
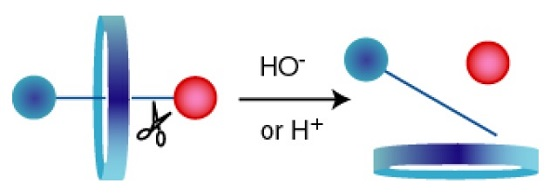
A Versatile Axle for the Construction of Disassemblage Rotaxanes

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
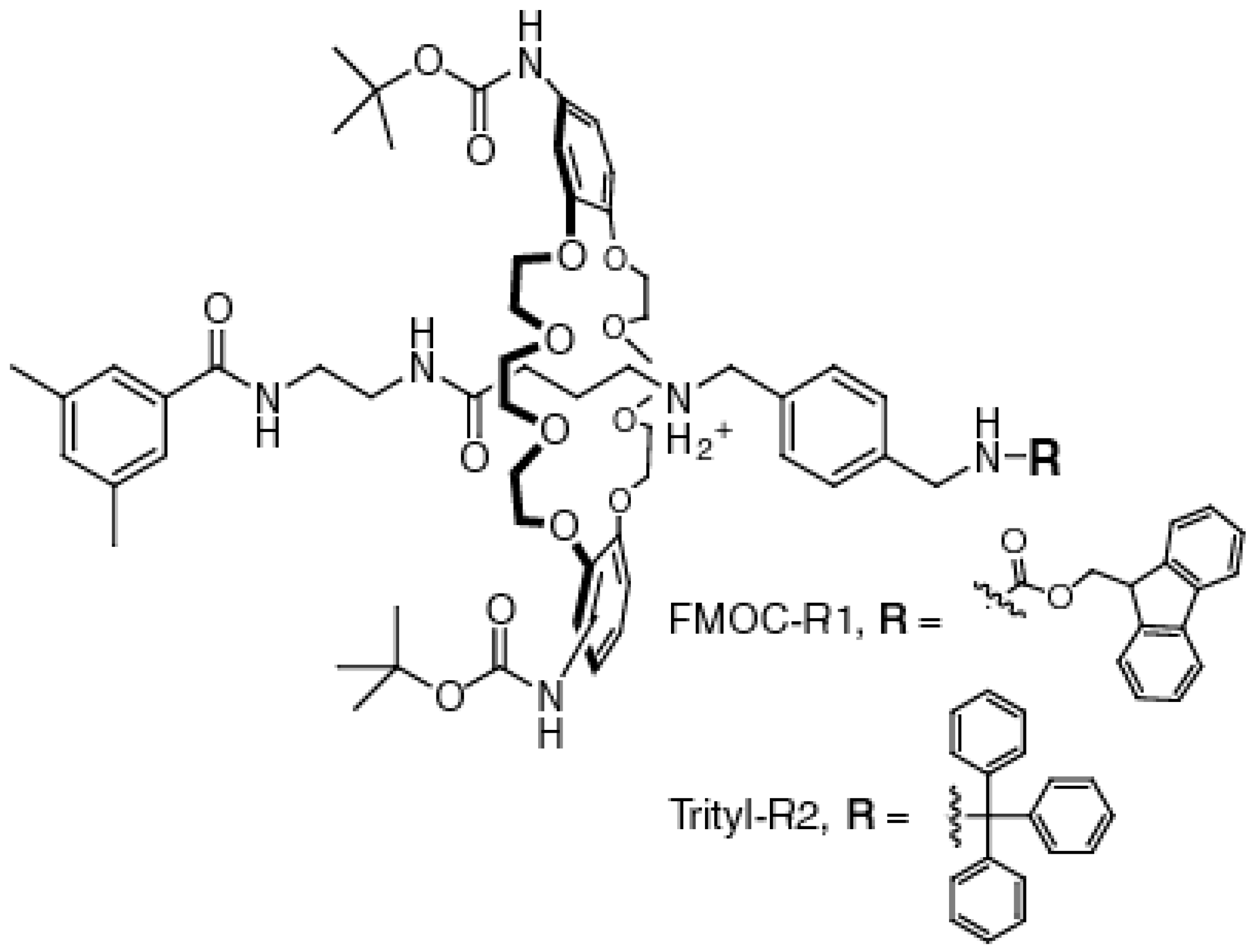
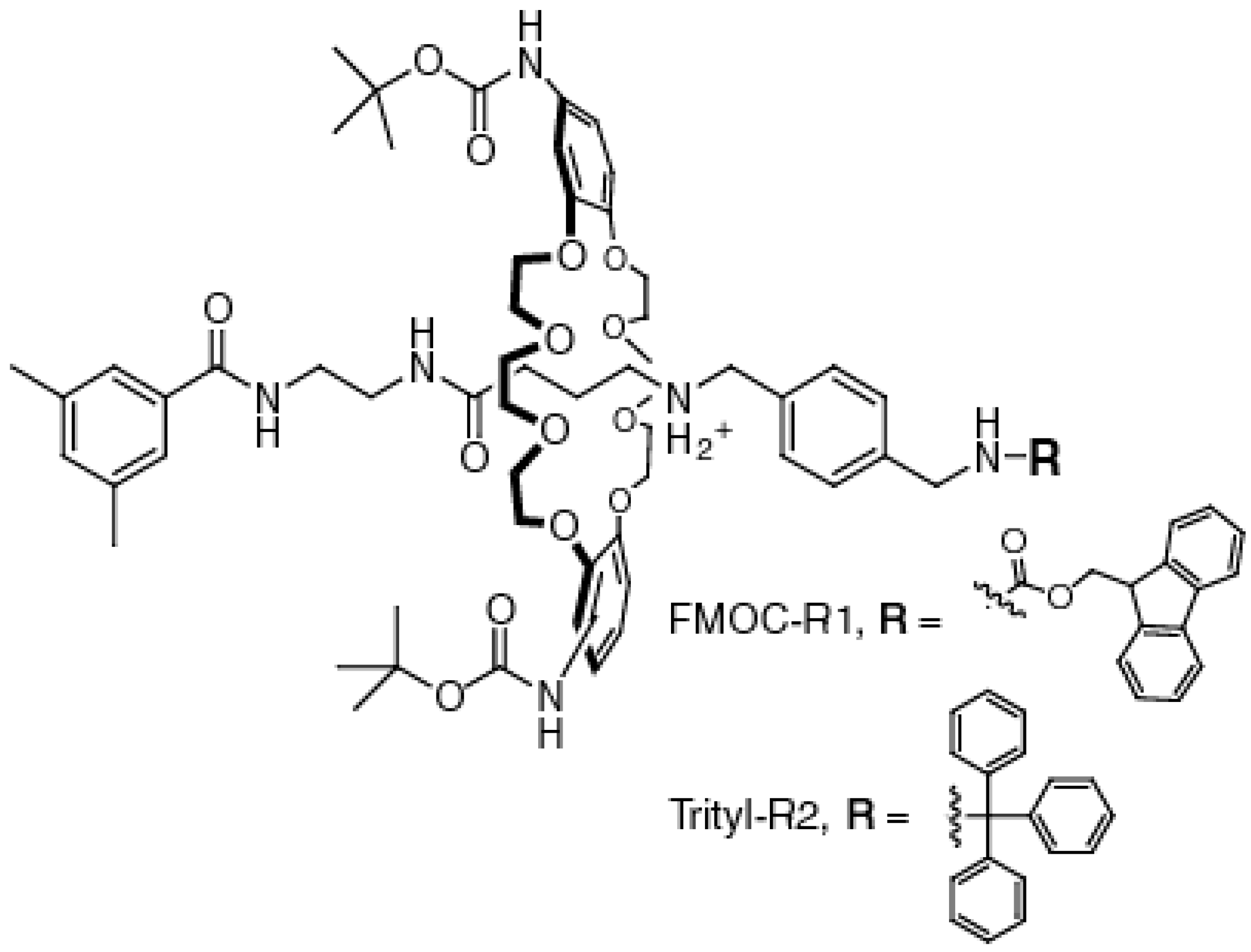
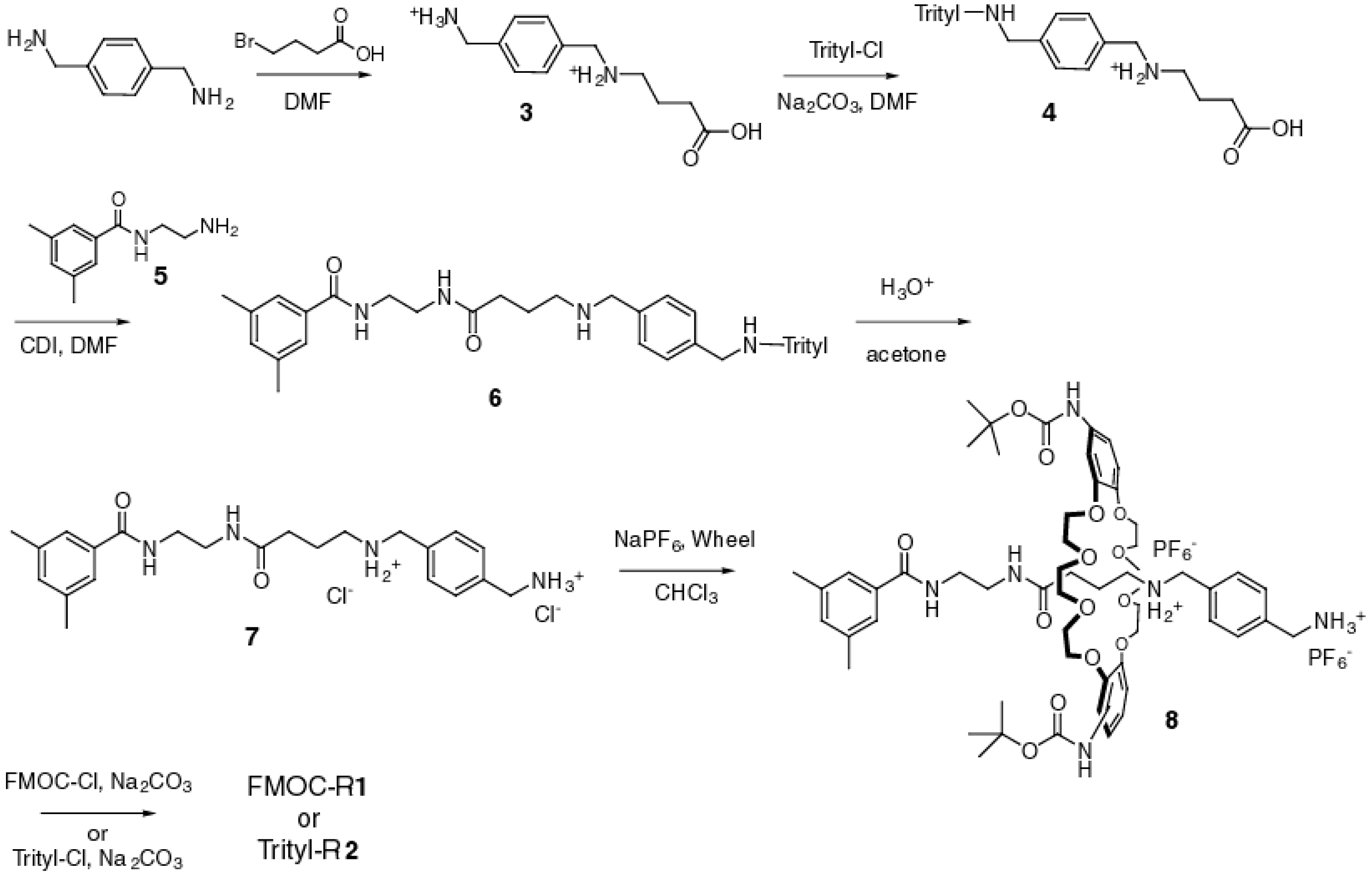
2.1. Synthesis of the [2]Rotaxanes
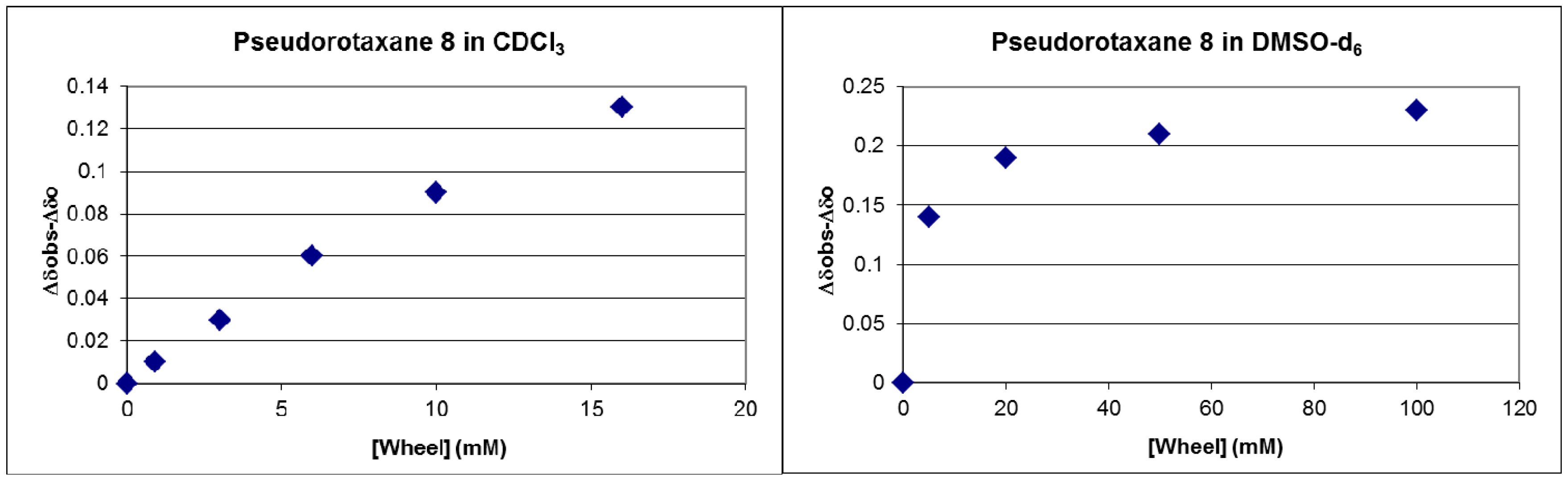
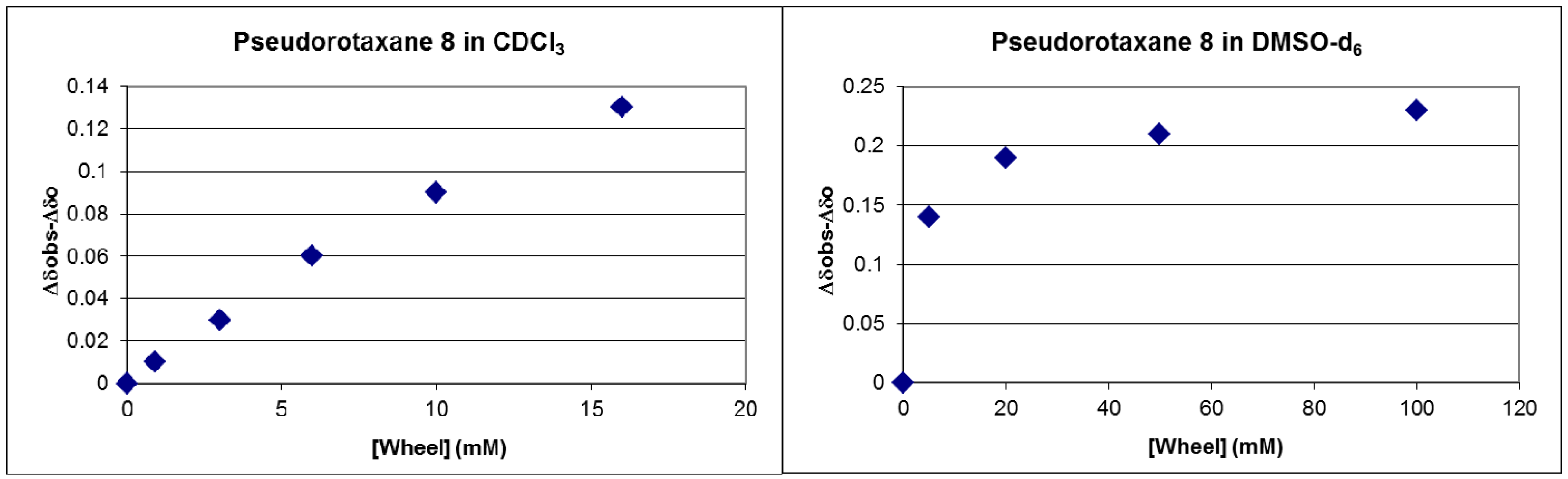
2.2. Measuring the Stability of Pseudorotaxane 8
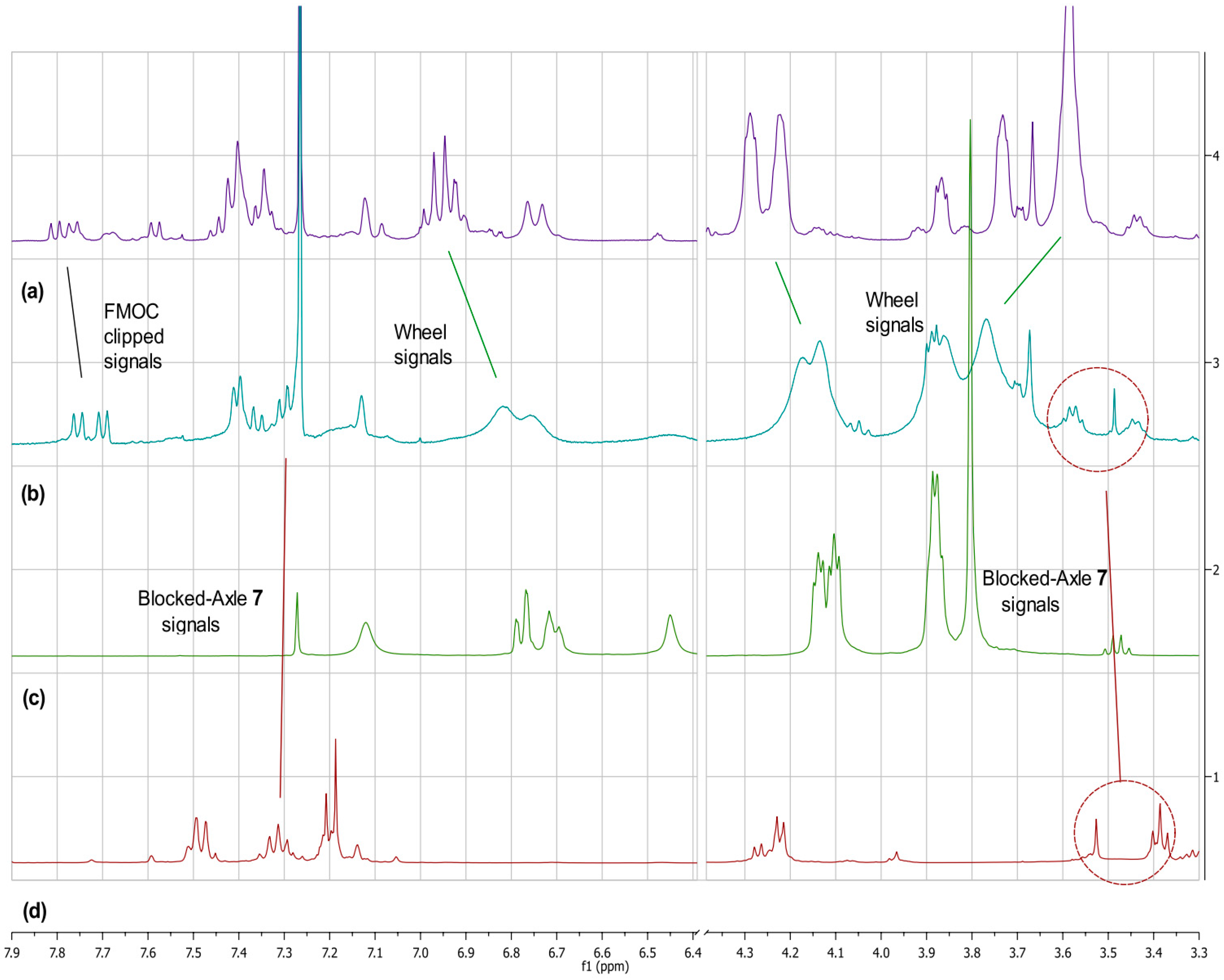
2.3. Investigating the Rate of Disassemblage
3. Materials and Methods
3.1. General Synthetic Protocol
3.2. Synthetic Procedures
3.3. Measuring the Association Constants
3.4. Observing Disassemblage
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yin, J.; Hu, Y.; Yoon, J. Fluorescent probes and bioimaging: Alkali metals, alkaline earth metals and pH. Chem. Soc. Rev. 2015, 44, 1460–4744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Hoshino, K. Molecular Sensors and Nanodevices: Principles, Designs and Applications in Biomedical