
Five New Biphenanthrenes from Cremastra appendiculata



Abstract
:
1. Introduction
2. Results and Discussion
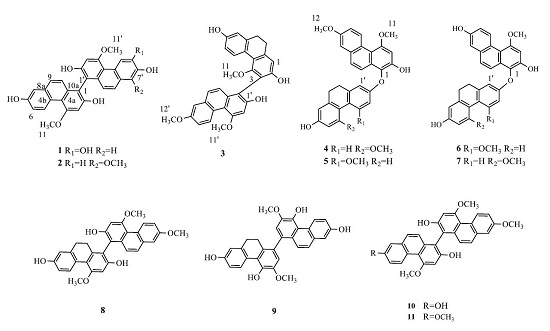
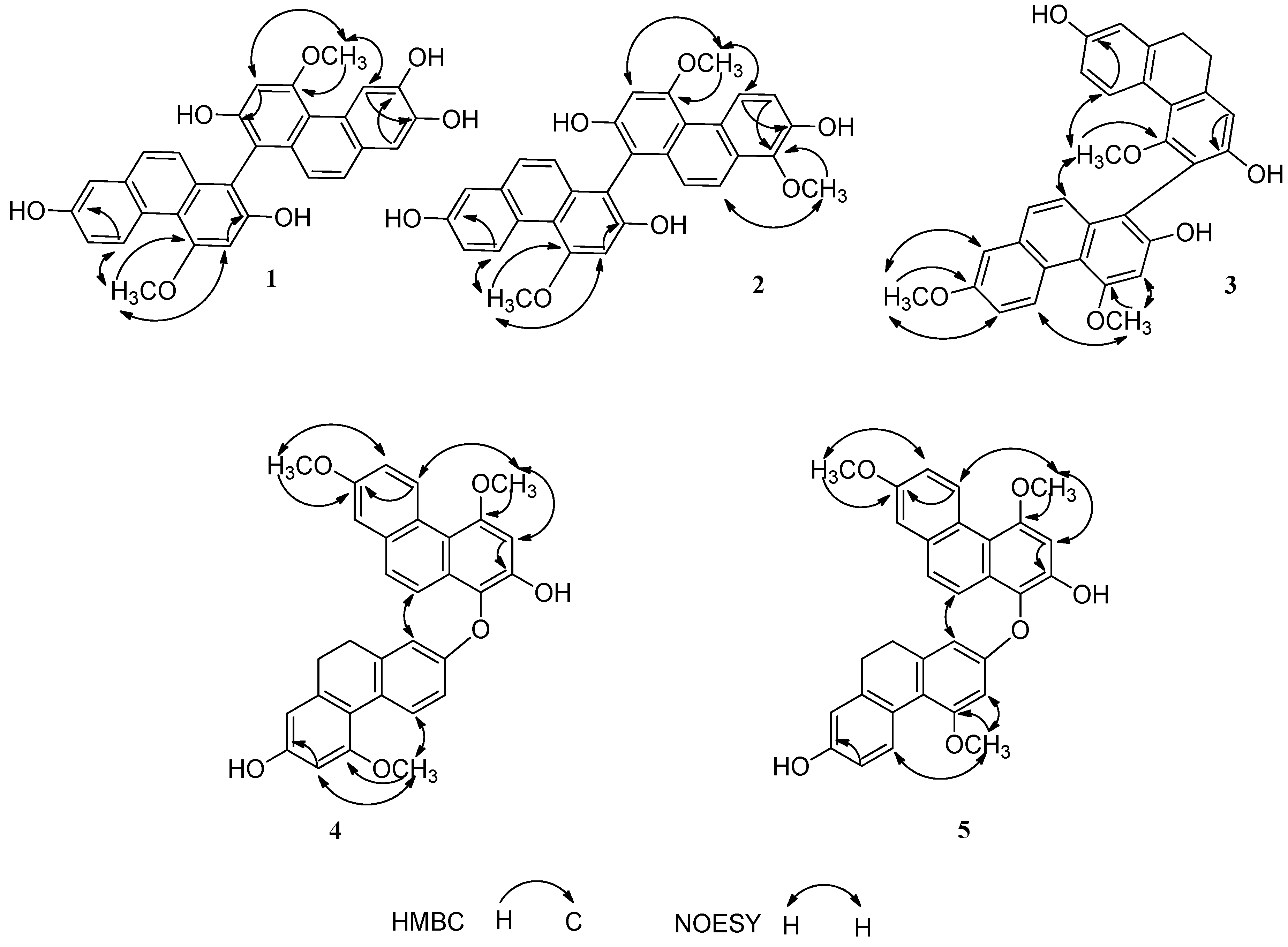
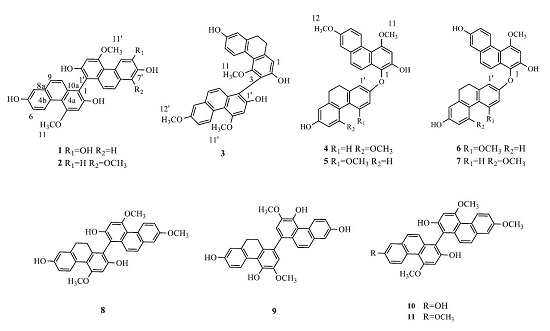
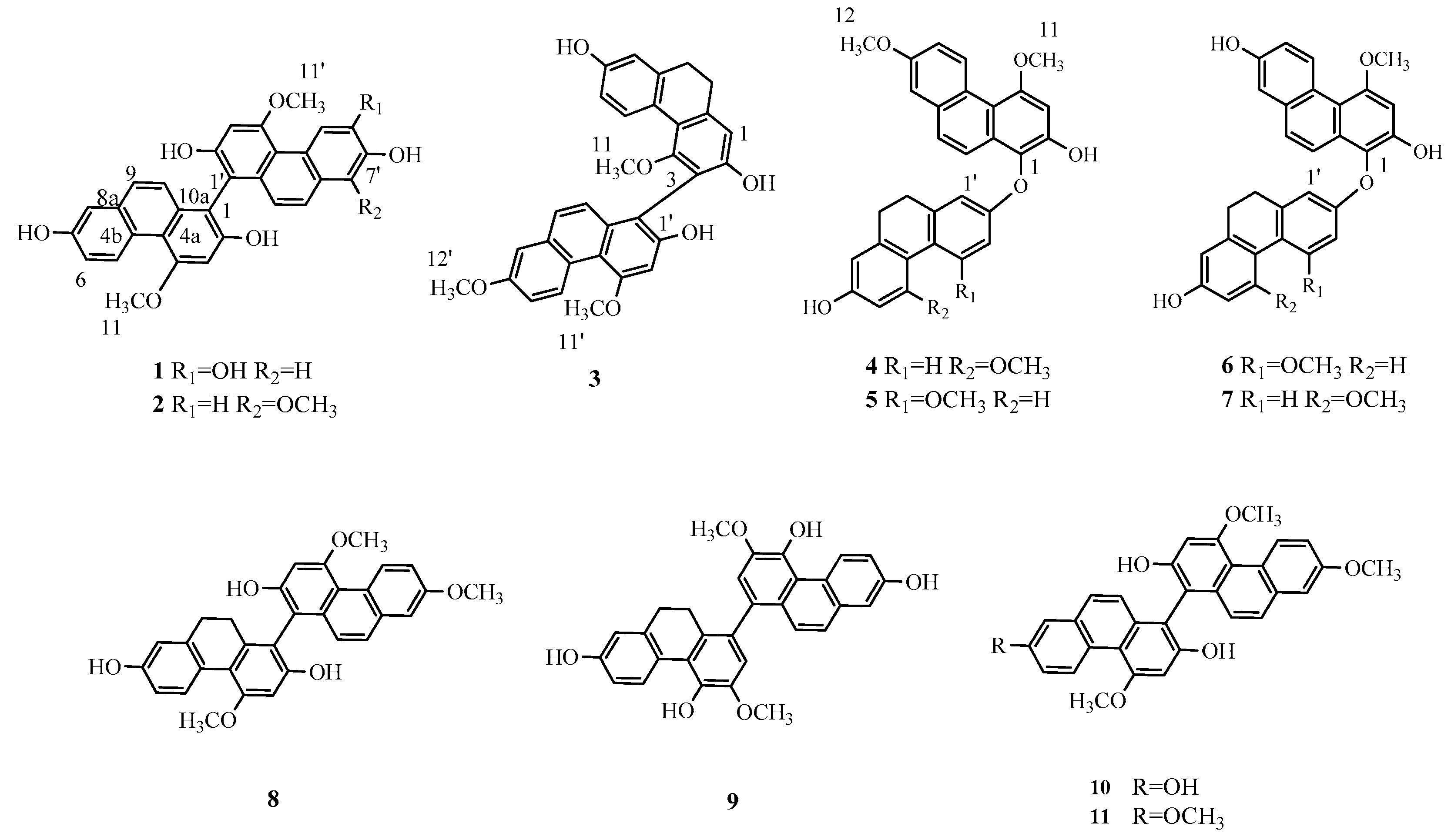
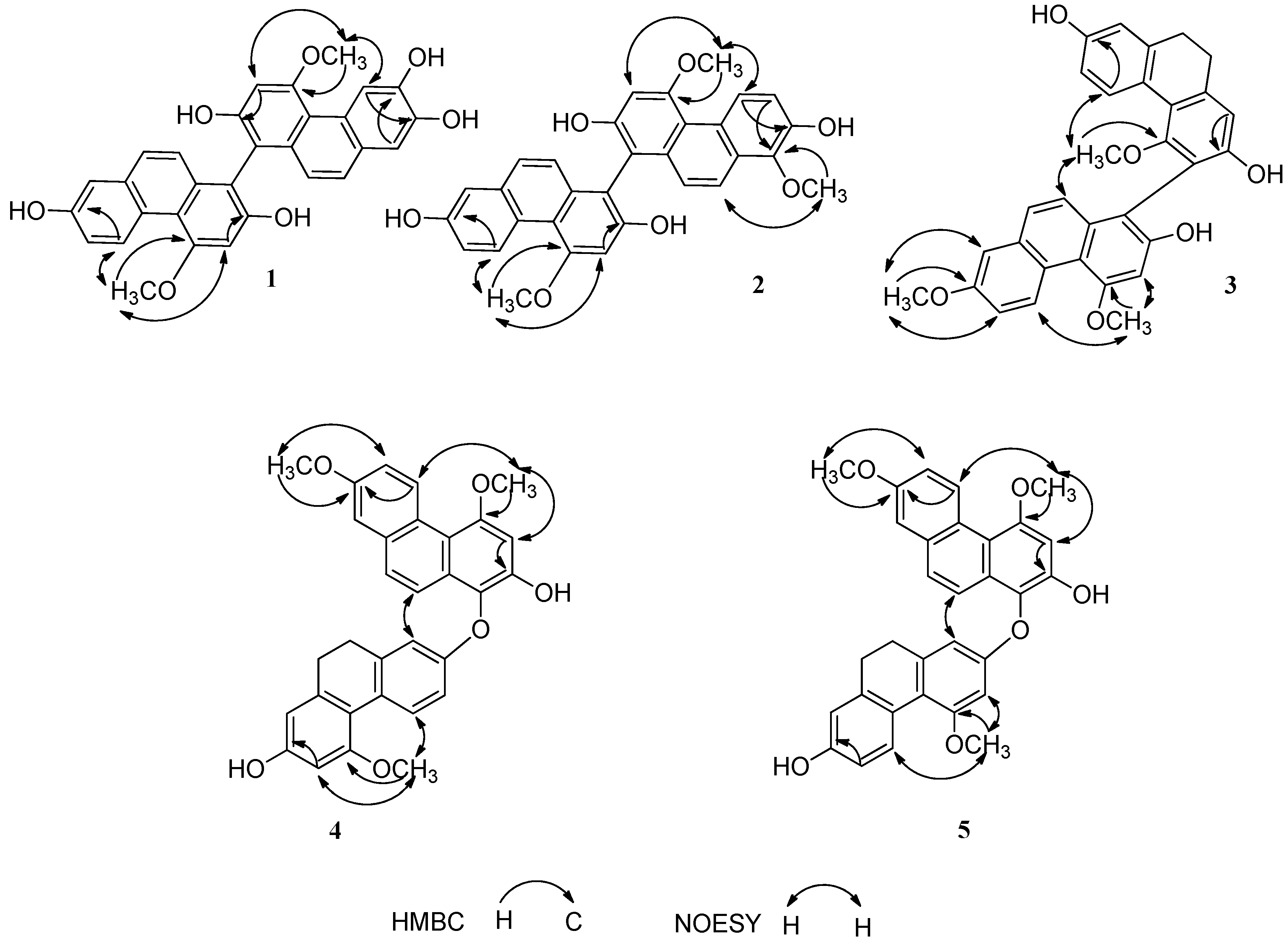
2.1. Structure Elucidation
2.2. Cytotoxicity Assay
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data
3.5. Cytotoxic Bioassay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kovács, A.; Vasas, A.; Hohmann, J. Natural phenanthrenes and their biological activity. Phytochemistry 2008, 69, 1084–1110. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Li, S.; Wang, S.J.; Wang, Y.H.; Yang, Y.C.; Shi, J.G.; He, L. Mono-, Bi-, and triphenanthrenes from the tubers of Cremastra appendiculata. J. Nat. Prod. 2006, 69, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guan, S.H.; Meng, Y.H.; Zhang, Y.B.; Cheng, C.R.; Shi, Y.Y.; Feng, R.H.; Zeng, F.; Wu, Z.Y.; Zhang, J.X.; et al. Phenanthrenes, 9,10-dihydrophenanthrenes, bibenzyls, with their derivatives, and malate or tartrate benzyl ester glucosides from tubers of Cremastra appendiculata. Phytochemistry 2013, 94, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Cai, L.; Tai, Z.G.; Zeng, X.H.; Ding, Z.T. Four new phenanthrenes from Monomeria barbata Lindl. Fitoterapia 