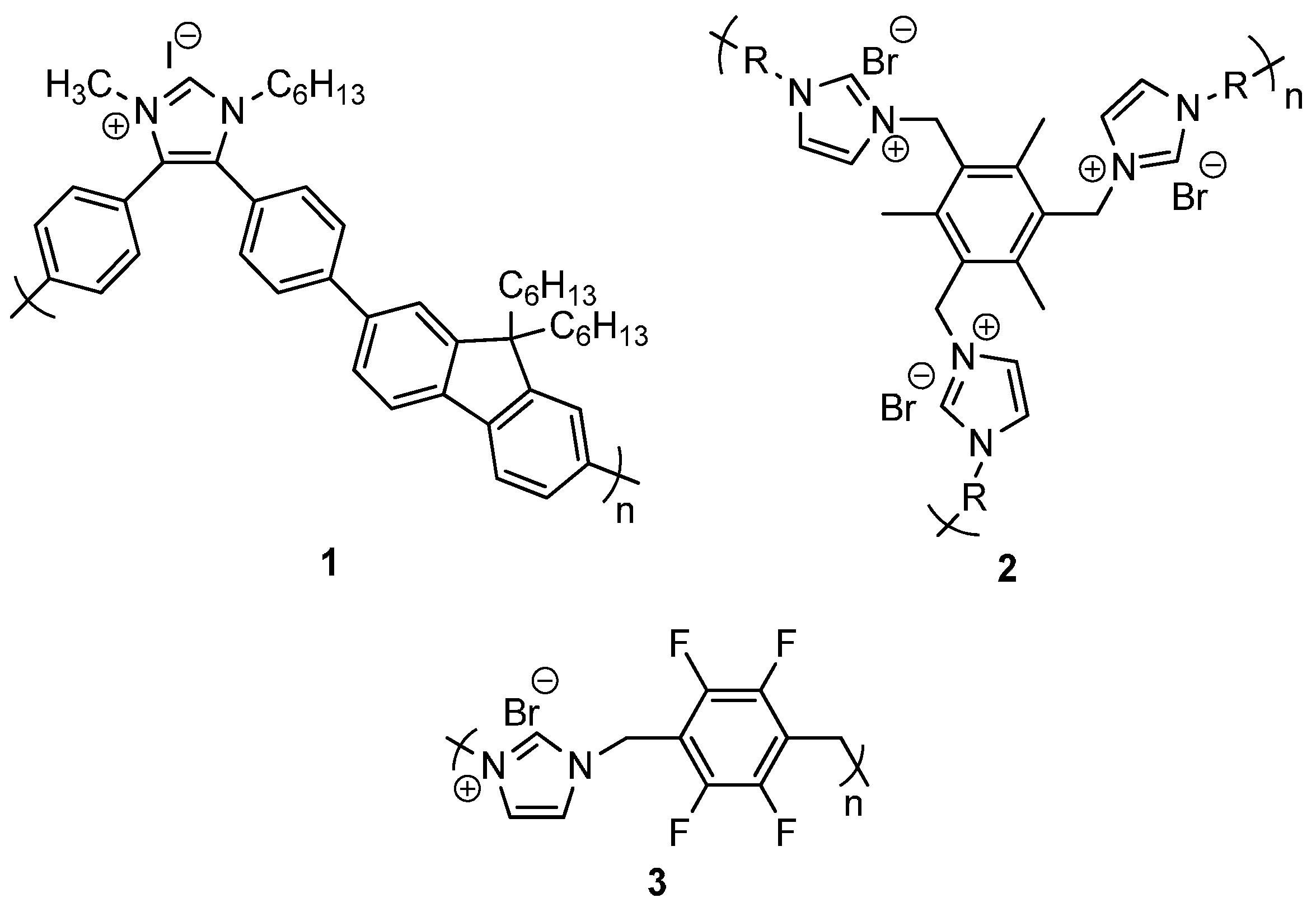
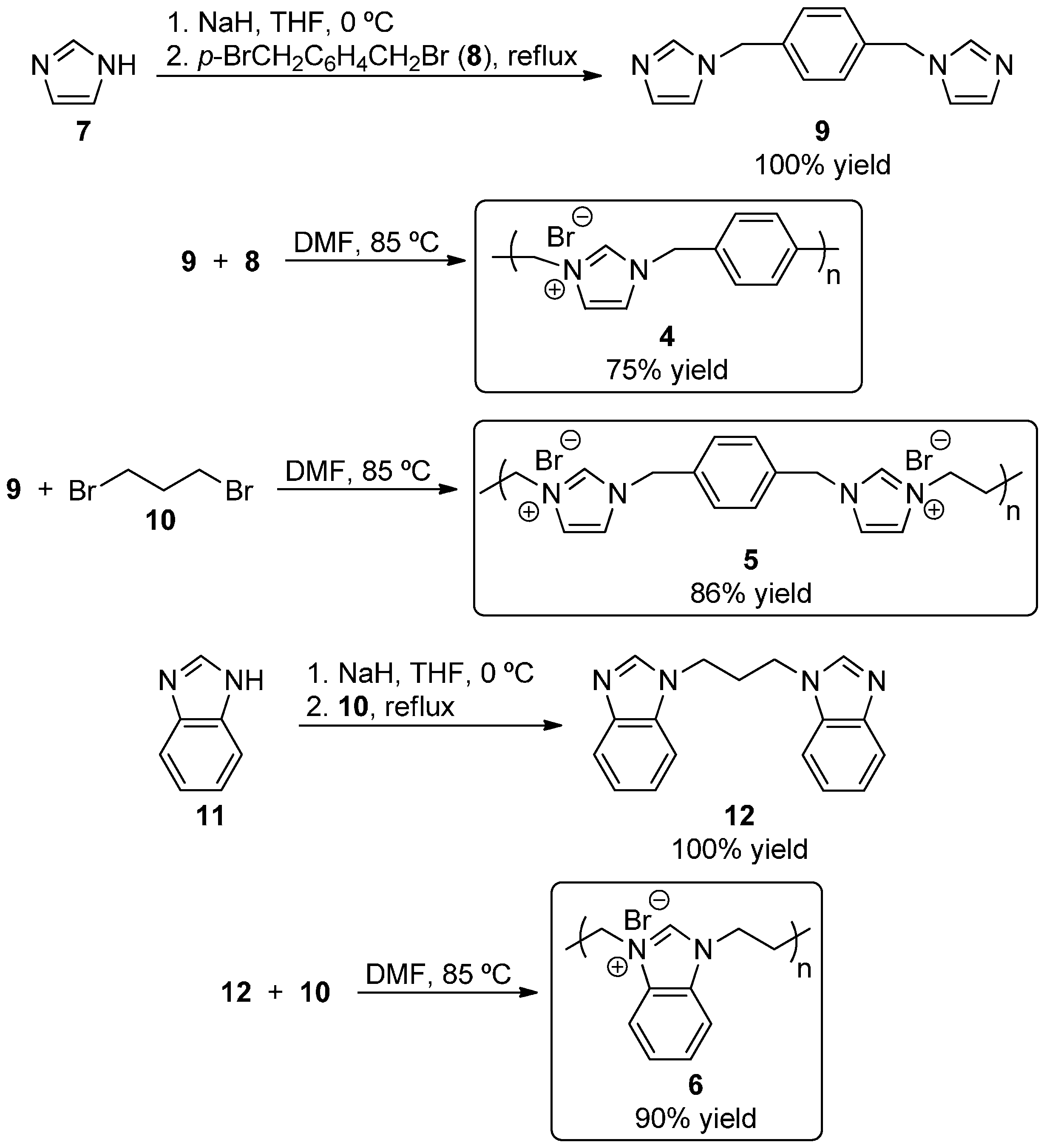
2.1. Synthesis of Self-Supported NHC Precursors
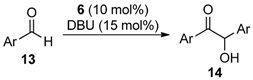
2.2. Self-Supported NHCs as Catalysts in Benzoin Condensation Reactions
2.3. Self-Supported NHCs as Catalysts in Redox Esterification Reactions of α,β-Unsaturated Aldehydes
3. Experimental Section
3.1. General Information
3.2. Synthesis of NHC Precursors
3.3. General Procedure for the Benzoin Condensation Reactions
3.4. Recycling of Polymer 5 from Benzoin Condensation Reactions
3.5. General Procedure for the Redox Esterification of α,β-Unsaturated Aldehydes
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Breslow, R. Rapid deuterium exchange in thiazolium salts. J. Am. Chem. Soc. 1957, 79, 1762–1763. [Google Scholar] [CrossRef]
- Breslow, R. On the mechanism of thiamine action. IV. Evidence from studies on model systems. J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar] [CrossRef]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, A.; Enders, D. N-heterocyclic carbene catalyzed domino reactions. Angew. Chem. Int. Ed. 2012, 51, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.; Hutson, G.E.; Cohen, D.T.; Scheidt, K.A. A continuum of progress: Applications of N-heterocyclic carbene catalysis in total synthesis. Angew. Chem. Int. Ed. 2012, 51, 11686–11698. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Churchill, G.; Smith, A.D. NHCs in asymmetric organocatalysis: Recent advances in azolium enolate generation and reactivity. Synthesis 2012, 44, 2295–2309. [Google Scholar]
- Ryan, S.J.; Candish, L.; Lupton, D.W. Acyl anion free N-heterocyclic carbene organocatalysis. Chem. Soc. Rev. 2013, 42, 4906–4917. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [PubMed]
- Mahatthananchai, J.; Bode, J.W. On the mechanism of N-heterocyclic carbene-catalyzed reactions involving acyl azoliums. Acc. Chem. Res. 2014, 47, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Canac, Y.; Lavallo, V.; Bertrand, G. Comparative reactivity of different types of stable cyclic and acyclic mono- and diamino carbenes with simple organic substrates. J. Am. Chem. Soc. 2014, 136, 5023–5030. