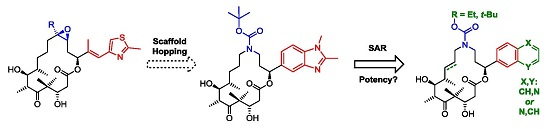
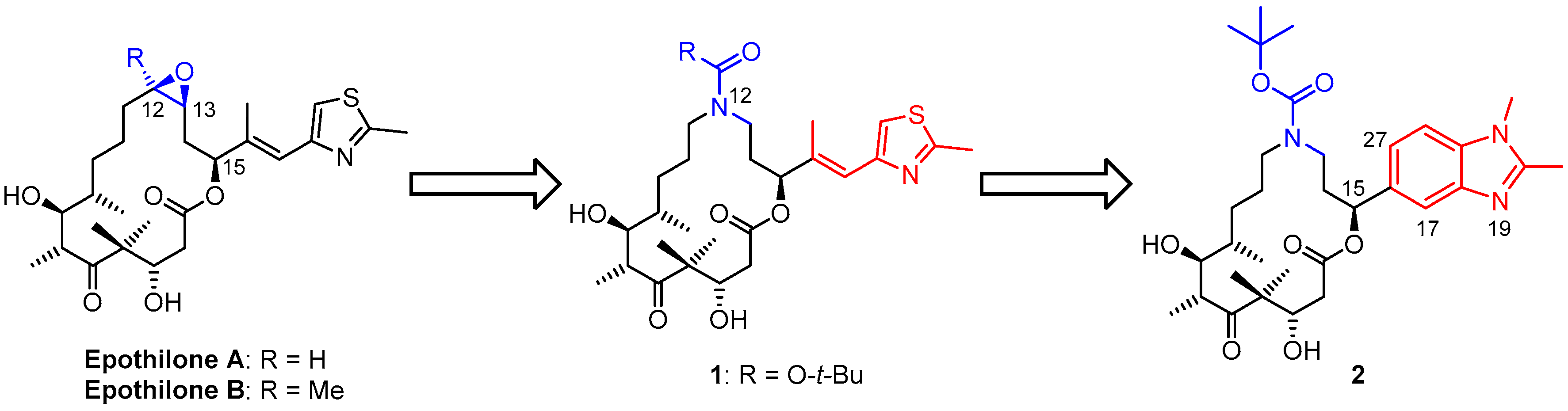
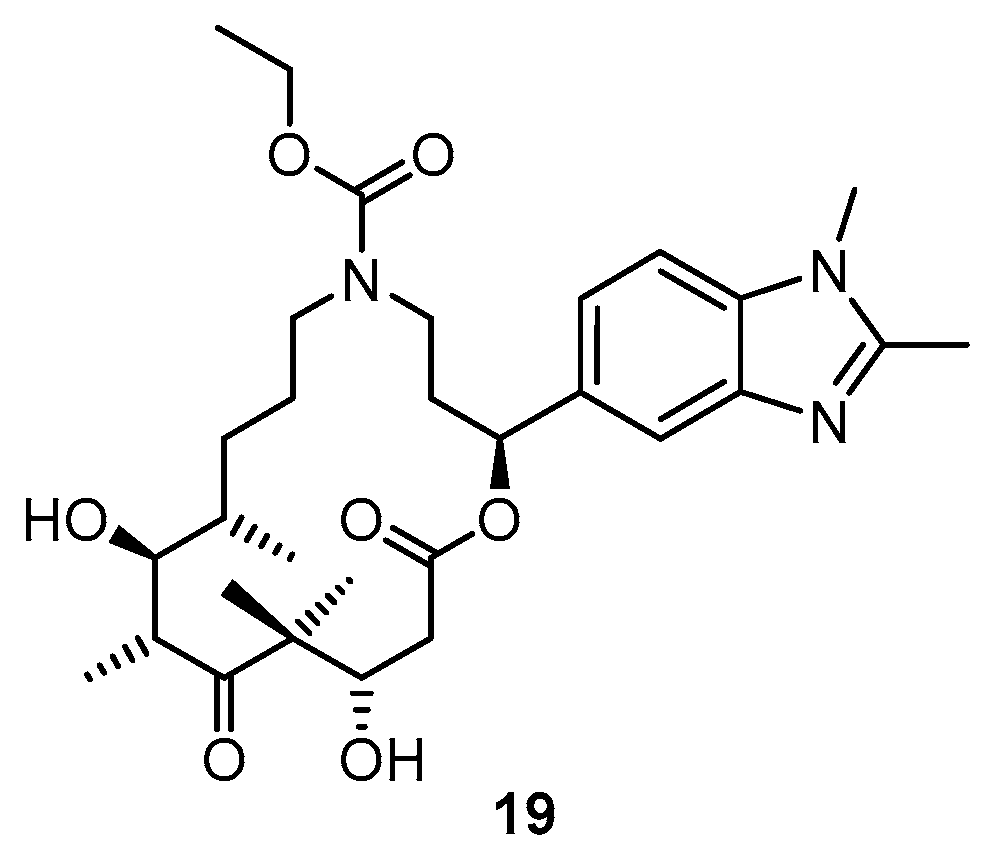
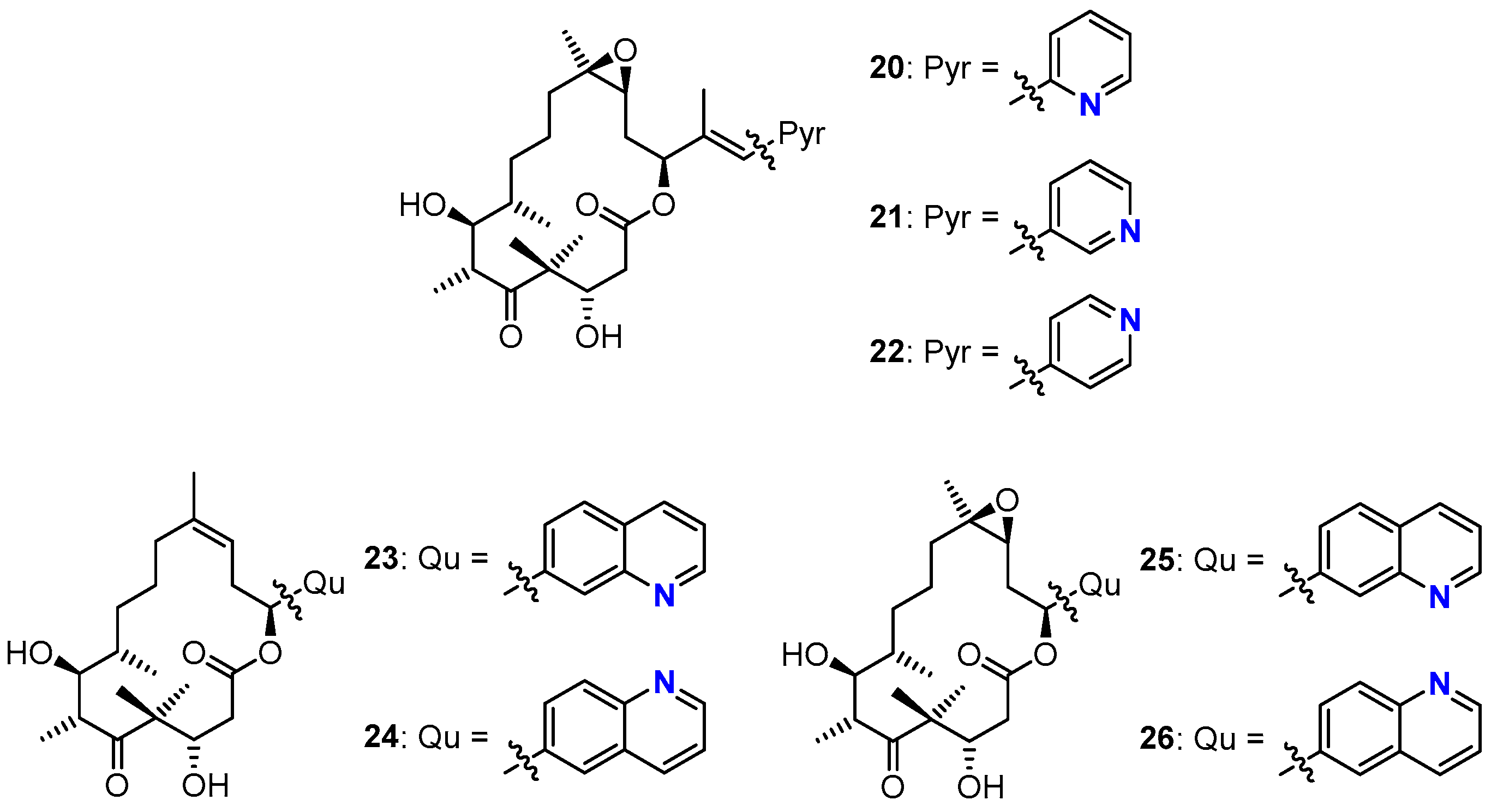
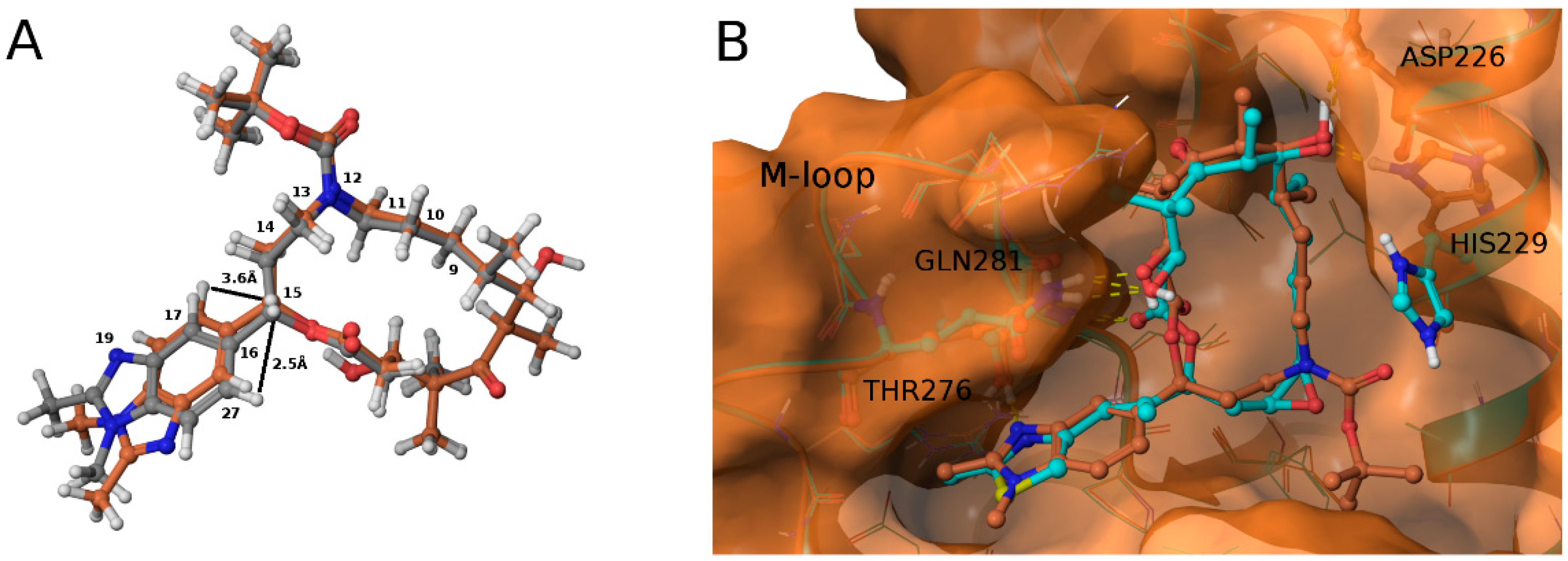
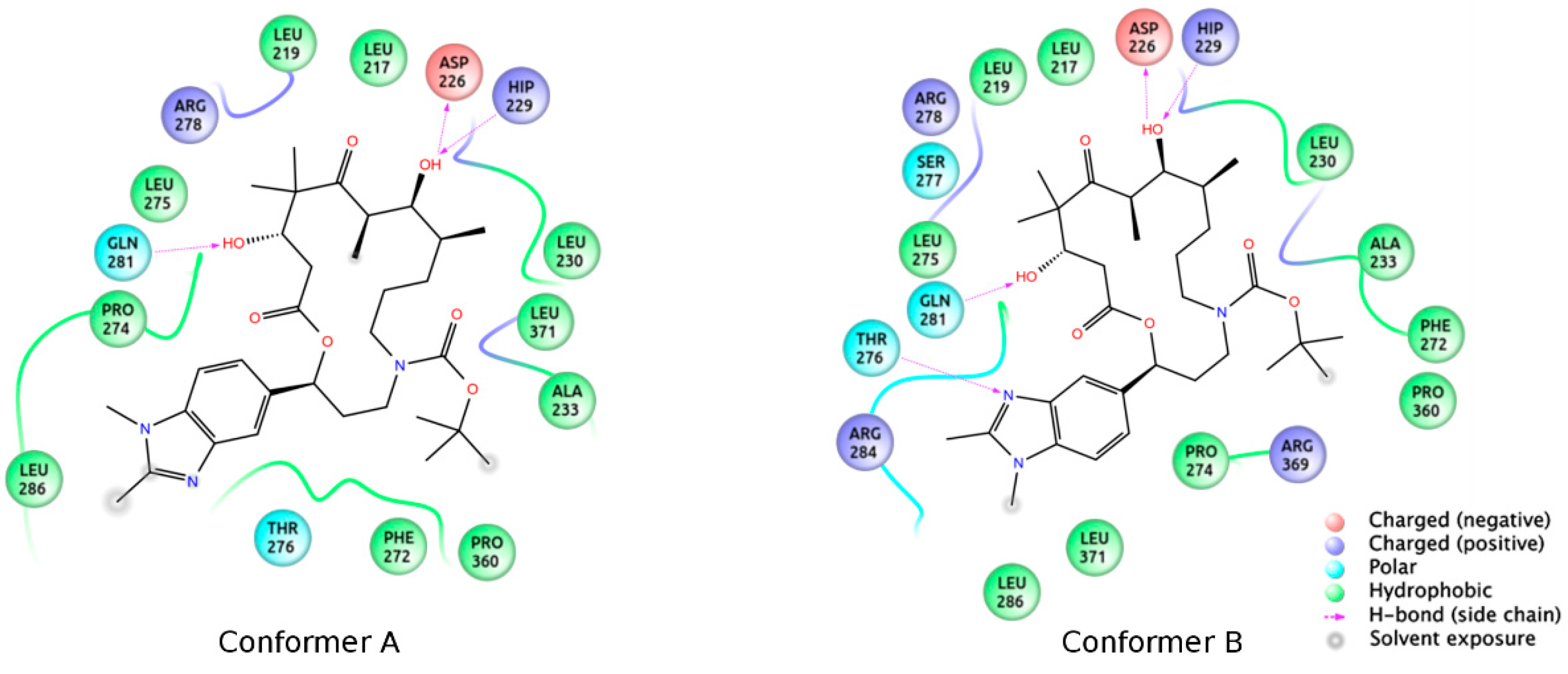
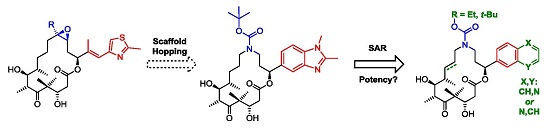
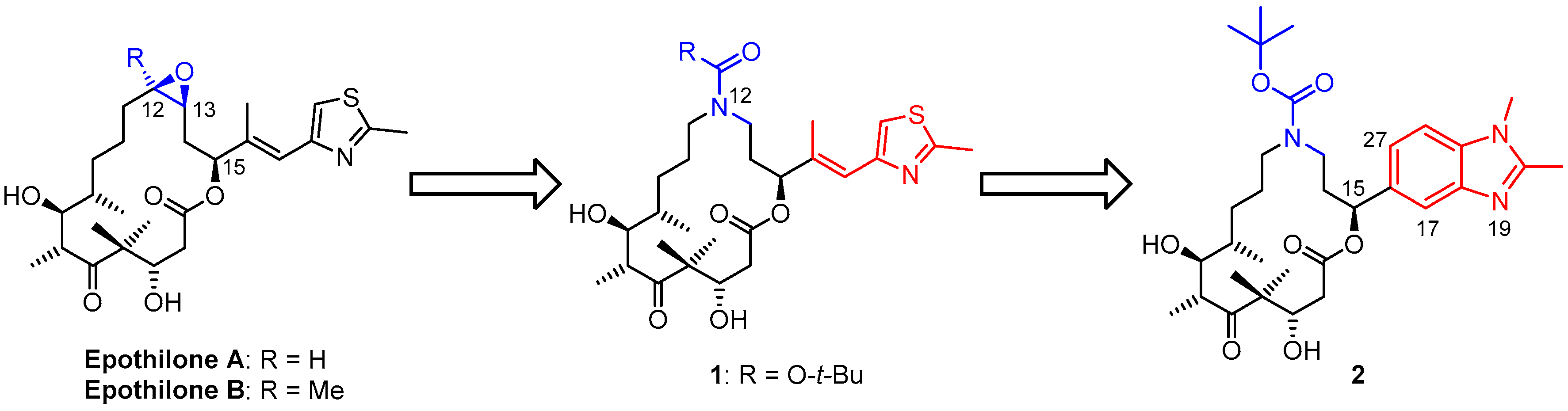
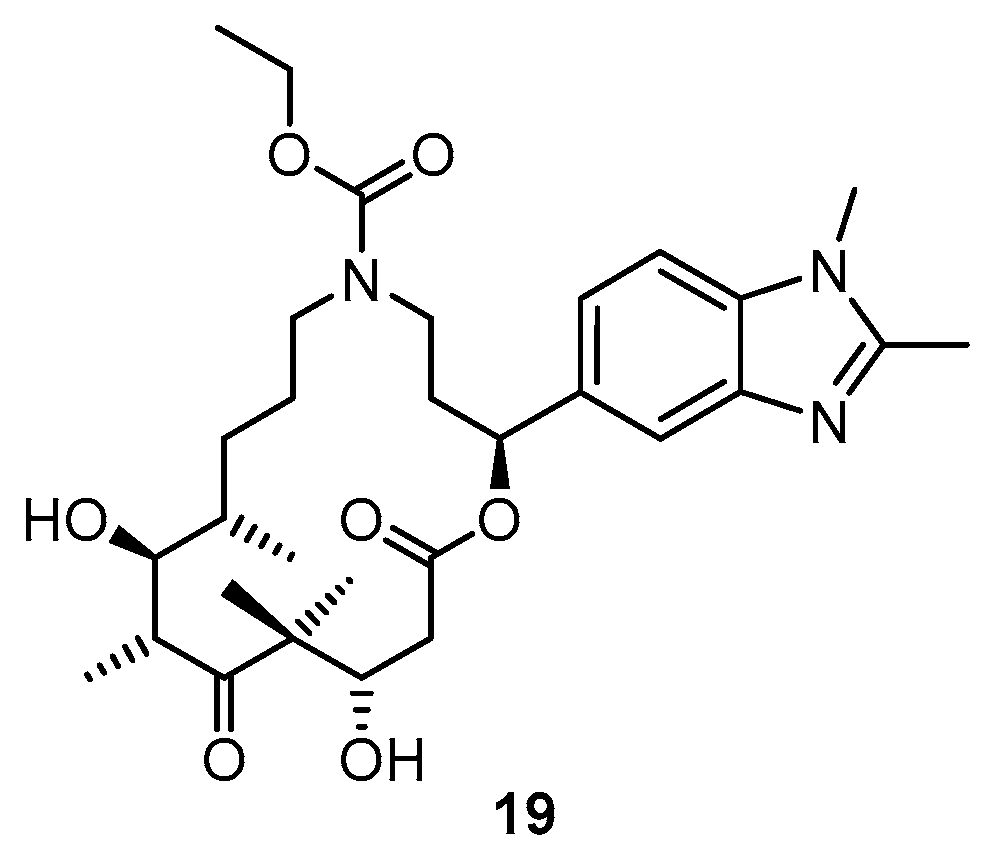
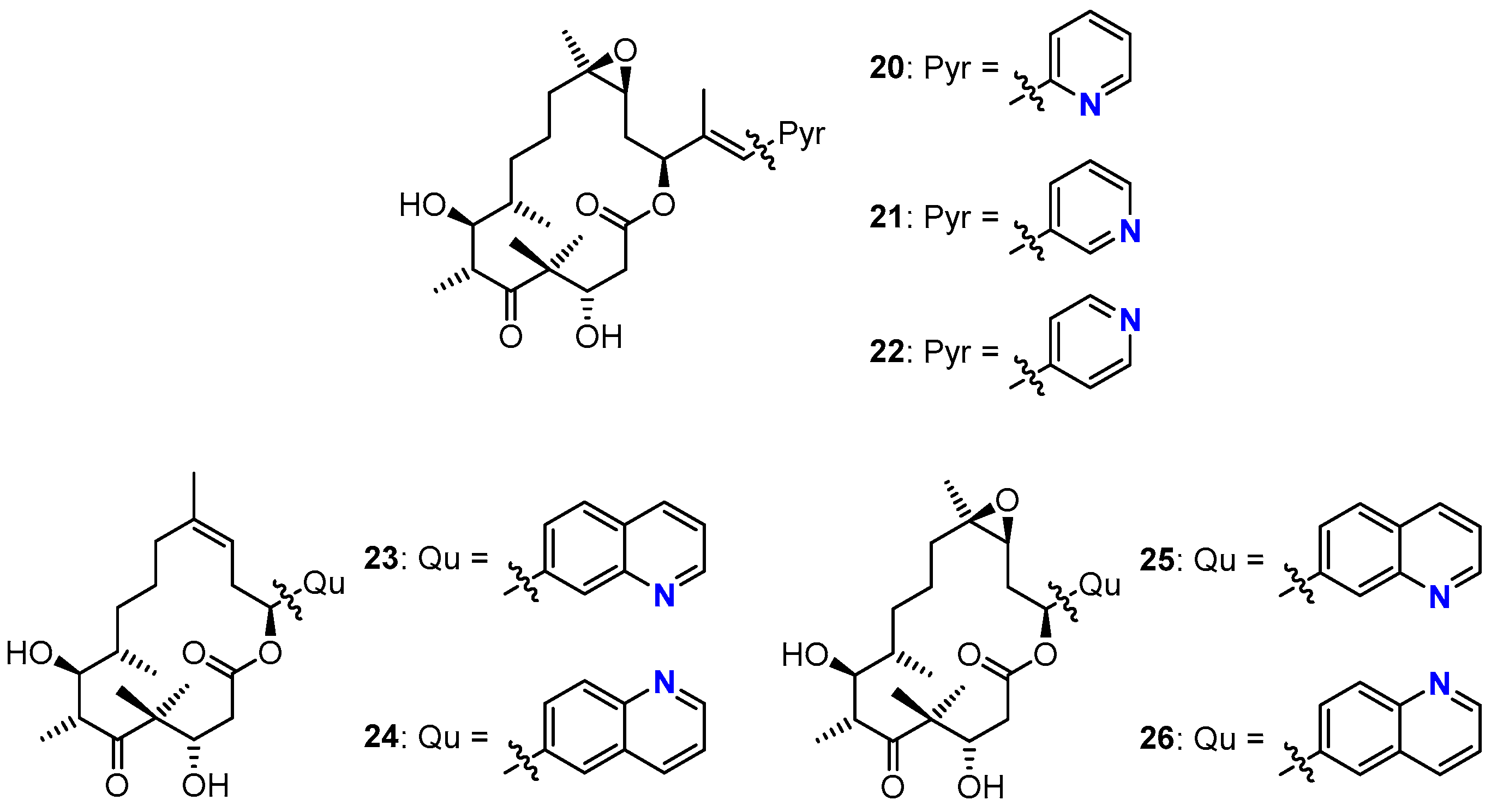
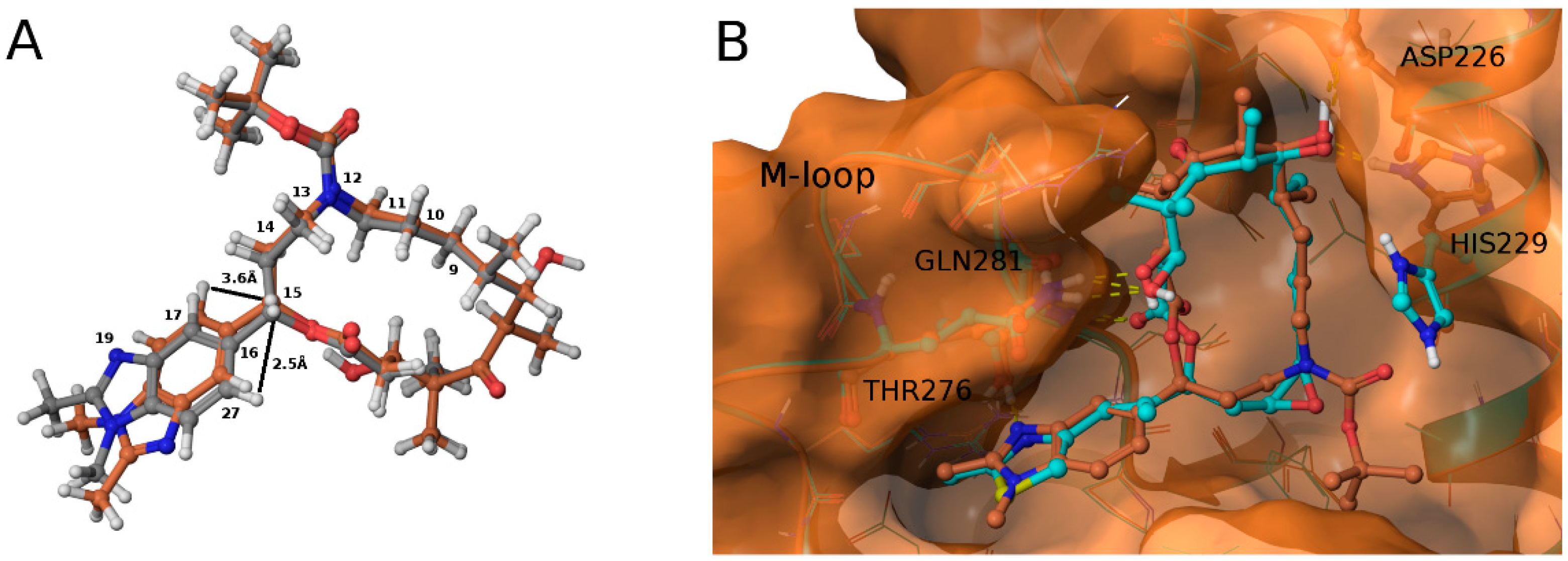
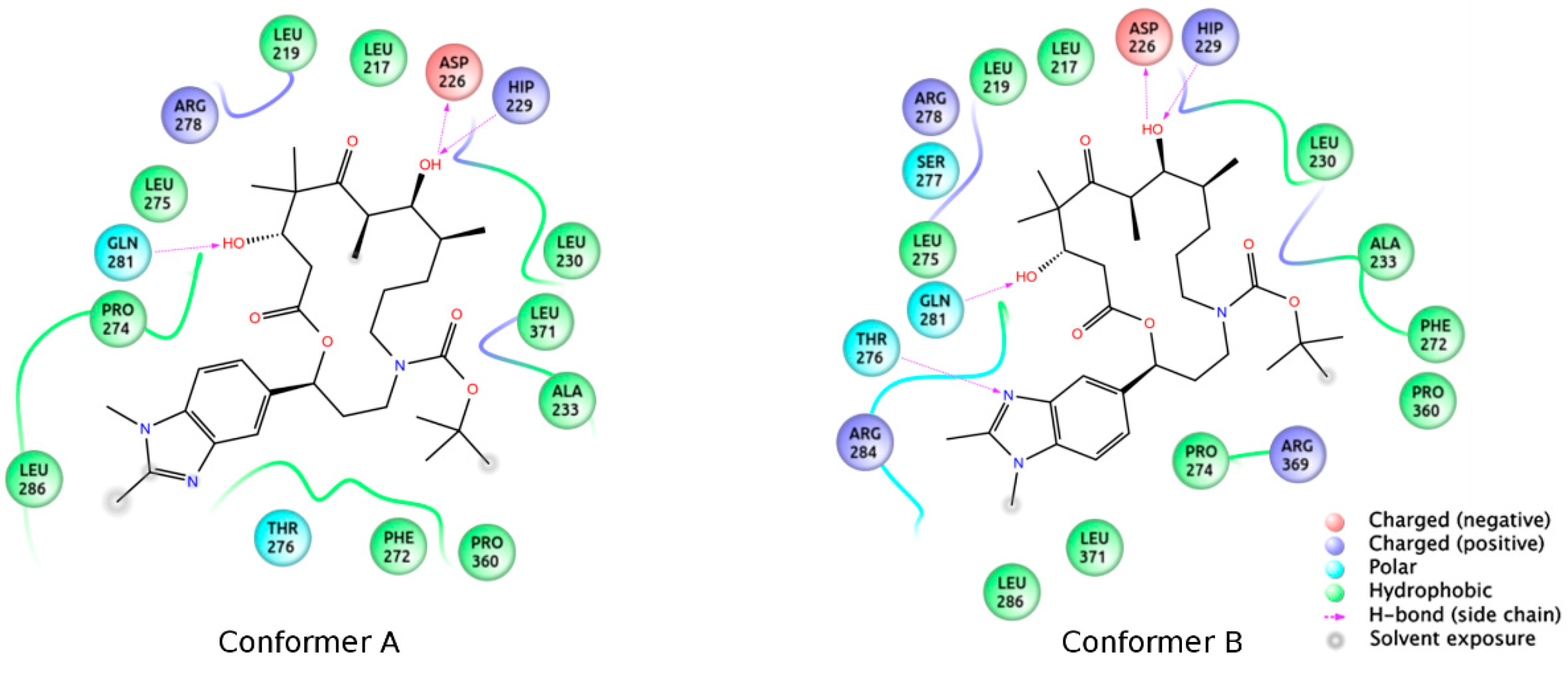
3.2. Chemistry
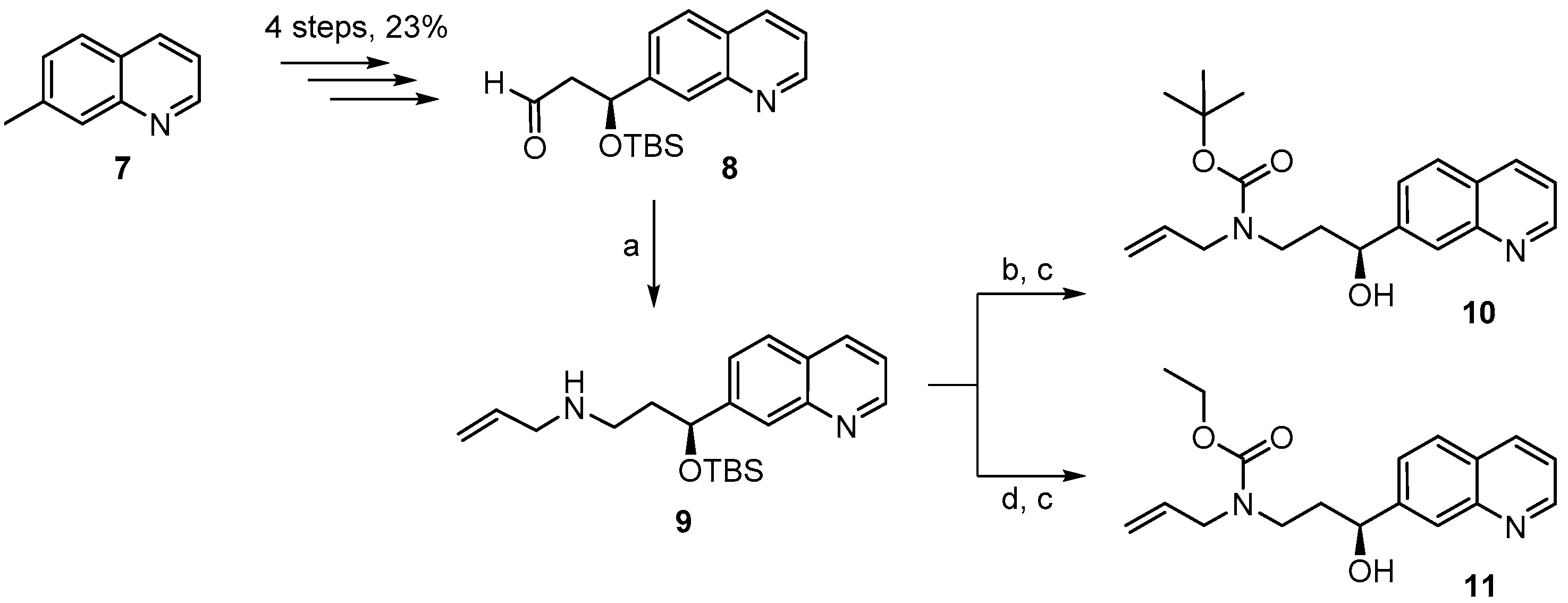
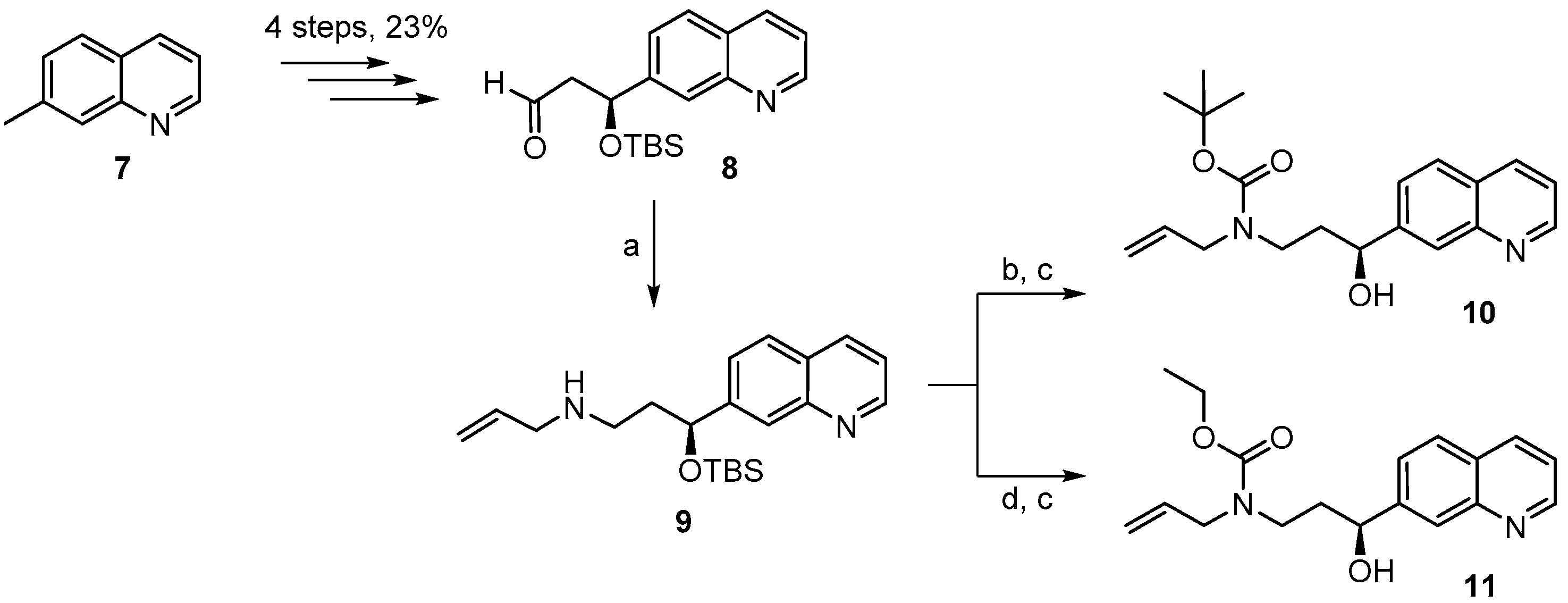
(S)-N-(3-(t-Butyldimethylsilyloxy)-3-(quinolin-7-yl)propyl) prop-2-en-1-amine (
9). To heat-activated molecular sieves (4 Å, 650 mg) was added a solution of aldehyde
8 [
25] (522 mg, 1.65 mmol) in 8 mL THF. To this solution were added 0.6 mL of allylamine (7.86 mmol) and the mixture was heated to 50 °C for 24 h. It was then filtered through a pad of dry Celite™, the residue was washed with THF and the combined filtrates were concentrated under reduced pressure to give a yellow oil. This material showed the expected mass for the imine and was directly used as such. For the reduction, 67 mg of NaBH
4 (1.7 mmol) were placed in a 10 mL two-necked flask at 0 °C and a solution of the crude imine in 3 mL MeOH was added (gas formation could be observed). After 20 min the reaction mixture was diluted with water and extracted with EtOAc. The combined organic fractions were washed with brine, dried over MgSO
4 and concentrated in vacuo. Purification of the residue by FC (hexane/EtOAc 4:1 → 1:1 + 1% Et
3N) furnished 445 mg (76%) of the desired amine
9 as a slightly yellow oil;
= −54.0° (
c = 1.37, CH
2Cl
2).