2010, 81, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.J.; Yu, H.; Qing, C.; Zhang, Y.L.; Liu, Y.; Chen, Y.G. Two new biphenanthrenes with cytotoxic activity from Bulbophyllum odoratissimum. Fitoterapia 2009, 80, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Tang, C.P.; Li, X.Q.; Ye, Y. Phochinenins A–F, dimeric 9,10-dihydrophenanthrene derivatives, from Pholidota chinensis. Helv. Chim. Acta 2008, 91, 2122–2129. [Google Scholar] [CrossRef]
- Apel, C.; Dumontet, V.; Lozach, O.; Meijer, L.; Guéritte, F.; Litaudon, M. Phenanthrene derivatives from Appendicula reflexa as new CDK1/cyclin B inhibitors. Phytochem. Lett. 2012, 5, 814–818. [Google Scholar] [CrossRef]
- Majumder, P.L.; Bandyopadhyay, S.; Pal, S. Rigidanthrin, a new dimeric phenanthrene derivative of the orchid Bulbophyllum rigidum. J. Indian Chem. Soc. 2008, 85, 1116–1123. [Google Scholar]
- Majumder, P.L.; Mukhoti, N.; Chattopadhyay, S. Agrostophyllanthrol and isoagrostophyllanthrol, two novel diastereomeric phenanthropyran derivatives from the orchid Agrostophyllum khasiyanum. J. Indian Chem. Soc. 2008, 85, 1315–1325. [Google Scholar] [CrossRef]
- Gutierrez, R.M.P.; Gonzalez, A.M.N.; Baez, E.G.; Diaz, S.L. Studies on the constituents of bulbs of the orchid Prosthechea michuacana and antioxidant activity. Chem. Nat. Compd. 2010, 46, 554–561. [Google Scholar] [CrossRef]