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Breugst, M.; Neudörfl, J.-M.; Sunoj, R.B.; Berkessel, A. Keto-Enol thermodynamics of Breslow intermediates. J. Am. Chem. Soc. 2016, 138, 5044–5051. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Chi, D.Y. Polymer-supported ionic liquids: Imidazolium salts as catalysts for nucleophilic substitution reactions including fluorinations. Angew. Chem. Int. Ed. 2004, 43, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Antonietti, M. Poly(ionic liquid)s: Polymers expanding classical property profiles. Polymer 2011, 52, 1469–1482. [Google Scholar] [CrossRef]
- Yuan, J.; Mecerreyes, D.; Antonietti, M. Poly(ionic liquid)s: An update. Prog. Polym. Sci. 2013, 38, 1009–1036. [Google Scholar] [CrossRef]
- Nishimura, N.; Ohno, H. 15th anniversary of polymerized ionic liquids. Polymer 2014, 55, 3289–3297. [Google Scholar] [CrossRef]
- Prakash, C.; Shinde, S.S.; Biradar, A.V. Tailor made ionic liquids: catalyst and media for organic transformations. Curr. Org. Chem. 2015, 19, 728–742. [Google Scholar]
- Zhou, H.; Zhang, W.-Z.; Wang, Y.-M.; Qu, J.-P.; Lu, X.-B. N-Heterocyclic carbene functionalized polymer for reversible fixation-release of CO2. Macromolecules 2009, 42, 5419–5421. [Google Scholar] [CrossRef]
- Pawar, G.M.; Buchmeiser, M.R. Polymer-supported carbon dioxide-protected N-heterocyclic carbenes: Synthesis and application in organo- and organometallic catalysis. Adv. Synth. Catal. 2010, 352, 917–928. [Google Scholar] [CrossRef]
- Pinaud, J.; Vignolle, J.; Gnanou, Y.; Taton, D. Poly(N-heterocyclic carbene)s and their CO2 adducts as recyclable polymer-supported organocatalysts for benzoin condensation and transesterification reactions. Macromolecules 2011, 44, 1900–1908. [Google Scholar] [CrossRef]
- Gondo, C.A.; Bode, J.W. Catalytic redox amidations of aldehydes with a polymer-supported peptide-N-heterocyclic carbene multifunctional catalyst. Synlett 2013, 24, 1205–1210. [Google Scholar] [CrossRef]
- Coupillaud, P.; Pinaud, J.; Guidolin, N.; Vignolle, J.; Fèvre, M.; Veaudecrenne, E.; Mecerreyes, D.; Taton, D. Poly(ionic liquid)s based on imidazolium hydrogen carbonate monomer units as recyclable polymer-supported N-heterocyclic carbenes: Use in organocatalysis. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4530–4540. [Google Scholar]