1H-NMR (400 MHz, CDCl
3): δ = 8.87 (dd,
J = 4.3 Hz, 1.7 Hz, 1H), 8.09 (dd,
J = 8.3 Hz, 1.2 Hz, 1H), 7.94 (br s, 1H), 7.75 (d,
J = 8.4 Hz, 1H), 7.54 (dd,
J = 8.4 Hz, 1.7 Hz, 1H), 7.33 (dd,
J = 8.2 Hz, 4.2 Hz, 1H), 5.85 (m, 1H), 5.10 (dq,
J = 17.2 Hz, 1.6 Hz, 1H), 5.02 (dq,
J = 10.3 Hz, 1.4 Hz, 1H), 4.97 (dd,
J = 7.3 Hz, 4.8 Hz, 1H), 3.17 (dt,
J = 6.1 Hz, 1.3 Hz, 2H), 2.66 (t,
J = 7.0 Hz, 2H), 2.01–1.84 (br m, 2H), 0.87 (s, 9H), 0.03 (s, 3H), −0.17 (s, 3H); N
H not visible.
13C-NMR (100 MHz, CDCl
3): δ = 150.5, 148.2, 147.0, 136.9, 135.8, 127.7, 127.4, 125.7, 124.9, 120.7, 115.7, 73.5, 52.5, 45.8, 40.6, 25.8 (3 × CH
3), 18.1, −4.6, −5.0. HRMS (ESIpos): calcd. for C
21H
33N
2OSi [M + H]
+: 357.2357, found: 357.2361.
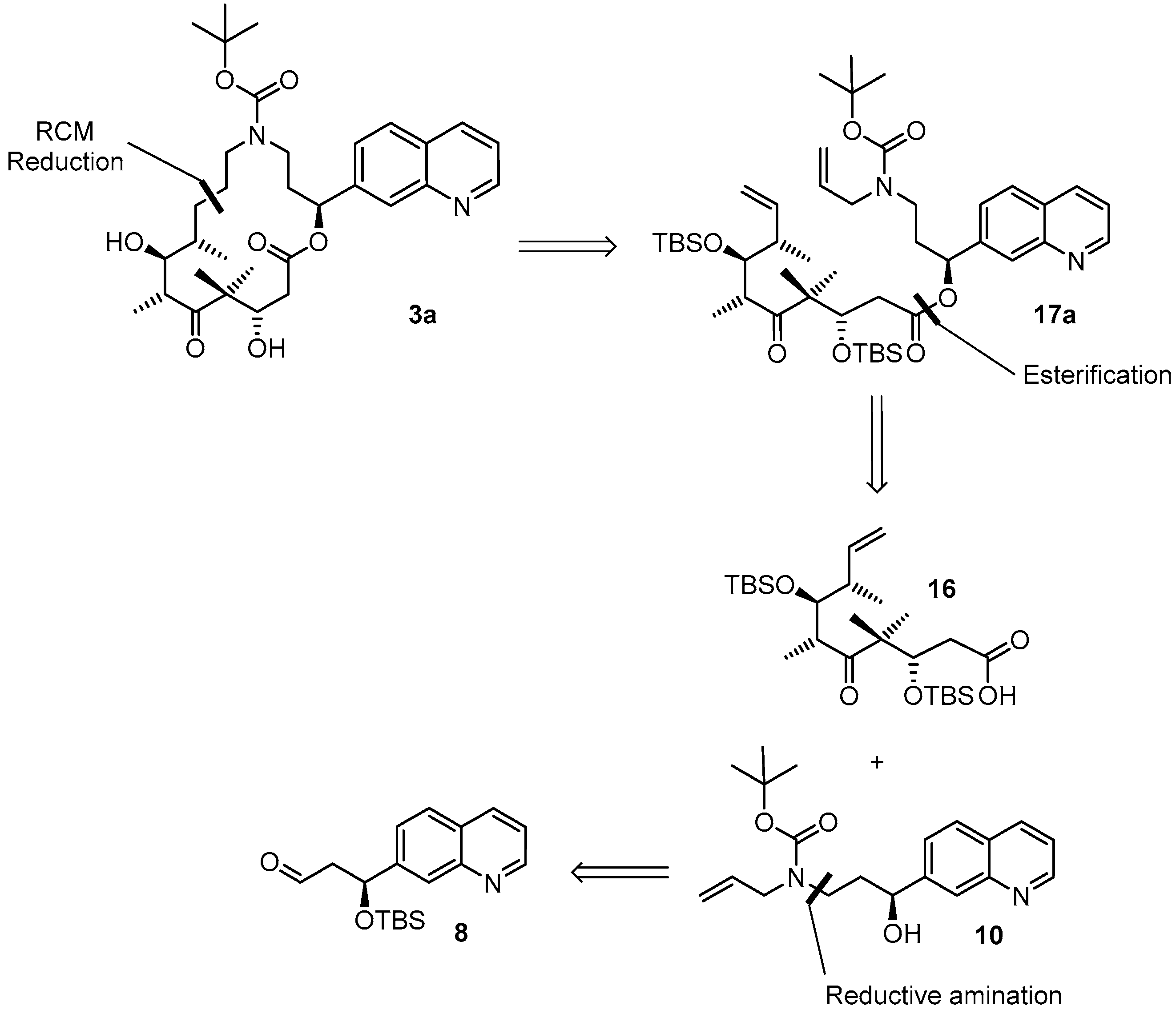
(S)-t-Butyl allyl(3-(t-butyldimethylsilyloxy)-3-(quinolin-7-yl)propyl)carbamate (9A). A solution of amine 9 (416 mg, 1.17 mmol) and Boc2O (390 mg, 1.75 mmol) was stirred at rt for 14 h. Ethanolamine (1 mL) was then added and the mixture was stirred for one additional hour at rt and then concentrated in vacuo. To the resulting crude product water was added and the mixture was extracted with Et2O and CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification of the residue by FC (hexane/EtOAc 10:1 → 1:1 + 1% Et3N) gave 460 mg (86%) of the desired carbamate 9A as colorless oil; Rf = 0.8 (hexane/EtOAc 1:1). = −36.2° (c = 1.50, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.84 (dd, J = 4.1 Hz, 1.5 Hz, 1H), 8.06 (d, J = 7.9 Hz, 1H), 7.94 (s, 1H), 7.72 (d, J = 8.4 Hz, 1H), 7.50 (dd, J = 8.5 Hz, 1.2 Hz, 1H), 7.29 (dd, J = 8.1 Hz, 4.2 Hz, 1H), 5.67 (m, 1H), 5.01 (m, 1H), 4.97 (dq, J = 7.7 Hz, 1.4 Hz, 1H), 4.84 (dd, J = 6.6 Hz, 4.7 Hz, 1H), 3.72 (m, 2H), 3.21 (m, 2H), 1.95 (m, 2H), 1.33 (s, 9H), 0.85 (s, 9H), 0.01 (s, 3H), −0.20 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 155.2, 150.4, 148.1, 146.5, 135.7, 134.1, 127.7, 127.4, 125.6, 124.7, 120.7, 116.3, 79.2, 73.0, 49.6, 43.6, 38.7, 28.3 (3 × CH3), 25.7 (3 × CH3), 18.0, −4.7, −5.1. HRMS (ESIpos): calcd. for C26H40N2O3SiNa [M + Na]+: 479.27004, found: 479.27007.
(S)-t-Butyl allyl(3-hydroxy-3-(quinolin-7-yl)propyl)carbamate (10). To a solution of silyl-ether 9A (200 mg, 0.44 mmol) in 5 mL THF were added 1.32 mL (1.32 mmol) of a TBAF-solution (1 M in THF) at rt and the mixture was stirred for 5 h. The reaction was quenched with saturated, aqueous NH4Cl solution and the mixture was extracted with EtOAc. The combined organic layers were washed with H2O, dried over MgSO4 and concentrated in vacuo. The residue was purified by FC (hexane/EtOAc 2:1 → 1:1) to give 152 mg (quantitative yield) of the desired free alcohol 10 as a colorless oil; Rf = 0.1 (hexane/EtOAc 2:1). = −1.94° (c = 1.05, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.85 (dd, J = 4.2 Hz, 1.4 Hz, 1H), 8.09 (dm, J = 8.1 Hz, 1H), 8.01 (br s, 1H), 7.76 (d, J = 8.5 Hz, 1H), 7.62 (m, 1H), 7.33 (dd, J = 8.1 Hz, 4.2 Hz, 1H), 5.78 (m, 1H), 5.14 (br s, 1H), 5.10 (d, J = 4.7 Hz, 1H), 4.82 (m, 1H), 4.72 (m, 1H, OH), 3.88 (dd, J = 15.7 Hz, 5.4 Hz, 2H), 3.72 (m, 1H), 3.12 (m, 1H), 2.02 (m, 1H), 1.81 (m, 1H), 1.44 (s, 9H). 13C-NMR (100 MHz, CDCl3): δ = 171.1, 150.4, 148.2, 146.0, 135.8, 133.9, 127.9, 127.4, 125.6, 124.8, 120.8, 116.7, 80.4, 69.9, 50.1, 43.4, 37.9, 28.3 (3 × CH3). HRMS (ESIpos): calcd. for C20H26N2O3SiNa [M + Na]+: 365.1836, found: 365.1834.
(S)-Ethyl allyl(3-(t-butyldimethylsilyloxy)-3-(quinolin-7-yl) propyl)carbamate (9B). To a solution of amine 9 (150 mg, 0.42 mmol) and K2CO3 (176 mg, 1.26 mmol) in 4 mL acetone was added a solution of ethyl chloroformate (70 mg, 0.63 mmol) in 1 mL acetone at 0 °C. The mixture was stirred at rt and the reaction was quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification of the residue by FC (hexane/EtOAc 2:1) gave 156 mg (87%) of the desired carbamate 9B as a yellow oil; Rf = 0.2 (hexane/EtOAc 4:1); 0.4 (hexane/EtOAc 2:1). = −41.8° (c = 1.79, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.88 (dd, J = 4.3 Hz, 1.6 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 7.95 (br s, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.53 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.35 (dd, J = 8.3 Hz, 4.3 Hz, 1H), 5.69 (m, 1H), 5.03 (m, 2H), 4.88 (dd, J = 7.1 Hz, 5.9 Hz, 1H), 4.06 (q, J = 7.2 Hz, 2H), 3.78 (m, 2H), 3.26 (m, 2H), 1.98 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 3H), −0.17 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 156.2, 150.6, 148.2, 146.4, 135.8, 133.9, 127.8, 127.5, 125.8, 124.8, 120.8, 116.8, 72.9, 61.1, 49.7, 43.6, 38.6, 25.8 (3 × CH3), 18.1, 14.6, −4.6, −5.1. IR (film): ῦ = 2931, 2857, 1698, 1472, 1417, 1384, 1250, 1092, 836, 776, 677. HRMS (ESIpos): calcd. for C24H36N2O3SiNa [M + Na]+: 451.2387, found: 451.2385.
(S)-Ethyl allyl(3-hydroxy-3-(quinolin-7-yl)propyl)carbamate (11). To a solution of silyl-ether 9B (154 mg, 0.36 mmol) in 4 mL THF were added 1.08 mL (1.08 mmol) TBAF-solution (1M in THF) at rt. The mixture was stirred at rt for 7 h; aqueous NH4Cl solution was then added and the solution was extracted with EtOAc. The combined organic layers were washed with H2O, dried over MgSO4 and concentrated in vacuo. The residue was purified by FC (hexane/EtOAc 2:1 → 1:1) to furnish 109 mg (96%) of the free alcohol 11 as a yellow oil; Rf = 0.1 (hexane/EtOAc 2:1). = −8.57° (c = 1.25, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.86 (dd, J = 4.3 Hz, 1.7 Hz, 1H), 8.10 (dd, J = 8.3 Hz, 1.0 Hz, 1H), 8.01 (br s, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 6.2 Hz, 1H), 7.34 (dd, J = 8.4 Hz, 4.3 Hz, 1H), 5.78 (m, 1H), 5.15 (br s, 1H), 5.11 (m, 1H), 4.85 (d, J = 8.6 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.95–3.73 (m, 3H), 3.19 (m, 1H), 2.05 (m, 1H), 1.86 (m, 1H), 1.23 (t, J = 7.1 Hz, 3H); OH-proton not visible. 13C-NMR (100 MHz, CDCl3): δ = 157.6, 150.5, 148.2, 145.9, 135.8, 133.5, 127.9, 127.4, 125.6, 124.8, 120.8, 117.0, 70.0, 61.9, 49.7, 43.6, 37.7, 14.6. IR (film): ῦ = 3392 (br), 2980, 2926, 1673, 1472, 1415, 1241, 1069, 839, 771, 677 cm−1. HRMS (ESIpos): calcd. for C18H22N2O3Na [M + Na]+: 337.1523, found: 337.1520.