- Qian, C.D.; Jiang, F.S.; Yu, H.S.; Shen, Y.; Fu, Y.H.; Cheng, D.Q.; Gan, L.S.; Ding, Z.S. Antibacterial biphenanthrenes from the Fibrous Roots of Bletilla Striata. J. Nat. Prod. 2015, 78, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Masae, Y.; Li, B.; Tomoko, K.; Keiko, I.; Shuzo, T.; Yuriko, Y.; Kenichi, T. Bisphenanthreneethers from Bletilla striata. Phytochemistry 1992, 31, 3985–3987. [Google Scholar]
- Liu, L.; Ye, J.; Li, P.; Tu, P.F. Chemical constituents of tubers of Cremastra appendiculata. Chin. J. Chin. Mater. Med. 2014, 39, 250–253. [Google Scholar]
- Editorial Committee of the Administration Bureau of Traditional Chinese Medicine. Chinese Materia Medica (Zhong Hua Ben Cao); Shanghai Science and Technology Press: Shanghai, China, 1999; Volume 6, p. 7828. [Google Scholar]
- Liu, L.; Li, J.; Zeng, K.W.; Li, P.; Tu, P.F. Three new phenanthrenes from Cremastra appendiculata (D. Don) Makino. Chin. Chem. Lett. 2013, 24, 737–739. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Zeng, K.W.; Li, P.; Tu, P.F. Five new benzylphenanthrenes from Cremastra appendiculata. Fitoterapia 2015, 103, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
| Proton | 1 a | 2 b | 3 b |
|---|---|---|---|
| 1 | 6.71 s | ||
| 3 | 6.97 s | 7.00 s | |
| 5 | 9.36 d (9.0) | 9.38 d (9.0) | 8.08 d (9.0) |