- Bortolini, O.; Cavazzini, A.; Dambruoso, P.; Giovannini, P.P.; Caciolli, L.; Massi, A; Pacifico, S.; Ragnoa, D. Thiazolium-functionalized polystyrene monolithic microreactors for continuous-flow umpolung catalysis. Green Chem. 2013, 15, 2981–2992. [Google Scholar] [CrossRef]
- Coupillaud, P.; Vignolle, J.; Mecerreyes, D.; Taton, D. Post-polymerization modification and organocatalysis using reactive statistical poly(ionic liquid)-based copolymers. Polymer 2014, 55, 3404–3414. [Google Scholar] [CrossRef]
- Powell, A.B.; Suzuki, Y.; Ueda, M.; Bielawski, C.W.; Cowley, A.H. A recyclable, self-supported organocatalyst based on a poly(N-heterocyclic carbene). J. Am. Chem. Soc. 2011, 133, 5218–5220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, L.; Patra, P.K.; Hu, D.; Ying, J.Y. Colloidal poly-imidazolium salts and derivatives. Nano Today 2009, 4, 13–20. [Google Scholar] [CrossRef]
- Tan, M.; Zhang, Y.; Ying, J.Y. Hydrosilylation of ketone and imine over poly-N-heterocyclic carbene particles. Adv. Synth. Catal. 2009, 351, 1390–1394. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, Y. Copper- and copper-N-heterocyclic carbene-catalyzed C-H activating carboxylation of terminal alkynes with CO2 at ambient conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 20184–20189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ying, J.Y. Main-chain organic frameworks with advanced catalytic functionalities. ACS Catal. 2015, 5, 2681–2691. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Zhao, Y.; Ji, G.; Zhang, H.; Yu, B.; Gao, X.; Liu, Z. Fluoro-functionalized polymeric ionic liquids: Highly efficient catalysts for CO2 cycloaddition to cyclic carbonates under mild conditions. Green Chem. 2014, 16, 3724–3728. [Google Scholar] [CrossRef]
- Xia, X.; Toy, P.H. Rasta resin-triphenylphosphine oxides and their use as recyclable heterogeneous reagent precursors in halogenation reactions. Beilstein J. Org. Chem. 2014, 10, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yang, Y.-C.; Toy, P.H. Rasta resin-TBD-catalyzed γ-selective Morita–Baylis–Hillman reactions of α,γ-disubstituted allenones. Synlett 2015, 26, 1732–1736. [Google Scholar] [CrossRef]