(S)-3-((t-Butyldimethylsilyl)oxy)-1-((3aS,6R,7aR)-8,8-dimethyl-2,2-dioxidotetrahydro-3H-3a,6-methano-benzo[c]isothiazol-1(4H)-yl)-3-(quinolin-6-yl)propan-1-one (
12A). To a solution of 4.53 g (10.90 mmol) of (
S)-1-((3a
S,6
R,7a
R)-8,8-dimethyl-2,2-dioxidotetrahydro-3
H-3a,6-methanobenzo[c]isothiazol-1(4
H)-yl)-3-hydroxy-3-(quinolin-6-yl)propan-1-one (3:1 mixture of diastereoisomers) [
25] in 75 mL of DMF were added 2.25 g (32.9 mmol) of imidazole and 2.52 g (16.4 mmol) of TBSCl and the reaction mixture was stirred at 40 °C for 16 h. The solution was then evaporated, CH
2Cl
2 and water were added, and the organic layer was separated. The aqueous solution was additionally extracted with CH
2Cl
2 and the combined organic extracts were dried over MgSO
4, and evaporated. The residue was purified by FC (hexane/EtOAc 3:2, three runs) to give 3.76 g (65%) of protected alcohol
12A as a light-yellow solid (single isomer) and 1.15 g (20%) of the corresponding 3
R-isomer; R
f = 0.7 (hexane/EtOAc 1:1).
1H-NMR (400 MHz, CDCl
3): δ = 8.86 (dd,
J = 4.3 Hz, 1H), 8.10 (dm,
J = 8.3 Hz, 1H), 8.03–8.01 (m, 1H), 7.75 (d,
J = 7.0 Hz, 1H), 7.75 (d,
J = 7.5 Hz, 1H), 7.35 (dd,
J = 8.3 Hz, 4.2 Hz, 1H), 5.39 (t,
J = 6.8 Hz, 1H), 3.73 (d,
J = 7.7 Hz, 1H), 3.33 (s, 2H), 3.22 (dd,
J = 14.9 Hz, 6.9 Hz, 1H), 3.14 (dd,
J = 14.9 Hz, 6.9 Hz, 1H), 1.92 (dd,
J = 13.6 Hz, 7.9 Hz, 1H), 1.83–1.64 (br, m, 3H), 1.62 (m, 1H), 1.26 (m, 2H), 0.84 (s, 9H), 0.78 (s, 3H), 0.45 (s, 3H), 0.05 (s, 3H), −0.17 (s, 3H).
13C-NMR (100 MHz, CDCl
3): δ = 168.9, 150.2, 148.1, 141.8, 136.1, 129.5, 128.3, 127.9, 125.0, 121.1, 71.9, 64.9, 52.9, 48.1, 47.5, 46.5, 44.5, 38.2, 32.8, 26.3, 25.7 (3 × CH
3), 20.0, 19.6, 18.1, −4.7, −5.0.
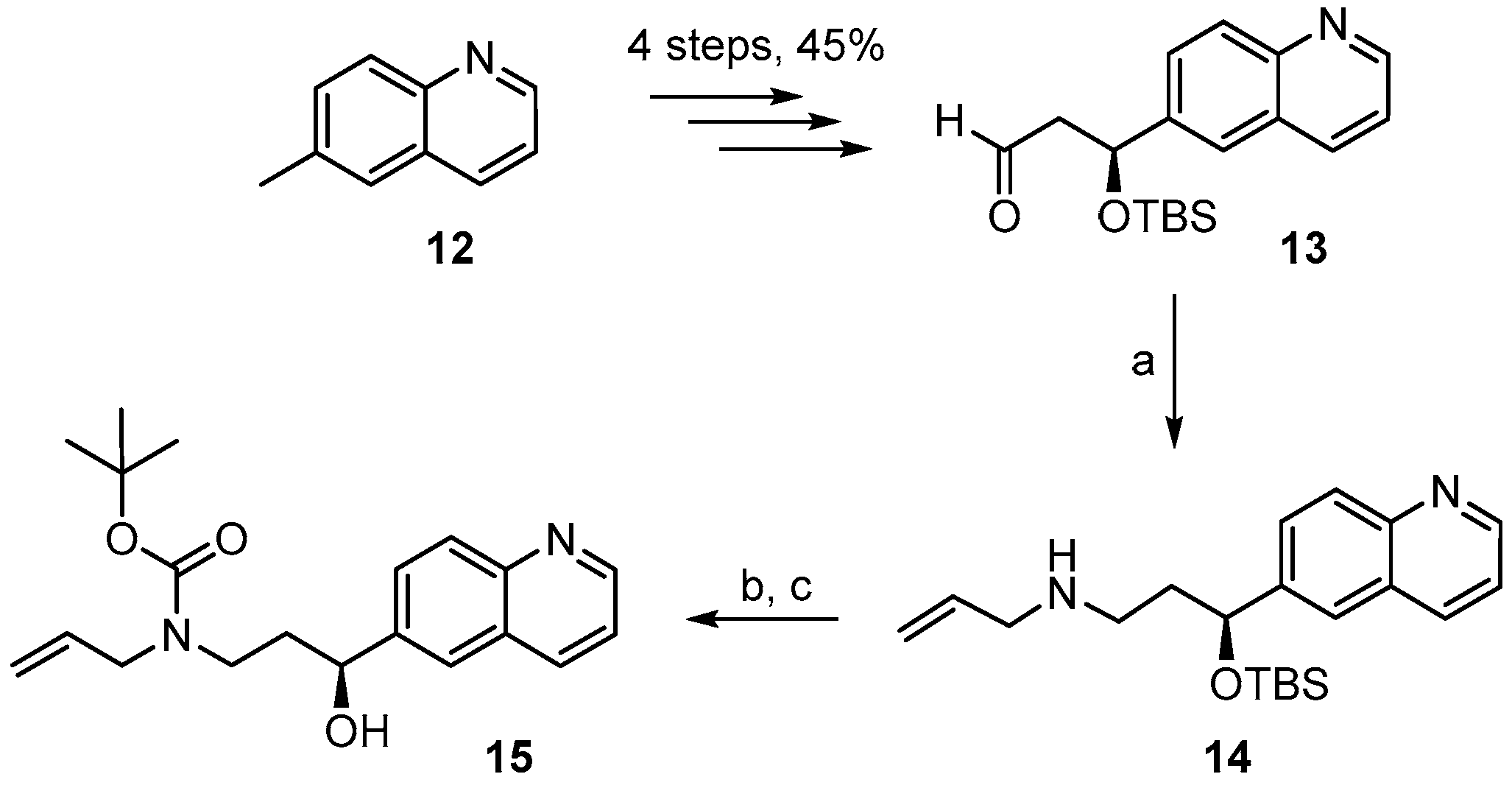
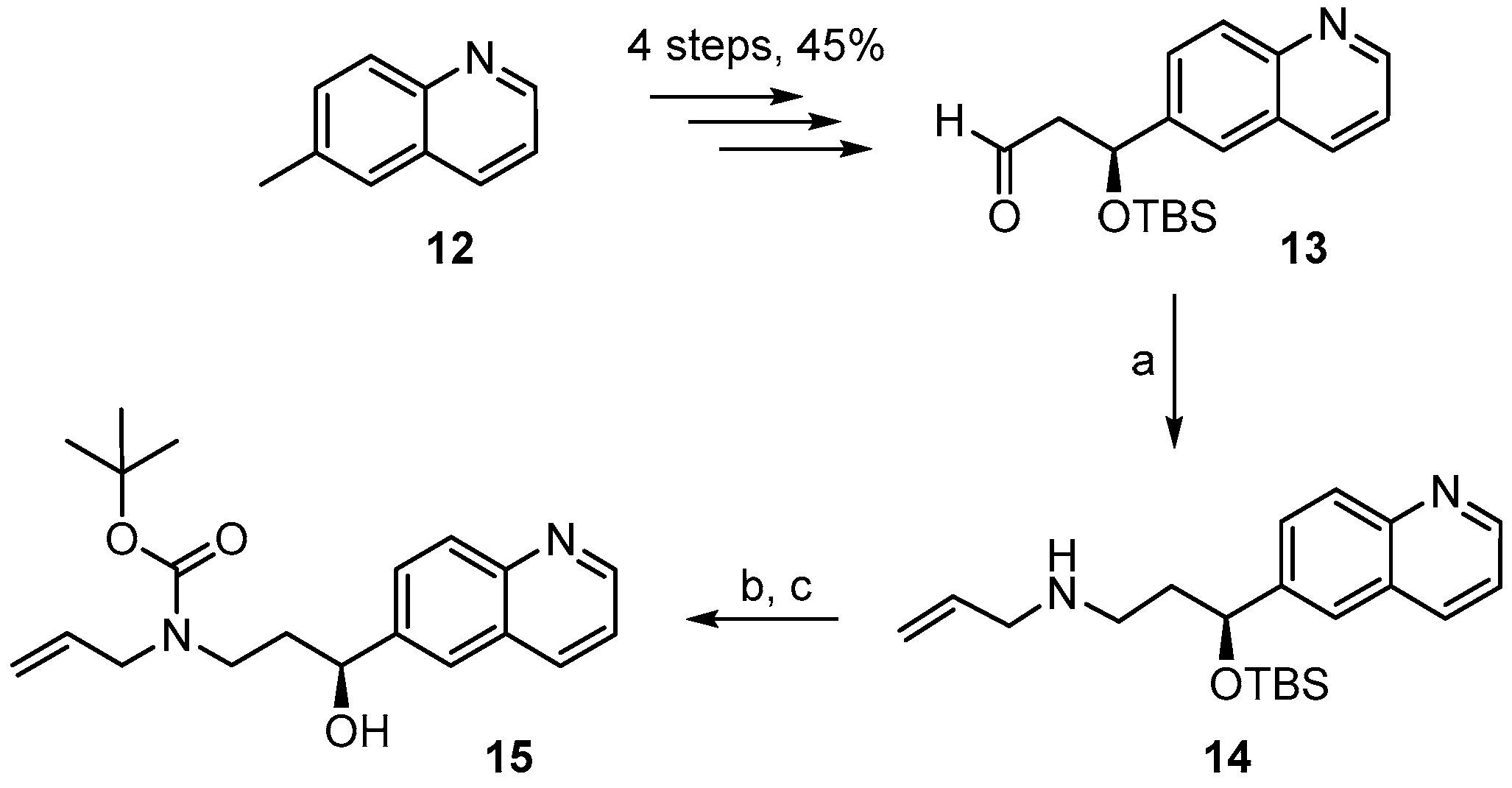
(S)-3-((t-Butyldimethylsilyl)oxy)-3-(quinolin-6-yl)propanal (13). To a solution of 3.76 g (7.11 mmol) of TBS-ether 12A in 35 mL of CH2Cl2 were added dropwise 18 mL of a 1M solution of DIBAL-H (18 mmol) at −78 °C under Ar over a period of 30 min. The mixture was then stirred at −78 °C for 6 h. Water was then added to quench the reaction, the mixture was diluted with additional water (200 mL), CH2Cl2 (200 mL) and 45 mL of 1 N NaOH. The organic layer was then separated and the aqueous solution was additionally extracted with CH2Cl2 (2 times). The combined organic extracts were washed with water, dried over MgSO4, and the solvent was evaporated. Purification of the residue by FC with hexane/EtOAc 1/1 (3 runs) gave 2.29 g (102%) of slightly impure aldehyde 13 as a yellow oil; Rf = 0.6 (hexane/EtOAc 1:1). = −76.2° (c = 1.09 in CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 9.77 (dd, J = 2.4 Hz, J = 2.0 Hz, 1H), 8.86 (dd, J = 4.3 Hz, 1.8 Hz, 1H), 8.10 (dm, J = 8.4 Hz, 1H), 8.06 (d, J = 8.7 Hz, 1H), 7.73 (m, 1H), 7.68 (dd, J = 8.7 Hz, 1.9 Hz, 1H), 7.36 (dd, J = 8.4 Hz, J = 4.2 Hz, 1H), 5.38 (dd, J = 8.0 Hz, J = 4.2 Hz, 1H), 2.91 (ddd, J = 15.9 Hz, J = 8 Hz, J = 2.5 Hz, 1H), 2.68 (ddd, J = 16.0 Hz, J = 4.3 Hz, J = 1.9 Hz, 1H), 0.84 (s, 9H), 0.03 (s, 3H), −0.17 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 200.6, 150.4, 147.9, 142.0, 136.0, 129.9, 127.9, 127.3, 124.1, 121.4, 70.3, 53.8, 25.6 (3 × CH3), 18.0, −4.7, −5.2. HRMS (ESIpos): calcd. for C18H25NO2SiNa [M + Na]+: 338.1547, found: 338.1539.
(S)-N-(3-(t-Butyldimethylsilyloxy)-3-(quinolin-6-yl)propyl)prop-2-en-1-amine (14). To heat-activated molecular sieves (4 Å, 600 mg) was added a solution of aldehyde 13 (250 mg, 0.79 mmol) in 5 mL THF. To this solution were added 0.3 mL of allylamine (3.93 mmol) and the mixture was heated to 50 °C for 24 h. It was then filtered through a pad of dry Celite™, the residue was washed with THF and the combined filtrates were concentrated under reduced pressure to give a yellow oil. For the reduction, 32 mg of NaBH4 (0.81 mmol) were placed in a 10 mL two-necked flask at 0 °C and a solution of the crude imine in 3 mL MeOH was added (gas formation could be observed). After 20 min the reaction mixture was diluted with water and extracted with EtOAc. The combined organic extracts were washed with brine, dried over MgSO4 and concentrated in vacuo. Purification of the residue by FC (hexane/EtOAc 4:1 → 1:1 + 1% Et3N) furnished 188 mg (67%) of the desired amine 14 as slightly yellow oil; = −58.1° (c = 1.29, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.82 (dd, J = 4.3 Hz, 1.8 Hz, 1H), 8.06 (dd, J = 8.4 Hz, 1.4 Hz, 1H), 8.02–8.00 (m, 1H), 7.65 (dd, J = 7.1 Hz, 2.1 Hz, 2H), 7.31 (dd, J = 8.3 Hz, 4.3 Hz, 1H), 5.82 (m, 1H), 5.07 (dq, J = 17.2 Hz, 1.6 Hz, 1H), 5.00 (dq, J = 10.3 Hz, 1.6 Hz, 1H), 4.93 (dd, J = 7.5 Hz, 4.7 Hz, 1H), 3.15 (dt, J = 6.0 Hz, 1.2 Hz, 2H), 2.63 (dd, J = 7.3 Hz, 6.7 Hz, 2H), 1.97–1.80 (br m, 2H), 0.85 (s, 9H), 0.01 (s, 3H), −0.21 (s, 3H); NH proton not visible. 13C-NMR (100 MHz, CDCl3): δ = 149.9, 147.7, 143.7, 136.8, 135.8, 129.3, 127.8, 127.8, 123.8, 121.0, 115.6, 73.3, 52.5, 45.7, 40.7, 25.7 (3 × CH3), 18.1, −4.7, −5.1. HRMS (ESIpos): calcd. for C21H32N2OSiNa [M + Na]+: 379.2176, found: 379.2178.
(S)-t-Butyl allyl(3-(t-butyldimethylsilyloxy)-3-(quinolin-6-yl)propyl)carbamate (14A). A solution of amine 14 (163 mg, 0.46 mmol) and Boc2O (154 mg, 0.69 mmol) in 7 mL CH2Cl2 was stirred at rt for 14 h. 1 mL of ethanolamine was then added and stirring was continued for one additional hour. The mixture was concentrated in vacuo, water was added to the residue, and the mixture was extracted with Et2O and CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification of the residue by FC (hexane/EtOAc 10:1 → 4:1 + 1% Et3N) gave 180 mg (86%) of the desired carbamate 14B as a colorless oil; Rf = 0.6 (hexane/EtOAc 2:1). = −37.5° (c = 1.29, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.83 (dd, J = 4.0 Hz, 1.6 Hz, 1H), 8.08 (dm, J = 8.2 Hz, 1H), 8.03 (d, J = 9.1 Hz, 1H), 7.67–7.64 (m, 2H), 7.33 (dd, J = 8.3 Hz, 4.2 Hz, 1H), 5.68 (m, 1H), 5.02 (q, J = 1.2 Hz, 1H), 4.99 (dq, J = 9.2 Hz, 1.5 Hz, 1H), 4.83 (dd, J = 7.0 Hz, 4.8 Hz, 1H), 3.72 (m, 2H), 3.20 (m, 2H), 1.95 (m, 2H), 1.34 (s, 9H), 0.86 (s, 9H), 0.01 (s, 3H), −0.20 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 155.3, 150.0, 147.7, 143.4, 135.9, 134.1, 129.4, 127.9, 127.6, 123.9, 121.1, 116.3, 79.3, 72.9, 49.7, 43.6, 38.8, 28.3 (3 × CH3), 25.7 (3 × CH3), 18.1, −4.7, −5.1. HRMS (ESIpos): calcd. for C26H40N2O3SiNa [M + Na]+: 479.27004, found: 479.2699.