Engineering; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Yang, Z.W.; Liu, X.R.; Zhao, S.S.; He, J.M. Chemically driven [2]rotaxane molecular shuttles. Prog. Chem. 2014, 26, 1899–1913. [Google Scholar]
- Xue, M.; Yang, Y.; Chi, X.D.; Yan, X.Z.; Huang, F.H. Development of pseudorotaxanes and rotaxanes: From synthesis to stimuli-responsive motions to applications. Chem. Rev. 2015, 115, 7398–7501. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tian, H. Bright functional rotaxanes. Chem. Soc. Rev. 2010, 39, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, S.; Zhang, J.; Zhu, K.; Li, N.; Huang, F. Benzo-21-crown-7/secondary dialkylammonium salt [2]pseudorotaxane- and [2]rotaxane-type threaded structures. Org. Lett. 2007, 9, 5553–5556. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Gibson, H.W. Polypseudorotaxanes and polyrotaxanes. Prog. Polym. Sci. 2005, 30, 982–1018. [Google Scholar] [CrossRef]
- Arumugaperumal, R.; Srinivasadesikan, V.; Raju, M.V.R.; Lin, M.C.; Shukla, T.; Singh, R.; Lin, H.C. Acid/Base and H2PO4− controllable high-contrast optical molecular switches with a novel BODIPY functionalized [2]rotaxane. ACS Appl. Mater. Interfaces 2015, 7, 26491–26503. [Google Scholar] [CrossRef] [PubMed]
- Langton, M.J.; Beer, P.D. Rotaxane and catenane host structures for sensing charged guest species. Acc. Chem. Res. 2014, 47, 1935–1949. [Google Scholar] [CrossRef] [PubMed]
- Girek, T. Cyclodextrin-based rotaxanes. J. Incl. Phenom. Macro. 2012, 74, 1–21. [Google Scholar] [CrossRef]
- Yang, W.L.; Li, Y.J.; Liu, H.B.A.; Chi, L.F.; Li, Y.L. Design and assembly of rotaxane-based molecular switches and machines. Small 2012, 8, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, Y. Switchable rotaxane system in response to external stimulus. J. Syn. Org. Chem. Jpn. 2011, 69, 62–72. [Google Scholar] [CrossRef]
- Willner, I.; Basnar, B.; Willner, B. From molecular machines to microscale motility of objects: Application as “smart materials”, sensors, and nanodevices. Adv. Funct. Mater. 2007, 17, 702–717. [Google Scholar] [CrossRef]