| 6 | 7.09 dd (9.0, 3.0) | 7.10 dd (9.0, 3.0) | 6.63 dd (9.0, 3.0) |
| 8 | 7.04 d (3.0) | 7.06 d (3.0) | 6.69 d (3.0) |
| 9 | 7.28 d (9.0) | 7.31 d (9.0) | 2.73–2.78 m c |
| 10 | 6.90 d (9.0) | 6.91 d (9.0) | 2.73–2.78 m c |
| 3′ | 6.92 s | 7.00 s | 6.96 s |
| 5′ | 8.96 s | 9.17 d (9.0) | 9.50 d (9.0) |
| 6′ | 7.20 d (9.0) | 7.17 dd (9.0 3.0) | |
| 8′ | 7.02 s | 7.25 d (3.0) | |
| 9′ | 7.20 d (9.0) | 7.66 d (9.0) | 7.53 d (9.0) |
| 10′ | 6.71 d (9.0) | 6.98 d (9.0) | 7.36 d (9.0) |
| 4-OCH3 | 4.10 s | 4.12 s | 3.14 s |
| 4′-OCH3 | 4.10 s | 4.14 s | 4.15 s |
| 7′-OCH3 | 3.92 s | ||
| 8′-OCH3 | 3.76 s |
| Carbon | 1 a | 2 b | 3 b |
|---|---|---|---|
| 1 | 111.3 | 111.0 | 112.3 |
| 2 | 153.3 | 153.5 | 156.0 |
| 3 | 99.7 | 99.7 c | 117.7 |
| 4 | 157.9 | 157.9 | 158.6 |
| 5 | 128.8 | 128.8 | 129.6 |
| 6 | 116.6 | 116.6 | 114.4 |
| 7 | 154.1 | 154.1 | 156.8 |
| 8 | 110.9 | 110.9 | 115.4 |
| 9 | 126.7 | 126.8 | 31.8 |
| 10 | 125.0 | 124.7 | 31.4 |
| 4a | 114.2 | 114.2 | 121.1 |
| 4b | 123.6 | 123.6 | 126.3 |
| 8a | 132.5 | 132.4 | 140.9 |
| 10a | 133.4 | 133.3 | 141.9 |
| 1′ | 110.6 | 111.0 | 110.9 |
| 2′ | 153.1 | 153.3 | 154.3 |
| 3′ | 99.0 | 99.8 c | 100.5 |
| 4′ | 157.7 | 157.8 | 160.5 |
| 5′ | 112.7 | 123.5 | 130.5 |
| 6′ | 145.6 | 117.4 | 117.2 |
| 7′ | 144.2 | 145.5 | 158.1 |
| 8′ | 111.7 | 140.8 | 109.5 |
| 9′ | 126.5 | 120.0 | 128.6 |