(S)-t-Butyl allyl[3-hydroxy-3-(quinolin-6-yl)propyl] carbamate (15). To a solution of silyl-ether 14B (200 mg, 0.44 mmol) in 5 mL THF were added 1.32 mL (1.32 mmol) TBAF-solution (1 M in THF) and the mixture was stirred at rt for 5 h. The reaction was quenched with saturated aqueous NH4Cl solution and the mixture was extracted with EtOAc. The combined organic layers were washed with H2O, dried over MgSO4 and concentrated in vacuo. The residue was purified by FC (hexane/EtOAc 2:1 → 1:1) to furnish 143 mg (95%) of the desired free alcohol 15 as a colorless oil; Rf = 0.2 (hexane/EtOAc 2:1). = −9.49° (c = 1.13, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.85 (dd, J = 4.2 Hz, 1.5 Hz, 1H), 8.12 (dd, J = 8.4 Hz, 1.2 Hz, 1H), 8.04 (d, J = 8.7 Hz, 1H), 7.84 (br s, 1H), 7.66 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 7.36 (dd, J = 8.2 Hz, 4.2 Hz, 1H), 5.80 (m, 1H), 5.16 (br s, 1H), 5.12 (d, J = 6.3 Hz, 1H), 4.80 (m, 2H; 1xOH), 3.91 (dd, J = 15.7 Hz, 5.4 Hz, 2H), 3.72 (m, 1H), 3.10 (m, 1H), 2.03 (m, 1H), 1.76 (m, 1H), 1.45 (s, 9H). 13C-NMR (100 MHz, CDCl3): δ = 171.13, 150.08, 147.71, 142.60, 136.07, 133.83, 129.35, 128.15, 127.72, 123.79, 121.18, 116.75, 80.53, 69.80, 50.12, 43.33, 38.09, 28.36 (3 × CH3). HRMS (ESIpos): calcd. for C20H26N2O3SiNa [M + Na]+: 365.1836, found: 365.1831.
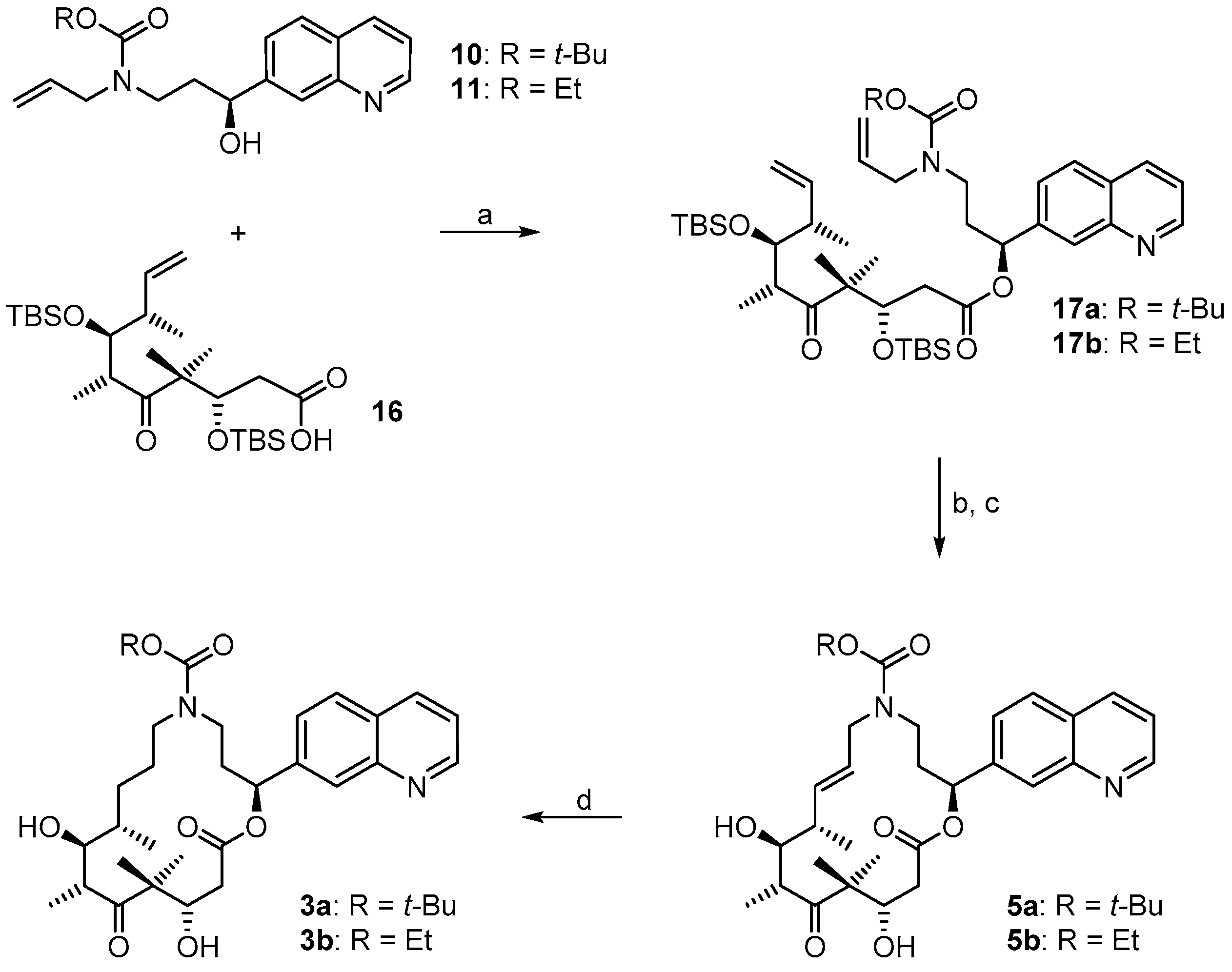
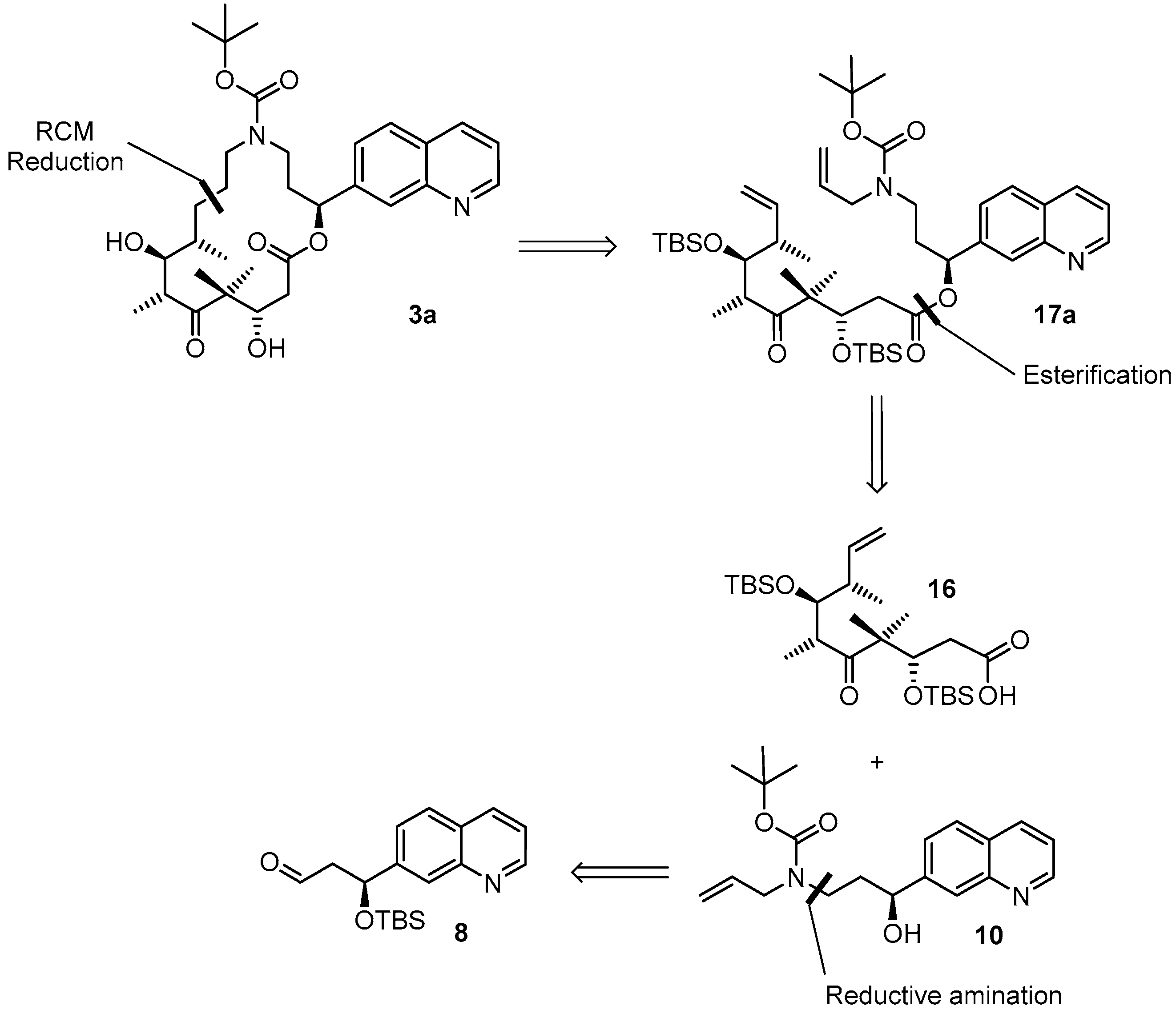
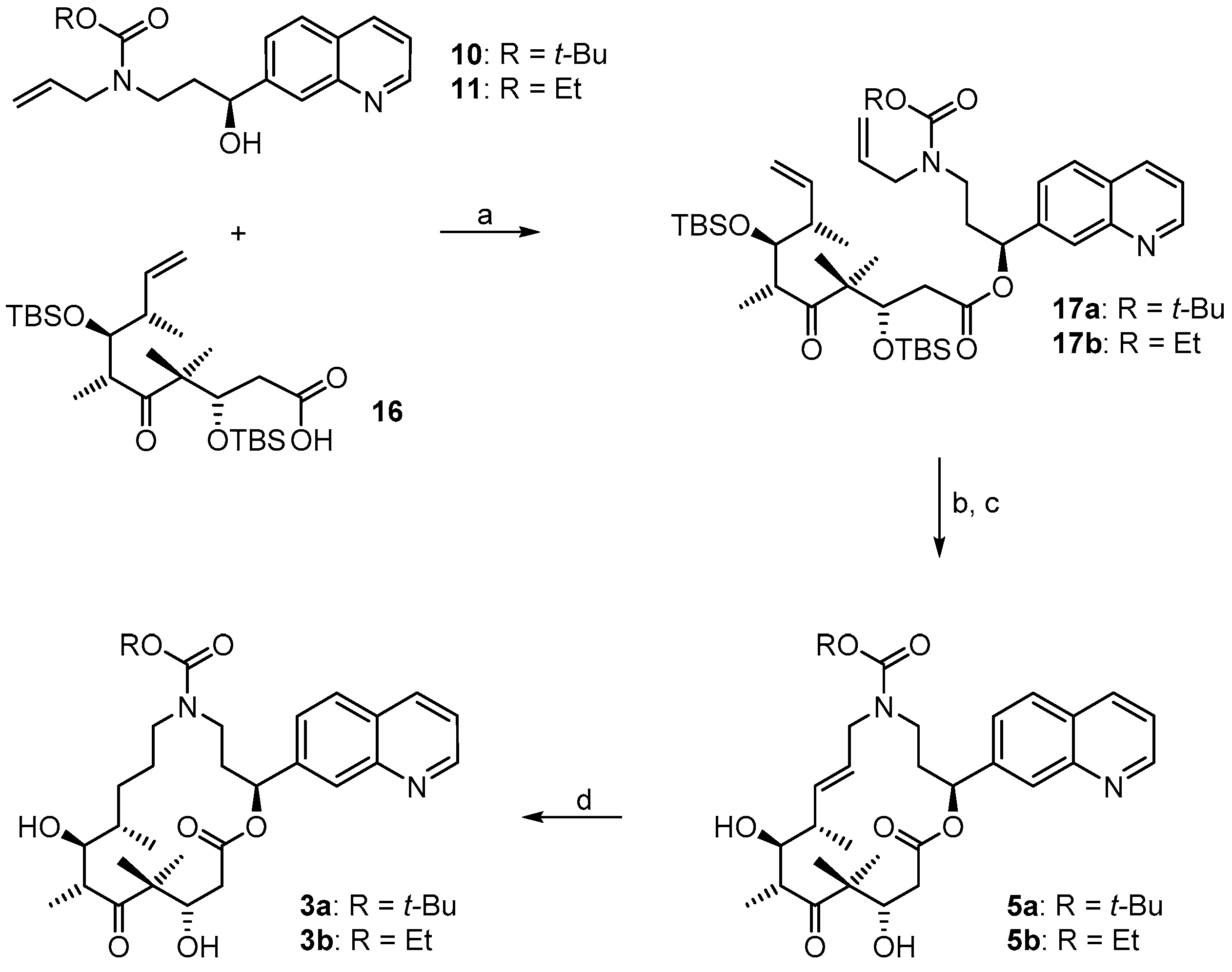
(3S,6R,7S,8S)-{(S)-3-[Allyl(t-butoxycarbonyl)amino]-1-(quinolin-7-yl)propyl}3,7-bis(t-butyldimethylsilyloxy)-4,4,6,8-tetra-methyl-5-oxodec-9-enoate (
17a). To a solution of alcohol 10 (115 mg, 0.34 mmol) in 3 mL CH
2Cl
2 were added sequentially 48 mg (0.39 mmol) of DMAP and 77 mg (0.39 mmol) of EDCI at 0 °C. After stirring for 5 min, a solution of 120 mg (0.24 mmol) of acid
16 [
29] in 2 mL CH
2Cl
2 was added, the cooling bath was removed and stirring was continued at rt for 5 h, when TLC analysis (hexane/EtOAc 4:1) indicated complete conversion. The mixture was then concentrated in vacuo and the resulting residue was purified by FC (hexane/EtOAc 4:1) to furnish 166 mg (84%) of the desired ester
17a as a colorless oil; R
f = 0.3 (hexane/EtOAc 4:1).
= −45.1° (
c = 1.01, CH
2Cl
2).
1H-NMR (400 MHz, CDCl
3): δ = 8.89 (dd,
J = 4.4 Hz, 1.3 Hz, 1H), 8.11 (d,
J = 8.4 Hz, 1H), 8.00 (br s, 1H), 7.78 (d,
J = 8.5 Hz, 1H), 7.50 (d,
J = 8.2 Hz, 1H), 7.37 (dd,
J = 8.4 Hz, 4.1 Hz, 1H), 5.86 (m, 2H), 5.69 (m, 1H), 5.06–4.98 (m, 3H), 4.94 (dm,
J = 17.3 Hz, 1H), 4.31 (dd,
J = 6.5 Hz, 3.2 Hz, 1H), 3.76 (dd,
J = 7.1 Hz, 2.0 Hz, 3H), 3.21 (m, 2H), 3.00 (qi,
J = 7.0 Hz, 1H), 2.50 (dd,
J = 17.1 Hz, 3.4 Hz, 1H), 2.34 (dd,
J = 17.1 Hz, 6.1 Hz, 1H), 2.22 (m, 1H), 2.12 (m, 1H), 2.02 (s, 1H), 1.37 (br s, 9H), 1.15 (br s, 3H), 1.00 (s, 3H), 0.98 (d,
J = 7.0 Hz, 3H), 0.94 (d,
J = 7.0 Hz, 3H), 0.87 (s, 9H), 0.86 (s, 9H), 0.05 (s, 3H), 0.02 (s, 3H), −0.01 (s, 3H), −0.01 (s, 3H).
13C-NMR (100 MHz, CDCl
3): δ = 217.8, 171.3, 155.2, 150.8, 148.1, 141.4, 139.9, 135.8, 134.1, 128.2, 127.9, 126.9, 125.1, 121.2, 115.3, 116.3, 79.6, 76.2, 74.2, 73.8, 53.3, 50.0, 46.1, 43.6, 43.5, 40.3, 34.9, 28.4 (3 × CH
3), 26.2 (3 × CH
3), 26.1 (3 × CH
3), 23.6, 19.7, 18.8, 18.5, 18.2, 15.2, −3.5, −3.9, −4.2, −4.7. Some of the peaks in the carbon spectrum were hardly visible due to peak broadening. IR (film): ῦ = 2929 (br), 2857, 1738, 1696, 1463, 1365, 1251, 1162, 987, 834, 775, 669 cm
−1. HRMS (ESIpos): calcd. for C
46H
76N
2O
7Si
2Na [M + Na]
+: 847.5083, found: 847.5088.