- Martinez-Cuezva, A.; Rodrigues, L.V.; Navarro, C.; Carro-Guillen, F.; Buriol, L.; Frizzo, C.P.; Martins, M.A.P.; Alajarin, M.; Berna, J. Dethreading of tetraalkylsuccinamide-based [2]rotaxanes for preparing benzylic amide macrocycles. J. Org. Chem. 2015, 80, 10049–10059. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C.F.; Lau, K.N.; Wong, W.Y. Revisiting the formation and tunable dissociation of a [2]pseudorotaxane formed by slippage approach. Int. J. Mol. Sci. 2016, 16, 8254–8265. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Ruiz, A.; Tiburcio, J. Electrostatic kinetic barriers in the threading/dethreading motion of a rotaxane-like complex. Org. Lett. 2015, 17, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Yan, X.; Huang, F. Reversible formation of a poly [3]rotaxane based on photo dimerization of an anthracene-capped [3]rotaxane. Chem. Comm. 2014, 50, 14105–14108. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, D.; Jean–François Morin, J.F. Recent advances in the synthesis of ammonium-based rotaxanes. Molecules 2010, 15, 3709–3730. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular-permeability in a human tumor xenograft-molecular-size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism Of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Moresco, R.M.; Matarrese, M.; Fazio, F. PET and SPET molecular imaging: focus on serotonin system. Curr. Top. Med. Chem. 2006, 6, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Crespi, F.; Croce, A.C.; Fiorani, S.; Masala, B.; Heidbreder, C.; Bottiroli, G. In vivo autofluorescence spectrofluorometry of central serotonin. J. Neurosci. Methods 2004, 140, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ashton, P.R.; Chrystal, E.J.T.; Glink, P.T.; Menzer, S.; Schiavo, C.; Spencer, N.; Stoddart, J.F.; Tasker, P.A.; White, A.J.P.; Williams, D.J. Pseudorotaxanes formed between secondary dialkylammonium salts and crown ethers. Chem. Eur. J. 1996, 2, 709–728. [Google Scholar] [CrossRef]
- Gasa, T.B.; Valente, C.; Stoddart, J.F. Solution-phase counterion effects in supramolecular and mechanostereochemical systems. Chem. Soc. Rev. 2011, 40, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Gibson, H.W. Ion pairing and host-guest complexation in low dielectric constant solvents. J. Am. Chem. Soc. 2003, 125, 7001–7004. [Google Scholar] [CrossRef] [PubMed]
- Späth, A.; König, B. Molecular recognition of organic ammonium ions in solution using synthetic receptors. Beilstein J. Org. Chem. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Smithrud, D.B. Facile synthesis of rotaxanes through condensation reactions of DCC-rotaxanes. Org. Lett. 2001, 16, 2485–2486. [Google Scholar]
- Ashton, P.R.; Bartsch, R.A.; Cantrill, S.J.; Hanes, R.E.; Hickingbottom, S.K.; Lowe, J.N.; Preece, J.A.; Stoddart, J.F.; Talanov, V.S.; Wang, Z.H. Secondary dibenzylammonium ion binding by [24]crown-8 and [25]crown-8 macrocycles. Tetrahedron Lett. 1999, 40, 3661–3664. [Google Scholar] [CrossRef]
- Jiang, W.; Schafer, A.; Mohr, P.C.; Schalley, C.A. Monitoring self-sorting by electrospray ionization mass spectrometry: Formation intermediates and error-correction during the self-assembly of multiply threaded pseudorotaxanes. J. Am. Chem. Soc. 2010, 132, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Conners, K.A. Binding Constants, the Measurement of Molecular Complex Stability; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Sample Availability: Disassemblage of FMOC-R1 in DMSO-d6 are available from authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powers, L.A.; Smithrud, D.B. A Versatile Axle for the Construction of Disassemblage Rotaxanes. Molecules 2016, 21, 1043. https://doi.org/10.3390/molecules21081043
Powers LA, Smithrud DB. A Versatile Axle for the Construction of Disassemblage Rotaxanes. Molecules. 2016; 21(8):1043. https://doi.org/10.3390/molecules21081043
Chicago/Turabian StylePowers, Lucas A., and David B. Smithrud. 2016. "A Versatile Axle for the Construction of Disassemblage Rotaxanes" Molecules 21, no. 8: 1043. https://doi.org/10.3390/molecules21081043
APA StylePowers, L. A., & Smithrud, D. B. (2016). A Versatile Axle for the Construction of Disassemblage Rotaxanes. Molecules, 21(8), 1043. https://doi.org/10.3390/molecules21081043