| 10′ | 121.5 | 124.7 | 126.5 |
| 4a′ | 113.8 | 114.2 | 116.7 |
| 4b′ | 124.4 | 124.3 | 126.6 |
| 8a′ | 125.6 | 126.2 | 134.5 |
| 10a′ | 133.8 | 133.4 | 135.2 |
| 4-OCH3 | 55.5 | 55.5 | 60.0 |
| 4′-OCH3 | 55.5 | 55.5 | 56.2 |
| 7′-OCH3 | 55.8 | ||
| 8′-OCH3 | 60.3 |
| Proton | 4 | 5 | 6 | 7 |
|---|---|---|---|---|
| 3 | 6.94 s | 7.04 s | 6.94 s | 6.94 s |
| 5 | 9.45 d (9.5) | 9.51 d (9.5) | 9.41 d (9.5) | 9.41 d (9.5) |
| 6 | 7.20 dd (9.5, 3.0) | 7.24 dd (9.5, 3.0) | 7.11 dd (9.5, 3.0) | 7.11 dd (9.5, 3.0) |
| 8 | 7.26 d (3.0) | 7.37 d (3.0) | 7.14 d (3.0) | 7.13 d (3.0) |
| 9 | 7.59 d (9.5) | 7.70 d (9.5) | 7.52 d (9.5) | 7.50 d (9.5) |
| 10 | 7.70 d (9.5) | 7.74 d (9.5) | 7.67 d (9.5) | 7.66 d (9.5) |
| 1′ | 6.67 d (3.0) | 6.20 d (3.0) | 6.24 d (3.0) | 6.66 d (3.0) |
| 3′ | 6.69 dd (9.5, 3.0) | 6.65 d (3.0) | 6.58 d (3.0) | 6.67 dd (9.5, 3.0) |
| 4′ | 8.05 d (9.5) | 8.02 d (9.5) | ||
| 5′ | 8.07 d (9.0) | 8.02 d (9.5) | ||
| 6′ | 6.38 d (3.0) | 6.69 dd (9.0,3.0) | 6.61 dd (9.5, 3.0) | 6.39 d (3.0) |
| 8′ | 6.29 d (3.0) | 6.68 d (3.0) | 6.60 d (3.0) | 6.29 d (3.0) |
| 9′ | 2.61–2.63 m a | 2.59–2.60 m | 2.59–2.63 m | 2.61–2.63 m a |
| 10′ | 2.61–2.63 m a | 2.53–2.56 m | 2.54–2.58 m | 2.61–2.63 m a |
| 4-OCH3 | 4.12 s | 4.16 s | 4.12 s | 4.12 s |
| 7-OCH3 | 3.91 s | 3.94 s | ||
| 4′-OCH3 | 3.81 s | 3.75 s | ||
| 5′-OCH3 | 3.80 s | 3.80 s |