(2S,9S,10S,11R,14S,E)-t-Butyl-10,14-bis(t-butyldimethyl-silyloxy)-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-7-ene-5-carboxylate (17aA). To a solution of diene 17a 63 mg (0.076 mmol) in 148 mL toluene was added Grubbs 2nd generation catalyst (10 mg, 0.012 mmol, 15 mol %) in 2 mL of toluene at reflux temperature. The reddish-yellow solution was stirred at reflux temperature for 30 min, when complete conversion had occurred according to MS analysis; the mixture was then cooled down with an ice bath. The solvent was removed in vacuo and the residue was purified by FC (hexane/EtOAc 10:1 → 4:1). Because the resulting product still had a brownish color, a second column chromatography was performed leading to 50 mg (82%) of the desired olefin 17aA as a greyish foam; Rf = 0.2 (hexane/EtOAc 4:1); Rf = 0.5 (hexane/EtOAc 2:1). = −11.2° (c = 1.00, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.90 (dd, J = 4.1 Hz, 1.0 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 8.08 (br s, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.55 (dd, J = 8.4 Hz, 1.6 Hz,1H), 7.37 (dd, J = 8.4 Hz, 4.2 Hz, 1H), 6.10 (d, J = 9.7 Hz, 1H), 5.37 (dd, J = 15.4 Hz, 6.3 Hz, 1H), 5.26 (dm, J = 15.6 Hz, 1H), 4.39 (m, 2H), 4.01 (dd, J = 8.7 Hz, 2.5 Hz, 1H), 3.64 (m, 1H), 3.25 (m, 2H), 2.99 (m, 1H), 2.60 (dd, J = 16.6 Hz, 6.3 Hz, 1H), 2.48 (m, 1H), 2.44–2.38 (m, 1H), 2.30 (m, 1H), 2.20 (m, 1H), 1.40 (s, 9H), 1.24 (s, 3H), 1.15 (s, 3H), 1.13 (d, J = 6.8 Hz, 3H), 1.04 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 0.83 (s, 9H), 0.12 (s, 3H), 0.06 (s, 6H), −0.03 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 216.5, 170.4, 155.6, 150.8, 148.2, 141.5, 135.7, 134.6, 128.2, 127.9, 127.0, 125.2, 124.5, 121.3, 79.6, 77.1, 73.4, 73.0, 54.3, 48.2, 43.8, 43.6, 42.8, 42.5, 34.5, 28.4 (3 × CH3), 25.9 (6 × CH3), 23.8, 19.0, 18.3, 18.2, 18.1, 12.8, −3.7, −4.3, −4.3, −4.9. Some of the peaks in the carbon spectrum were hardly visible, due to peak broadening. IR (film): ῦ = 2929 (br), 2857, 1739, 1472, 1365, 1252, 1160, 1082, 988, 836, 775, 670 cm−1. HRMS (ESIpos): calcd. for C44H72N2O7Si2Na [M + Na]+: 819.4770, found: 847.4766.
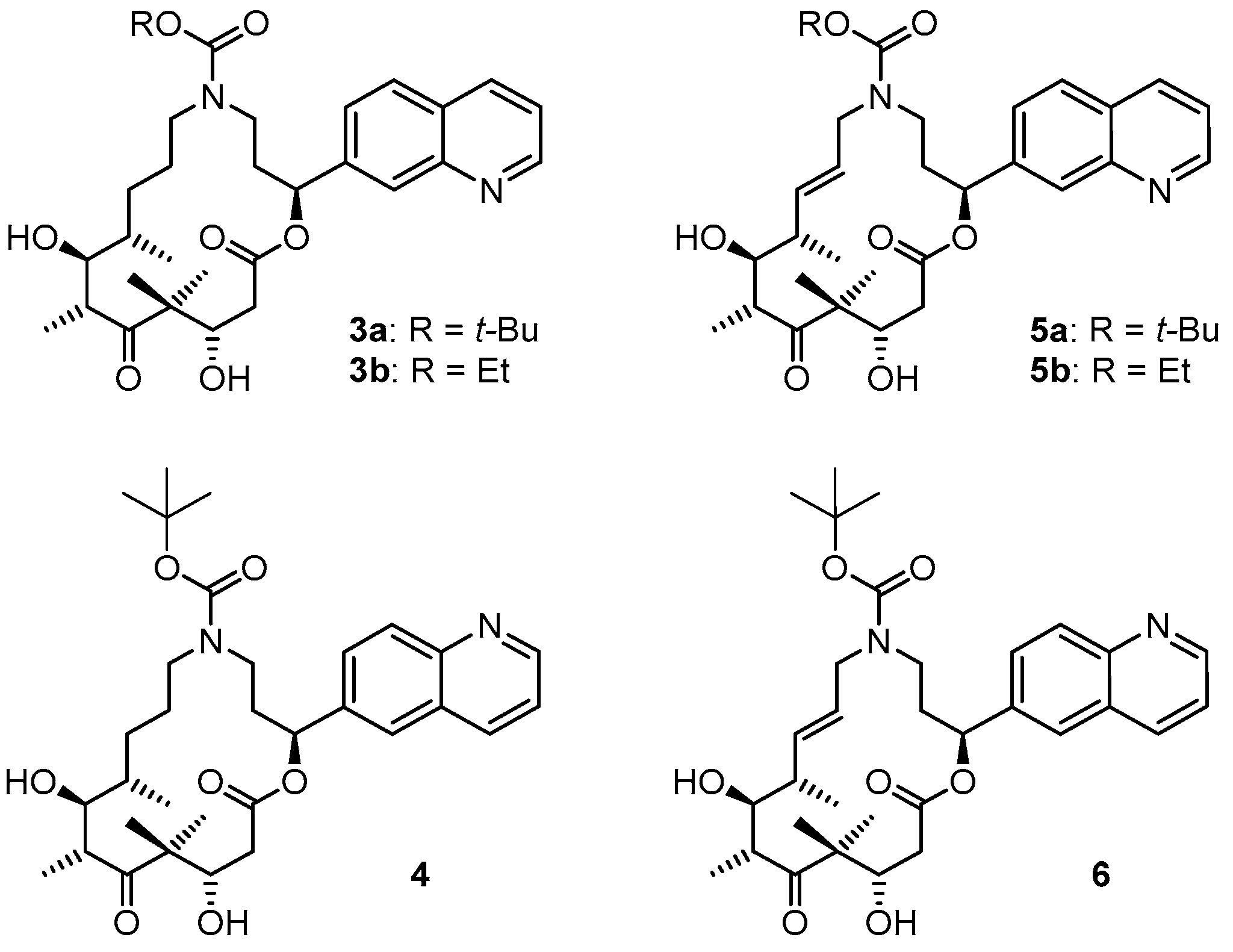
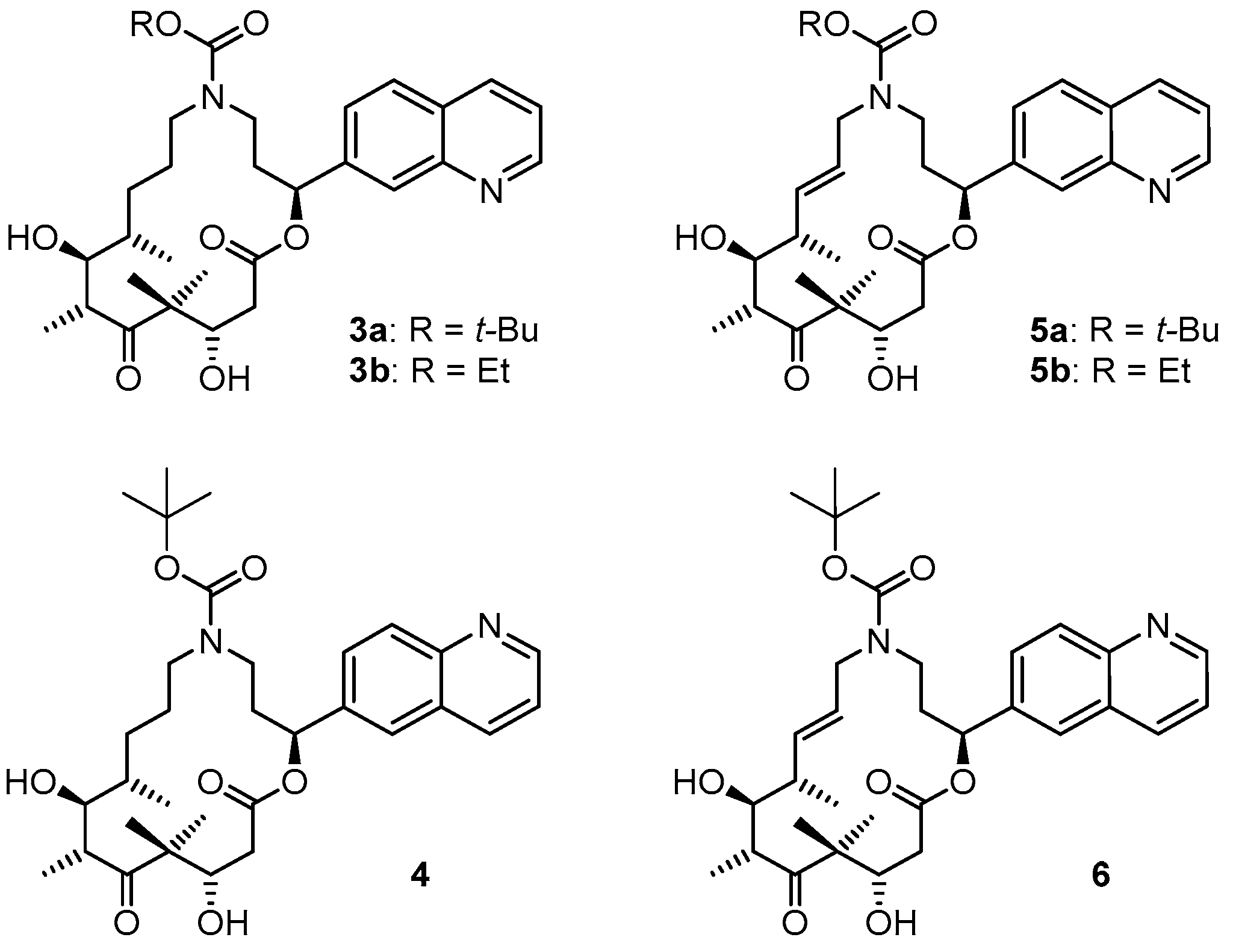
(2S,9S,10S,11R,14S,E)-t-Butyl-10,14-dihydroxy-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-7-ene-5-carboxylate (5a). To a solution of bis-TBS-ether 17aA (116 mg, 0.15 mmol) in 6 mL THF in a 50 mL plastic tube were added 1.3 mL of pyridine and 2 mL of HF∙pyridine (HF∙pyridine dropwise) at 0 °C. After 15 min, the cooling bath was removed and the mixture was stirred at rt for 2 h. As significant amounts of starting material were still detectable at this point by MS analysis, another 1.3 mL of HF∙pyridine were added; 2 h later the conversion to the mono-TBS-protected product was nearly complete. Another 0.8 mL of HF∙pyridine were added and stirring was continued. Subsequent to this, samples of the reaction mixture were analyzed by MS every 30 min. After 7 h only small amounts of the bis-TBS-protected product were still detectable. To avoid BOC-cleavage, the reaction was quenched at this point. Thus, the reaction mixture was added dropwise to 35 mL of a cooled saturated, aqueous NaHCO3 solution (strong gas formation). The solution was then extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by FC (CH2Cl2/MeOH 200:1 → 20:1) followed by preparative RP-HPLC on a Waters Symmetry® C18 5 μm (Waters, Milford, MA, USA), 19 mm × 100 mm column, employing an CH3CN/H2O gradient (CH3CN/H2O 40:60 → 90:10) at a flow rate of 25 mL/min to furnish 57 mg (67%) of the macrolactone 5a as a white solid; Rf = 0.3 (EtOAc). = −107.4° (c = 1.00, CH2Cl2). 1H-NMR (500 MHz, 318 K, DMSO-d6): δ = 8.89 (dd, J = 4.2 Hz, 1.7 Hz, 1H), 8.33 (dm, J = 8.4 Hz, 1H), 8.04 (br s, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.62 (dd, J = 8.4 Hz, 1.5 Hz, 1H), 7.50 (dd, J = 8.2 Hz, 4.2 Hz, 1H), 5.93 (m, 2H), 5.37 (m, 1H), 5.17 (d, J = 6.6 Hz, 1H, OH), 4.53 (m, 2H), 4.01 (dd, J = 14.9 Hz, 5.3 Hz, 1H), 3.68 (dd, J = 15.2 Hz, 7.6 Hz, 1H), 3.56 (m, 1H), 3.28 (m, 2H), 3.12 (m, 1H), 2.45–2.38 (m, 2H), 2.20 (m, 1H), 2.09 (m, 1H), 2.02 (m, 1H), 1.30 (s, 9H), 1.19 (s, 3H), 1.12 (d, J = 6.6 Hz, 3H), 1.04 (d, J = 6.8 Hz, 3H), 0.93 (s, 3H). 13C-NMR (125 MHz, 318 K, DMSO-d6): δ = 216.6, 169.7, 154.3, 150.5, 147.4, 141.6, 135.6, 135.5, 128.0, 127.0, 125.5, 125.1, 124.2, 121.2, 78.5, 74.4, 73.3, 69.2, 53.4, 48.3, 43.8, 41.0, 40.4, 38.5, 34.3, 27.8 (3 × CH3), 19.9, 18.9, 17.3, 14.8. Some of the carbon signals were hardly visible, due to peak broadening. IR (film): ῦ = 3472 (br), 2929 (br), 1740, 1687, 1415, 1365, 1251, 1173, 1042, 982 cm−1. HRMS (ESIpos): calcd. for C32H44N2O7Na [M + Na]+: 591.3041, found: 591.3042.
(2S,9S,10S,11R,14S)-t-Butyl-10,14-dihydroxy-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-ane-5-carboxylate (3a). To a yellow suspension of macrolactone 5a (9 mg, 0.016 mmol) and dipotassium azadicarboxylate (PADA) (500 mg, 2.58 mmol) in 5 mL CH2Cl2 was added a solution of AcOH (0.3 mL, 5.25 mmol) in 3 mL CH2Cl2 dropwise via syringe pump at rt over a period of 2.5 h. After stirring for 18 h, the white suspension was filtered through a plug of Celite™, the residue was washed with CH2Cl2 and the combined filtrates were concentrated in vacuo. The resulting crude mixture was again dissolved in 5 mL of CH2Cl2 together with 500 mg (2.58 mmol) PADA and another 0.3 mL (5.25 mmol) AcOH were added as a solution in 3 mL CH2Cl2 dropwise via syringe pump. After 18 h, the reaction mixture was again filtered. The reaction/work-up-sequence was repeated until a reasonable conversion to the reduced product could be observed by HPLC. Altogether 3.5 g (18 mmol) of PADA and 2.1 mL (36.7 mmol) of AcOH were used, the overall reaction time was 7 days. After the final work up, the resulting crude mixture was purified by preparative RP-HPLC on a Waters Symmetry® C18 5 μm, 19 mm × 100 mm column, employing an CH3CN/H2O gradient (CH3CN/H2O 40:60 → 90:10) at a flow rate of 25 mL/min, to yield 6 mg (66%) of the desired azathilone 3a as a white solid; Rf = 0.2 (CH2Cl2/MeOH 50:1). = −8.7° (c = 1.00, CH2Cl2). 1H-NMR (500 MHz, 318 K, DMSO-d6): δ = 8.89 (dd, J = 4.1 Hz, 1.6 Hz, 1H), 8.33 (dm, J = 8.3 Hz, 1H), 8.05 (br s, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.63 (dd, J = 8.4 Hz, 1.4 Hz, 1H), 7.50 (dd, J = 8.3 Hz, 4.2 Hz, 1H), 5.93 (m, 1H), 5.26 (d, J = 7.6 Hz, 1H, OH), 4.26 (d, J = 6.4 Hz, 1H, OH), 4.19 (m, 1H), 3.43 (m, 1H), 3.37 (m, 1H), 3.23 (m, 2H), 3.10 (m, 1H), 2.96 (m, 1H), 2.42 (dd, J = 14.4 Hz, 10.6 Hz, 1H), 2.11 (m, 2H), 1.55 (m, 1H), 1.43 (m, 1H), 1.33 (s, 9H), 1.32 (s, 3H), 1.30–1.16 (m, 4H), 1.02 (d, J = 6.7 Hz, 3H), 0.93 (s, 3H), 0.90 (d, J = 6.8 Hz, 3H). 13C-NMR (125 MHz, 318 K, DMSO-d6): δ = 217.8, 170.5, 154.3, 150.6, 147.5, 141.7, 135.5, 128.0, 127.0, 125.1, 124.3, 121.2, 78.1, 74.0, 73.1, 71.3, 52.7, 47.0, 44.2, 42.2, 38.6, 36.0, 35.5, 27.9 (3 × CH3), 27.0, 24.7, 20.5, 20.0, 17.2, 14.1. Some of the carbon signals were hardly visible, due to peak broadening. IR (film): ῦ = 3442 (br), 2922 (br), 1735, 1687, 1457, 1418, 1366, 1291, 1253, 1160, 1072, 1047, 976, 837 cm−1. HRMS (ESIpos): calcd. for C32H46N2O7Na [M + Na]+: 593.3197, found: 593.3201.