- Xia, X.; Toy, P.H. Polyethyleneimine-supported triphenylphosphine and its use as a highly loaded bifunctional polymeric reagent in chromatography-free one-pot Wittig reactions. Synlett 2015, 26, 1737–1743. [Google Scholar] [CrossRef]
- Ma, S.; Toy, P.H. Chromatography-free esterification reactions using a bifunctional polymer. Synlett 2016, 27, 1207–1210. [Google Scholar]
- Yang, Y.-C.; Toy, P.H. Self-supported ligands as a platform for catalysis: Use of a polymeric oxime in a recyclable palladacycle precatalyst for Suzuki–Miyaura reactions. Synlett 2014, 25, 1319–1324. [Google Scholar]
- Marti, J.; López-Calahorra, F.; Bofill, J.M. A theoretical study of benzoin condensation. J. Mol. Struct. THEOCHEM 1995, 339, 179–194. [Google Scholar] [CrossRef]
- Moore, J.L.; Silvestri, A.P.; de Alaniz, J.R.; DiRocco, D.A.; Rovis, T. Mechanistic investigation of the enantioselective intramolecular Stetter reaction: Proton transfer is the first irreversible step. Org. Lett. 2011, 13, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Scheidt, K.A. Conversion of α,β-unsaturated aldehydes into saturated esters: An umpolung reaction catalyzed by nucleophilic carbenes. Org. Lett. 2005, 7, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Maki, B.E.; Chan, A.; Scheidt, K.A. Protonation of homoenolate equivalents generated by N-heterocyclic carbenes. Synthesis 2008, 40, 1306–1315. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
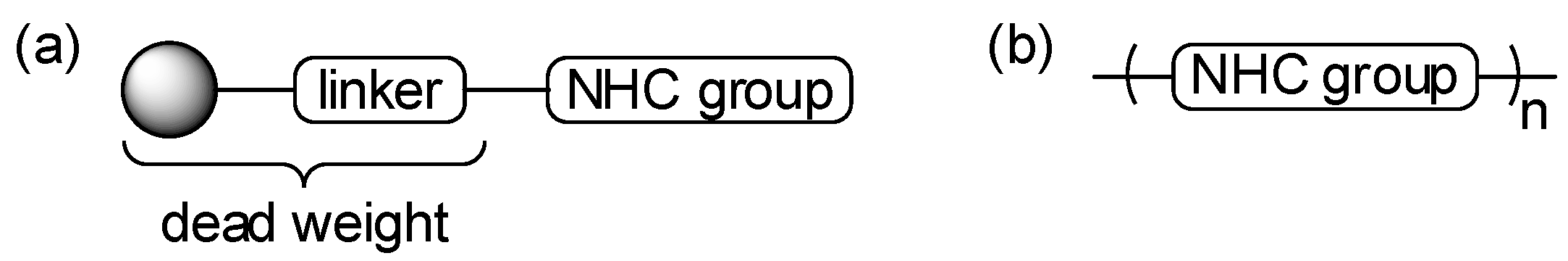

| Entry | NHC-HBr | Solvent | Time (h) | Isolated Yield (%) |
|---|---|---|---|---|
| 1 | -- | DMSO (2 mL) | 18 | 0 |
| 2 | 4 | DMSO (2 mL) | 18 | 0 |
| 3 | 5 | DMSO (2 mL) | 18 | 74 |
| 4 | 6 | DMSO (2 mL) | 18 | 82 |
| 5 | 6 | THF (2 mL) | 18 | 41 |
| 6 | 6 | DMF (2 mL) | 5 | 84 |
| 7 | 6 | DMF (1 mL) | 5 | 92 |
| 8 | 6 | DMF (1 mL) b | 5 | 0 |
| 9 | 15 | DMF (1 mL) | 5 | 7 |

| Entry | Substrate | Time (h) | Product | Isolated Yield (%) |
|---|---|---|---|---|
| 1 | Ar = 4-Cl-C6H4- (13b) | 12 | Ar = 4-Cl-C6H4- (14b) | 94 |
| 2 | Ar = 4-Br-C6H4- (13c) | 12 | Ar = 4-Br-C6H4- (14c) | 88 |