| Carbon | 4 | 5 | 6 | 7 |
|---|---|---|---|---|
| 1 | 130.9 | 130.0 | 130.8 | 131.0 |
| 2 | 148.6 | 148.0 | 148.0 | 148.0 |
| 3 | 101.3 | 101.1 | 101.1 | 101.2 |
| 4 | 157.6 | 157.4 | 157.7 | 157.7 |
| 5 | 130.3 | 130.2 | 130.6 | 130.6 |
| 6 | 117.5 | 117.7 | 117.9 | 117.8 |
| 7 | 158.3 | 158.1 | 155.7 | 155.9 |
| 8 | 109.6 | 109.6 | 112.6 | 112.5 |
| 9 | 129.1 | 129.3 | 129.2 | 129.1 |
| 10 | 121.5 | 121.3 | 121.4 | 121.5 |
| 4a | 116.4 | 116.1 | 116.8 | 116.8 |
| 4b | 126.2 | 125.8 | 125.5 | 125.5 |
| 8a | 134.4 | 134.1 | 134.4 | 134.8 |
| 10a | 129.4 | 129.0 | 129.3 | 129.3 |
| 1′ | 114.7 | 107.3 | 107.9 | 114.9 |
| 2′ | 158.3 | 159.1 | 159.6 | 158.5 |
| 3′ | 113.5 | 99.6 | 99.6 | 113.6 |
| 4′ | 129.9 | 158.8 | 159.3 | 130.1 |
| 5′ | 159.3 | 130.2 | 130.4 | 159.3 |
| 6′ | 99.3 | 113.6 | 113.8 | 99.4 |
| 7′ | 158.0 | 156.5 | 156.6 | 158.0 |
| 8′ | 108.4 | 115.1 | 115.2 | 108.5 |
| 9′ | 31.6 a | 31.4 b | 30.8 c | 30.8 d |
| 10′ | 31.2 a | 30.6 b | 31.2 c | 31.4 d |
| 4a′ | 128.2 | 118.5 | 118.9 | 128.3 |
| 4b′ | 116.3 | 125.3 | 125.9 | 116.6 |
| 8a′ | 142.0 | 140.3 | 140.9 | 142.2 |
| 10a′ | 140.5 | 141.3 | 142.0 | 140.7 |
| 4-OCH3 | 56.3 | 56.4 | 56.4 | 56.4 |
| 7-OCH3 | 55.8 | 55.7 | ||
| 4′-OCH3 | 56.0 | 56.1 | ||
| 5′-OCH3 | 56.0 | 56.0 |
| Compounds | Cell Lines | ||
|---|---|---|---|
| HCT-116 | Hela | MDA-MB-231 | |