(3S,6R,7S,8S)-((S)-3-(Allyl(ethoxycarbonyl)amino)-1-(quinolin-7-yl)propyl) 3,7-bis(t-butyl-dimethylsilyloxy)-4,4,6,8-tetra-methyl-5-oxodec-9-enoate (
17b). To a solution of alcohol
11 (19 mg, 0.06 mmol) in 2 mL CH
2Cl
2 were added sequentially 8.2 mg (0.067 mmol) of DMAP and 13.1 mg (0.067 mmol) of EDCI at 0 °C. After stirring for 5 min, a solution of 21 mg (0.042 mmol) of acid
16 [
29] in 2 mL CH
2Cl
2 was added, the cooling bath was removed and stirring was continued at rt for 5 h, when TLC analysis (hexane/EtOAc 4:1) indicated complete conversion. The mixture was then concentrated in vacuo and the resulting residue was purified by FC (hexane/EtOAc 4:1) to furnish 28 mg (84%) of the desired ester
17b as a colorless oil; R
f = 0.1 (hexane/EtOAc 4:1).
= −37.9° (
c = 1.00, CH
2Cl
2).
1H-NMR (400 MHz, CDCl
3): δ = 8.89 (dd,
J = 4.3 Hz, 1.0 Hz, 1H), 8.11 (d,
J = 8.1 Hz, 1H), 8.01 (br s, 1H), 7.78 (d,
J = 8.3 Hz, 1H), 7.51 (d,
J = 7.7 Hz, 1H), 7.37 (dd,
J = 8.4 Hz, 4.1 Hz, 1H), 5.90–5.82 (m, 2H), 5.70 (m, 1H), 5.07 (m, 1H), 5.04 (dm,
J = 11.5 Hz, 1H), 4.99 (dd,
J = 10.6 Hz, 1.2 Hz, 1H), 4.94 (dm,
J = 17.5 Hz, 1H), 4.31 (dd,
J = 6.4 Hz, 3.4 Hz, 1H), 4.06 (q,
J = 7.1 Hz, 2H), 3.80 (m, 2H), 3.75 (dd,
J = 7.2 Hz, 2.0 Hz, 1H), 3.26 (m, 2H), 3.00 (qi,
J = 7.1 Hz, 1H), 2.50 (dd,
J = 16.9 Hz, 3.5 Hz, 1H), 2.35 (dd,
J = 16.7 Hz, 6.4 Hz, 1H), 2.23 (m, 1H), 2.14 (m, 1H), 2.02 (m, 1H), 1.16 (br t,
J = 7.1 Hz, 3H), 1.15 (s, 3H), 1.00 (s, 3H), 0.98 (d,
J = 7.2 Hz, 3H), 0.94 (d,
J = 7.0 Hz, 3H), 0.87 (s, 9H), 0.85 (s, 9H), 0.05 (s, 3H), 0.00 (s, 3H), −0.01 (s, 3H), −0.01 (s, 3H).
13C-NMR (100 MHz, CDCl
3): δ = 217.8, 171.3, 156.1, 150.7, 148.1, 141.5, 139.9, 135.7, 133.7, 128.2, 127.9, 126.9, 125.0, 121.2, 116.8, 115.3, 76.2, 74.0, 73.7, 61.3, 53.3, 49.9, 46.1, 43.4, 43.0, 40.3, 34.8, 26.2 (3 × CH
3), 26.0 (3 × CH
3), 23.6, 19.7, 18.8, 18.5, 18.2, 15.2, 14.6, −3.6, −3.9 −4.2, −4.7. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening. IR (film): ῦ = 2955, 2931, 2855, 1739, 1699, 1472, 1416, 1384, 1252, 1171, 988, 836, 774 cm
−1. HRMS (ESIpos): calcd. for C
44H
72N
2O
7Si
2Na [M + Na]
+: 819.4770, found: 819.4772.
(2S,9S,10S,11R,14S,E)-Ethyl 10,14-bis(t-butyldimethyl-silyloxy)-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-7-ene-5-carboxylate (17bA). To a solution of diene 17b (25 mg, 0.031 mmol) in 63 mL toluene was added Grubbs 2nd generation catalyst (4.0 mg, 0.0047 mmol, 15 mol %) in 2 mL of toluene at reflux temperature. The reddish-yellow solution was stirred at reflux temperature for 30 min, when complete conversion had occurred according to MS analysis; the mixture was then cooled down with an ice bath. The solvent was removed in vacuo and the residue was purified by FC (hexane/EtOAc 4:1) to furnish 23 mg (96%) of the desired olefin 17bA as a brown oil. = −8.6° (c = 1.00, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.91 (dm, J = 3.9 Hz, 1H), 8.12 (d, J = 8.4 Hz, 1H), 8.10 (s, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.56 (dd, J = 8.5 Hz, 1.7 Hz, 1H), 7.38 (dd, J = 8.5 Hz, 4.0 Hz, 1H), 6.11 (d, J = 10.5 Hz, 1H), 5.38 (dd, J = 15.7 Hz, 6.3 Hz, 1H), 5.30–5.23 (m, 1H), 4.47–4.38 (m, 2H), 4.10 (m, 2H), 4.00 (dd, J = 8.7 Hz, 2.9 Hz, 1H), 3.71 (m, 1H), 3.29 (m, 2H), 2.99 (m, 1H), 2.60 (dd, J = 16.8 Hz, 6.2 Hz, 1H), 2.48 (m, 1H), 2.43–2.38 (m, 1H), 2.32–2.16 (m, 2H), 1.24 (s, 3H), 1.20 (br t, J = 7.1 Hz, 3H), 1.15 (s, 3H), 1.13 (d, J = 6.8 Hz, 3H), 1.04 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 0.83 (s, 9H), 0.12 (s, 3H), 0.07 (s, 6H), −0.04 (s, 3H). 13C-NMR (100 MHz, CDCl3): δ = 216.3, 170.4, 150.8, 148.2, 144.3, 141.5, 135.7, 135.0, 128.1, 127.9, 127.2, 125.5, 124.2, 121.3, 77.2, 73.3, 73.0, 61.3, 54.3, 48.7, 43.8, 43.3, 42.9, 42.5, 34.3, 25.9 (6xCH3), 23.9, 19.1, 18.4, 18.2, 18.1, 14.7, 13.1, −3.7, −4.2 (2 × CH3), −4.9. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening (especially the quaternary C-atoms). IR (film): ῦ = 2931, 2855, 2354, 1739, 1695, 1472, 1424, 1385, 1250, 1216, 1084, 988, 836, 774 cm−1. HRMS (ESIpos): calcd. for C42H68N2O7Si2Na [M + Na]+: 791.4457, found: 791.4464.
(2S,9S,10S,11R,14S,E)-Ethyl 10,14-dihydroxy-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-7-ene-5-carboxylate (5b). To a solution of bis-TBS-ether 17bA (22 mg, 0.029 mmol) in 1.5 mL THF in a 12 mL plastic tube were added 0.25 mL of pyridine and 0.4 mL of HF∙pyridine (HF∙pyridine dropwise) at 0 °C. After 15 min, the cooling bath was removed and the mixture was stirred at rt for 2 h. As significant amounts of starting material were still detectable at this point by MS analysis, another 0.3 mL of HF∙pyridine were added; 4 h later the conversion was still incomplete and another 0.1 mL of HF∙pyridine were added. 3 h later MS analysis indicated the presence of product and only traces of the mono-TBS-deprotected species. The reaction mixture was then added dropwise to a cooled saturated, aqueous NaHCO3 solution (strong gas formation). The solution was then extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by FC (CH2Cl2/MeOH 100:1 → 20:1) followed by preparative RP-HPLC on a Waters Symmetry® C18 5 μm, 19 mm × 100 mm column, employing an CH3CN/H2O gradient (CH3CN/H2O 20:80 → 95:5) at a flow rate of 25 mL/min to furnish 8 mg (51%) of the macrolactone 5b as a white solid; Rf = 0.6 (CH2Cl2/MeOH 10:1). = −132.7° (c = 1.00, CH2Cl2). 1H-NMR (500 MHz, 318 K, DMSO-d6): δ = 8.90 (dd, J = 4.2 Hz, 1.7 Hz, 1H), 8.33 (dd, J = 8.4 Hz, 1.4 Hz, 1H), 8.05 (d, J = 0.9 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.63 (dd, J = 8.4 Hz, 1.7 Hz, 1H), 7.51 (dd, J = 8.3 Hz, 4.2 Hz, 1H), 5.95 (m, 2H), 5.38 (m, 1H), 5.15 (d, J = 6.6 Hz, 1H, OH), 4.56 (d, J = 6.4 Hz, 1H, OH), 4.52 (m, 1H), 4.08 (dd, J = 14.8 Hz, 5.2 Hz, 1H), 3.98 (q, J = 7.0 Hz, 2H), 3.71 (dd, J = 15.1 Hz, 7.8 Hz, 1H), 3.56 (dt, J = 7.0 Hz, 3.2 Hz, 1H), 3.37 (m, 1H), 3.27 (qi, J = 7.1 Hz, 1H), 2.47–2.39 (m, 2H), 2.22 (m, 1H), 2.10 (m, 1H), 2.04 (m, 2H), 1.18 (s, 3H), 1.12 (d, J = 6.7 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H), 1.04 (d, J = 6.8 Hz, 3H), 0.93 (s, 3H). 13C-NMR (125 MHz, 318 K, DMSO-d6): δ = 216.6, 169.8, 155.1, 150.6, 147.4, 141.6, 135.8, 135.5, 128.0, 127.0, 125.3, 125.2, 124.3, 121.2, 74.4, 73.3, 69.2, 60.4, 53.4, 48.4, 43.9, 41.1, 39.5, 38.5, 34.1, 19.9, 19.0, 17.4, 14.9, 14.3. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening (especially the quaternary C-atoms). IR (film): ῦ = 3446 (br), 2976, 2927, 1739, 1689, 1468, 1421, 1383, 1288, 1251, 1221, 1181, 1046, 983, 890, 838, 771 cm−1. HRMS (ESIpos): calcd. for C30H40N2O7Na [M + Na]+: 563.2728, found: 563.2728.
(2S,9S,10S,11R,14S)-Ethyl 10,14-dihydroxy-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-ane-5-carboxylate (3b). To a mixture of macrolactone 5b (4 mg, 0.0074 mmol) and dipotassium azadicarboxylate (PADA) (1.0 g, 5.15 mmol) in 5 mL CH2Cl2 was added a solution of AcOH (0.6 mL, 5.25 mmol) in 2.5 mL CH2Cl2 dropwise via syringe pump at rt over a period of 2.5 h. After stirring for 1 d, the mixture was filtered through a plug of Celite™, the filtrate was concentrated in vacuo and the residue was again treated with 1 g of PADA and 0.6 mL of AcOH as described above. After 2 days, analytical RP-HPLC showed the presence of 60% starting material and 40% product. The mixture was worked up as described above and the crude product mixture was again treated with 1 g of PADA and 0.6 mL of AcOH as described above and the mixture was stirred at room temperature for another 2.5 days. After this time only 40% of starting material were present in the reaction mixture. A last work-up/treatment with 1 g of PADA and 0.6 mL of AcOH cycle and stirring for another day resulted in a starting material/product ratio of ~25:75. The mixture was filtered, washed with H2O, extracted with CH2Cl2, dried over MgSO4 and concentrated. The resulting crude product mixture was purified by semi-preparative HPLC (Waters Symmetry® C18 5 μm, 7.8 mm × 100 mm column; gradient from CH3CN/H2O 20:80 to 90:10; 3 mL/min flow rate) to provide 1 mg (25%) of re-isolated starting material 5b and 2.6 mg (65%) of the desired macrolactone 3b both as white solids. Rf = 0.2 (CH2Cl2/MeOH 50:1); Rf = 0.6 (CH2Cl2/MeOH 10:1). = −9.6° (c = 1.00, CH2Cl2). 1H NMR (500 MHz, 318 K, DMSO-d6): δ = 8.89 (dd, J = 4.2 Hz, 1.7 Hz, 1H), 8.33 (dm, J = 8.5 Hz, 1H), 8.06 (br s, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.64 (dd, J = 8.6 Hz, 1.8 Hz, 1H), 7.50 (dd, J = 8.2 Hz, 4.1 Hz, 1H), 5.93 (dd, J = 7.1 Hz, 4.2 Hz, 1H), 5.25 (d, J = 7.4 Hz, 1H, OH), 4.27 (d, J = 6.3 Hz, 1H, OH), 4.19 (m, 1H), 3.97 (q, J = 7.0 Hz, 2H), 3.58 (m, 1H), 3.51 (m, 1H), 3.43 (m, 1H), 3.22–3.15 (m, 3H), 3.01 (m, 1H), 2.40 (dd, J = 14.7 Hz, 10.7 Hz, 1H), 2.12 (m, 2H), 1.57 (m, 1H), 1.45 (m, 1H), 1.39–1.30 (m, 1H), 1.32 (s, 3H), 1.27–1.22 (m, 2H), 1.12 (t, J = 7.0 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H), 0.93 (s, 3H), 0.90 (d, J = 6.9 Hz, 3H). 13C-NMR (125 MHz, 318 K, DMSO-d6): δ = 217.7, 170.5, 155.1, 150.6, 147.4, 141.7, 135.5, 128.0, 127.0, 125.2, 124.4, 121.2, 73.9, 73.0, 71.5, 60.2, 52.7, 46.9, 44.2, 42.1, 38.6, 36.0, 35.1, 26.9, 24.6, 20.5, 20.0, 17.2, 14.4, 14.1. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening. Several of the carbon signals were also duplicated, which we ascribe to the existence of rotamers. In these cases only the signal for the major rotamer is listed. IR (film): ῦ = 3408 (br), 2922 (br), 2851, 1735, 1687, 1457, 1428, 1386, 1290, 1249, 1214, 1148, 1048, 977, 837 cm−1. HRMS (ESIpos): calcd. for C30H43N2O7 [M + H]+: 543.3065, found: 543.3064.