| 3 | Ar = 4-MeO-C6H4- (13d) | 12 | Ar = 4-MeO-C6H4- (14d) | 82 |
| 4 | Ar = 3,4,5-(MeO)3 -C6H2- (13e) | 5 | Ar = 3,4,5-(MeO)3 -C6H2- (14e) | 82 |
| 5 | Ar = 4-CF3-C6H4- (13f) | 12 | Ar = 4-CF3-C6H4- (14f) | 78 |
| 6 | Ar = 2-Napth- (13g) | 5 | Ar = 2-Napth- (14g) | 88 |
| Cycle | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Isolated Yield (%) | 92 | 92 | 91 | 90 | 92 |
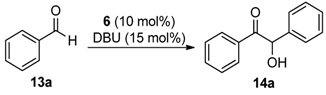
| Entry | 6 (mol %) | Additive (1 equiv.) | Time (h) | Isolated Yield (%) |
|---|---|---|---|---|
| 1 | 10 | -- | 4 | 44 |
| 2 | 10 | phenol | 48 | 86 |
| 3 | 10 | 4-nitrophenol | 48 | 91 |
| 4 | 20 | 4-nitrophenol | 3 | 90 |
| 5 b | 20 | 4-nitrophenol | 8 | 72 |
| 6 c | 20 | 4-nitrophenol | 8 | 51 |
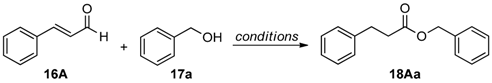
| Entry | Ar | R | Time (h) | Product | Isolated Yield (%) |
|---|---|---|---|---|---|
| 1 | Ph- (16A) | 4-Br-C6H4CH2- (17b) | 7 | 18Ab | 94 |
| 2 | Ph- (16A) | 2-Br-C6H4CH2- (17c) | 12 | 18Ac | 92 |
| 3 | Ph- (16A) | 2-Cl-C6H4CH2- (17d) | 12 | 18Ad | 94 |
| 4 | Ph- (16A) | PhCH(CH3)- (17e) | 20 | 18Ae | 30 |
| 5 | Ph- (16A) | menthyl- (17f) | 48 | 18Af | 30 |
| 6 b | Ph- (16A) | -- | 48 | 18Ag | 7 |
| 7 | 4-Br-C6H4- (16B) | Bn- (17a) | 4 | 18Ba | 89 |
| 8 | 4-CN-C6H4- (16C) | Bn- (17a) | 3 | 18Ca | 89 |
| 9 | 4-tBu-C6H4- (16D) | Bn- (17a) | 3 | 18Da | 90 |
| 10 | 4-MeO-C6H4- (16E) | Bn- (17a) | 5 | 18Ea | 79 |
| 11 | 2-furanyl- (16F) | Bn- (17a) | 7 | 18Fa | 89 |

{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Toy, P.H. Self-Supported N-Heterocyclic Carbenes and Their Use as Organocatalysts. Molecules 2016, 21, 1100. https://doi.org/10.3390/molecules21081100
Ma S, Toy PH. Self-Supported N-Heterocyclic Carbenes and Their Use as Organocatalysts. Molecules. 2016; 21(8):1100. https://doi.org/10.3390/molecules21081100
Chicago/Turabian StyleMa, Shuang, and Patrick H. Toy. 2016. "Self-Supported N-Heterocyclic Carbenes and Their Use as Organocatalysts" Molecules 21, no. 8: 1100. https://doi.org/10.3390/molecules21081100
APA StyleMa, S., & Toy, P. H. (2016). Self-Supported N-Heterocyclic Carbenes and Their Use as Organocatalysts. Molecules, 21(8), 1100. https://doi.org/10.3390/molecules21081100