| 1 | 92.24 ± 8.41 | 38.08 ± 5.16 | >100 |
| 2 | 63.62 ± 3.40 | 44.15 ± 4.31 | 65.55 ± 4.07 |
| 3 | 26.53 ± 6.14 | 19.94 ± 2.07 | 15.80 ± 3.31 |
| 4 | 33.03 ± 4.50 | 24.71 ± 3.21 | 56.14 ± 4.33 |
| 5 | >100 | 39.17 ± 6.59 | >100 |
| 6 | 41.15 ± 6.31 | >100 | >100 |
| 7 | 86.49 ± 9.59 | >100 | >100 |
| 8 | 32.33 ± 4.77 | 64.81 ± 5.72 | 45.46 ± 2.91 |
| 9 | 15.01 ± 1.90 | >100 | 11.09 ± 2.89 |
| 10 | 14.05 ± 2.25 | 17.43 ± 3.07 | 13.86 ± 3.33 |
| 11 | 14.39 ± 0.26 | 13.23 ± 1.95 | 12.13 ± 0.38 |
| paclitaxel | 1.05 ± 0.23 | 0.09 ± 0.01 | 0.016 ± 0.003 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Li, J.; Zeng, K.-W.; Jiang, Y.; Tu, P.-F. Five New Biphenanthrenes from Cremastra appendiculata. Molecules 2016, 21, 1089. https://doi.org/10.3390/molecules21081089
Liu L, Li J, Zeng K-W, Jiang Y, Tu P-F. Five New Biphenanthrenes from Cremastra appendiculata. Molecules. 2016; 21(8):1089. https://doi.org/10.3390/molecules21081089
Chicago/Turabian StyleLiu, Liang, Jun Li, Ke-Wu Zeng, Yong Jiang, and Peng-Fei Tu. 2016. "Five New Biphenanthrenes from Cremastra appendiculata" Molecules 21, no. 8: 1089. https://doi.org/10.3390/molecules21081089
APA StyleLiu, L., Li, J., Zeng, K.-W., Jiang, Y., & Tu, P.-F. (2016). Five New Biphenanthrenes from Cremastra appendiculata. Molecules, 21(8), 1089. https://doi.org/10.3390/molecules21081089