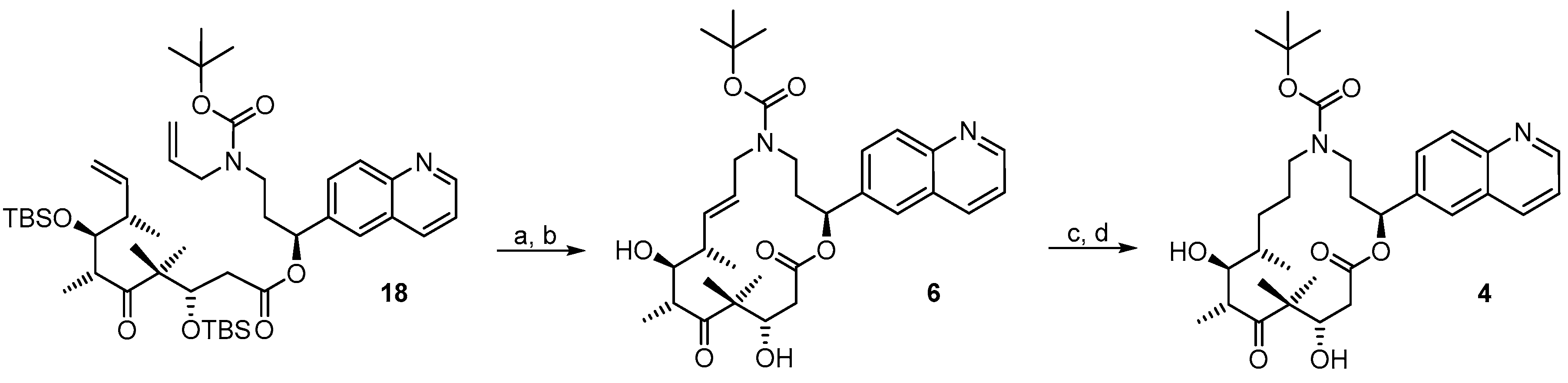
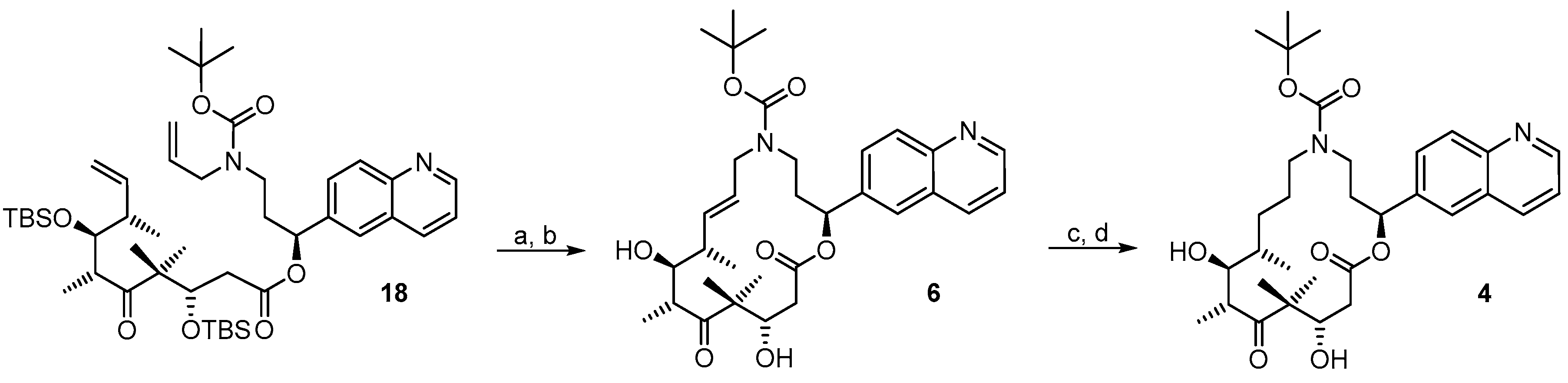
(3S,6R,7S,8S)-((S)-3-(Allyl(ethoxycarbonyl)amino)-1-(quinolin-6-yl)propyl) 3,7-bis(t-butyl-dimethylsilyloxy)-4,4,6,8-tetra-methyl-5-oxodec-9-enoate (
18). To a solution of alcohol
15 (48 mg, 0.14 mmol) in 3 mL CH
2Cl
2 were added sequentially 20 mg (0.16 mmol) of DMAP and 32 mg (0.16 mmol) of EDCI at 0 °C. After stirring for 5 min, a solution of 50 mg (0.1 mmol) of acid
16 [
29] in 2 mL CH
2Cl
2 was added, the cooling bath was removed and stirring was continued at rt for 5 h, when TLC analysis (hexane/EtOAc 4:1) indicated complete conversion. The mixture was then concentrated in vacuo and the resulting residue was purified by FC (hexane/EtOAc 4:1) to furnish 74 mg (90%) of the desired ester
18 as a colorless oil; R
f = 0.4 (hexane/EtOAc 4:1).
= −42.0° (
c = 1.00, CH
2Cl
2).
1H-NMR (400 MHz, CDCl
3): δ = 8.88 (dm,
J = 4.4 Hz, 1H), 8.12 (dm,
J = 8.8 Hz, 1H), 8.07 (d,
J = 8.8 Hz, 1H), 7.75 (br s, 1H), 7.66 (dd,
J = 8.8 Hz, 1.7 Hz, 1H), 7.38 (dd,
J = 8.1 Hz, 4.3 Hz, 1H), 5.85 (m, 2H), 5.70 (m, 1H), 5.07–4.98 (m, 3H), 4.94 (dm,
J = 17.6 Hz, 1H), 4.28 (dd,
J = 6.0 Hz, 3.7 Hz, 1H), 3.75 (dd,
J = 7.0 Hz, 2.0 Hz, 3H), 3.21 (m, 2H), 3.02 (qi,
J = 7.0 Hz, 1H), 2.50 (dd,
J = 17.1 Hz, 3.4 Hz, 1H), 2.32 (dd,
J = 17.1 Hz, 6.3 Hz, 1H), 2.21 (m, 1H), 2.11 (m, 1H), 2.04 (m, 1H), 1.38 (br s, 9H), 1.15 (s, 3H), 0.99 (d,
J = 7.0 Hz, 3H), 0.98 (s, 3H), 0.94 (d,
J = 7.0 Hz, 3H), 0.87 (s, 9H), 0.85 (s, 9H), 0.05 (s, 3H), 0.00 (s, 3H), −0.01 (s, 3H), −0.01 (s, 3H).
13C-NMR (100 MHz, CDCl
3): δ = 217.9, 171.3, 155.2, 150.6, 148.0, 139.8, 138.1, 136.2, 134.1, 130.0, 128.0, 127.6, 125.7, 121.4, 116.6, 115.3, 79.7, 76.2, 74.1, 73.9, 53.3, 49.9, 46.1, 43.5, 43.5, 40.4, 34.7, 28.4 (3 × CH
3), 26.2 (3 × CH
3), 26.0 (3 × CH
3), 23.6, 19.8, 18.7, 18.5, 18.2, 15.1, −3.6, −3.9, −4.3, −4.7. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening. IR (film): ῦ = 2929 (br), 2857, 1739, 1697, 1463, 1365, 1251, 1169, 988, 836, 776, 669 cm
−1. HRMS (ESIpos): calcd. for C
46H
76N
2O
7Si
2Na [M+Na]
+: 847.5083, found: 847.5080.
(2S,9S,10S,11R,14S,E)-Ethyl 10,14-bis(t-butyldimethyl-silyloxy)-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-6-yl)-1-oxa-5-azacyclohexadec-7-ene-5-carboxylate (18A). To a solution of diene 18 (73 mg, 0.088 mmol) in 148 mL toluene was added Grubbs 2nd generation catalyst (10 mg, 0.012 mmol, 15 mol %) in 2 mL of toluene at reflux temperature. The reddish-yellow solution was stirred at reflux temperature for 30 min, when complete conversion had occurred according to MS analysis; the mixture was then cooled down with an ice bath. The solvent was removed in vacuo and the residue was purified by FC (hexane/EtOAc 10:1 → 4:1) to furnish 58 mg (82%) of the desired olefin 18A as a light brown foam; Rf = 0.3 (hexane/EtOAc 4:1); Rf = 0.6 (hexane/ EtOAc 2:1). = −7.2° (c = 1.00, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ = 8.89 (d, J = 3.0 Hz, 1H), 8.12 (d, J = 7.9 Hz, 1H), 8.07 (d, J = 8.9 Hz, 1H), 7.81 (d, J = 1.8 Hz, 1H), 7.71 (dd, J = 8.9 Hz, 1.8 Hz, 1H), 7.39 (dd, J = 8.3 Hz, 4.1 Hz, 1H), 6.08 (d, J = 10.7 Hz, 1H), 5.34 (dd, J = 15.7 Hz, 6.3 Hz, 1H), 5.29–5.22 (m, 1H), 4.47–4.24 (m, 2H), 4.01 (dd, J = 8.7 Hz, 2.5 Hz, 1H), 3.64 (m, 1H), 3.24 (dd, J = 15.9 Hz, 1.3 Hz, 2H), 2.99 (m, 1H), 2.58 (dd, J = 16.8 Hz, 6.1 Hz, 1H), 2.48 (m, 1H), 2.37 (dd, J = 16.9 Hz, 2.5 Hz, 1H), 2.30–2.13 (m, 2H), 1.40 (s, 9H), 1.25 (s, 3H), 1.15 (s, 3H), 1.13 (d, J = 6.8 Hz, 3H), 1.04 (d, J = 7.0 Hz, 3H), 0.88 (s, 9H), 0.84 (s, 9H), 0.13 (s, 3H), 0.07 (s, 6H), −0.04 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 216.4, 170.4, 155.5, 150.7, 148.0, 138.4, 136.1, 134.6, 129.9, 128.0, 127.9, 125.7, 124.5, 121.4, 79.6, 77.2, 73.3, 72.9, 54.3, 48.3, 43.7, 43.5, 43.0, 42.6, 34.5, 28.4 (3 × CH3), 25.9 (6 × CH3), 23.9, 18.9, 18.4, 18.2, 18.1, 12.7, −3.7, −4.3, −4.3, −4.9. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening. IR (film): ῦ = 2928 (br), 2857, 1738, 1693, 1472, 1365, 1251, 1162, 1082, 988, 836, 775, 666 cm−1. HRMS (ESIpos): calcd. for C44H72N2O7Si2Na [M + Na]+: 819.4770, found: 847.4773.
(2S,9S,10S,11R,14S,E)-Ethyl 10,14-dihydroxy-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-6-yl)-1-oxa-5-azacyclohexadec-7-ene-5-carboxylate (6). To a solution of bis-TBS-ether 18A (56 mg, 0.07 mmol mmol) in 2.5 mL THF in a 12 mL plastic tube were added 0.5 mL of pyridine and 1.5 mL of HF∙pyridine (HF∙pyridine dropwise) at 0 °C. After 20 min, the cooling bath was removed and the mixture was stirred at rt for 2 h. As significant amounts of starting material and mono-protected intermediate were still detectable at this point by MS analysis, another 0.3 mL of HF∙pyridine were added; 4 h later the reaction mixture was added dropwise to a cooled saturated, aqueous NaHCO3 solution (strong gas formation). The solution was then extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by FC (CH2Cl2/MeOH 200:1 → 20:1) followed by preparative RP-HPLC (twice) on a Waters Symmetry® C18 5 μm, 19 mm × 100 mm column, employing an CH3CN/H2O gradient (CH3CN/H2O 40:60 → 90:10) at a flow rate of 25 mL/min to furnish 22 mg (55%) of the macrolactone 6 as a white solid; Rf = 0.4 (EtOAc). = −111.3° (c = 1.00, CH2Cl2). 1H-NMR (500 MHz, 318 K, DMSO-d6): δ = 8.88 (dd, J = 4.1 Hz, 1.7 Hz, 1H), 8.31 (dm, J = 8.5 Hz, 1H), 8.00 (d, J = 8.7 Hz, 1H), 7.97 (d, J = 1.6 Hz, 1H), 7.77 (dd, J = 8.7 Hz, 1.8 Hz, 1H), 7.52 (dd, J = 8.4 Hz, 4.2 Hz, 1H), 5.95 (dd, J = 15.6 Hz, 5.2 Hz, 1H), 5.91 (dd, J = 7.3 Hz, 3.4 Hz, 1H), 5.36 (m, 1H), 5.19 (d, J = 6.5 Hz, 1H, OH), 4.55 (d, J = 6.3 Hz, 1H), 4.53–4.50 (m, 1H), 4.00 (dd, J = 14.8 Hz, 5.4 Hz, 1H), 3.68 (dd, J = 14.7 Hz, 7.6 Hz, 1H), 3.56 (m, 1H), 3.29 (m, 2H), 3.09 (m, 1H), 2.45 (dd, J = 15.6 Hz, 4.8 Hz, 1H), 2.38 (dd, J = 15.4 Hz, 8.2 Hz, 1H), 2.18 (m, 1H), 2.10 (m, 1H), 2.02 (m, 1H), 1.31 (br s, 9H), 1.22 (s, 3H), 1.11 (d, J = 6.7 Hz, 3H), 1.05 (d, J = 6.9 Hz, 3H), 0.93 (s, 3H). 13C-NMR (125 MHz, 318 K, DMSO-d6): δ = 216.5, 169.7, 154.2, 150.3, 147.0, 138.4, 136.0, 135.8, 128.8, 127.3, 127.2, 125.3, 124.1, 121.5, 78.5, 74.4, 73.2, 69.3, 53.5, 48.4, 43.7, 41.0, 39.3, 38.4, 34.3, 27.8 (3 × CH3), 20.3, 18.7, 17.1, 14.6. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening. IR (film): ῦ = 3383 (br), 2925 (br), 1734, 1687, 1462, 1413, 1366, 1254, 1164, 1010, 985, 836; HRMS (ESIpos): calcd. for C32H44N2O7Na [M + Na]+: 591.3041, found: 591.3048.
(2S,9S,10S,11R,14S)-Ethyl 10,14-dihydroxy-9,11,13,13-tetramethyl-12,16-dioxo-2-(quinolin-7-yl)-1-oxa-5-azacyclohexadec-ane-5-carboxylate (4). To a yellow suspension of macrolactone 6 (9 mg, 0.016 mmol) and dipotassium azadicarboxylate (PADA) (307 mg, 1.6 mmol) in 1 mL MeOH and 0.2 mL CH2Cl2 was added a solution of AcOH (0.18 mL) in 4 mL MeOH dropwise via syringe pump at rt over a period of 4 h. After stirring for 14 h, the mixture was filtered through a plug of Celite™, the filtrate was concentrated in vacuo and the residue was redissolved in 1 mL MeOH and 0.2 mL CH2Cl2 and another 307 mg of PADA were added. Then a solution of acetic acid (0.18 mL) in 4 mL MeOH was added via syringe pump at rt over a period of 2 h. After this time the white suspension was heated to 50 °C for 5 h. At this point, only a traces of product were detectable by HPLC. The mixture was again filtered through a plug of Celite™ and concentrated in vacuo. The residue was redissolved in 4 mL of MeOH together with 1 g of PADA and a solution of acetic acid (0.6 mL) in 3 mL MeOH was added via syringe pump over a period of 2 h. The mixture was stirred for 14 h and then filtered, the filtrate was concentrated, the residue was dissolved in 4 mL of CH2Cl2 together with 1 g of PADA and a solution of AcOH (0.6 mL) in 3 mL CH2Cl2 was added via syringe pump over a 2 h period. The mixture was stirred at room temperature for 2 days and then worked up as above. The residue was dissolved in EtOAc and the solution was washed with water. After drying over MgSO4 the organic solution was concentrated in vacuo and the residue was purified twice by preparative RP-HPLC (Waters Symmetry® C18 5 μm, 19 mm × 100 mm column, gradient of CH3CN/H2O from 20:80 to 90:10 at a 25 mL/min flow rate) and finally be semi-preparative RP-HPLC (Waters Symmetry® C18 5 μm, 7.8 mm × 100 mm column, gradient of CH3CN/H2O from 20:80 to 90:10 at a 3 mL/min flow rate), to furnish 5 mg (55%) of the desired azathilone 4 and 1.6 mg (18%) of starting material 4 both as white solids; Rf = 0.1 (CH2Cl2/MeOH 50:1). = −17.2° (c = 1.00, CH2Cl2). 1H-NMR (500 MHz, 318 K, DMSO-d6): δ = 8.88 (dd, J = 4.2 Hz, 1.7 Hz, 1H), 8.32 (dd, J = 8.4 Hz, 1.4 Hz, 1H), 8.00 (d, J = 5.9 Hz, 1H), 7.99 (s, 1H), 7.78 (dd, J = 8.7 Hz, 1.8 Hz, 1H), 7.53 (dd, J = 8.2 Hz, 4.1 Hz, 1H), 5.91 (m, 1H), 5.27 (d, J = 7.5 Hz, 1H, OH), 4.27 (d, J = 6.7 Hz, 1H, OH), 4.22 (m, 1H), 3.43 (m, 1H), 3.35 (m, 1H), 3.23 (m, 2H), 3.09 (m, 1H), 2.98 (m, 1H), 2.40 (dd, J = 14.5 Hz, 10.7 Hz, 1H), 2.11 (m, 2H), 1.56 (m, 1H), 1.46 (m, 1H), 1.33 (s, 12H), 1.32–1.22 (m, 4H), 1.02 (d, J = 6.8 Hz, 3H), 0.94 (s, 3H), 0.90 (d, J = 6.8 Hz; 3H); 13C-NMR (125 MHz, 318 K, DMSO-d6): δ = 217.6, 170.4, 154.3, 150.3, 147.0, 138.5, 135.8, 128.8, 127.4, 127.3, 124.1, 121.5, 78.1, 73.8, 73.0, 71.3, 52.8, 46.9, 44.1, 42.1, 38.6, 35.9, 35.3, 27.9 (3 × CH3), 27.0, 24.4, 20.7, 19.8, 17.2, 13.8. Some of the signals in the carbon spectrum were hardly visible, due to peak broadening. IR (film): ῦ = 3390 (br), 2926 (br), 1733, 1684, 1467, 1420, 1366, 1290, 1253, 1148, 1026, 1010, 977, 837 cm−1. HRMS (ESIpos): calcd. for C32H46N2O7Na [M + Na]+: 593.3197, found: 593.3196.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}