
Design, Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Selective PI3Kβ Inhibitors

Abstract
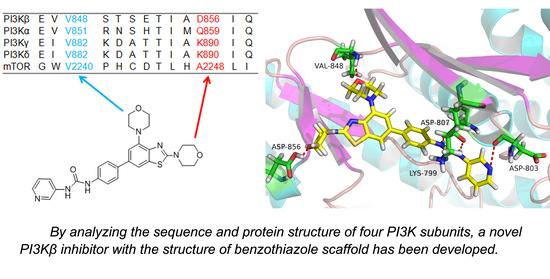
:
1. Introduction
2. Results and Discussion
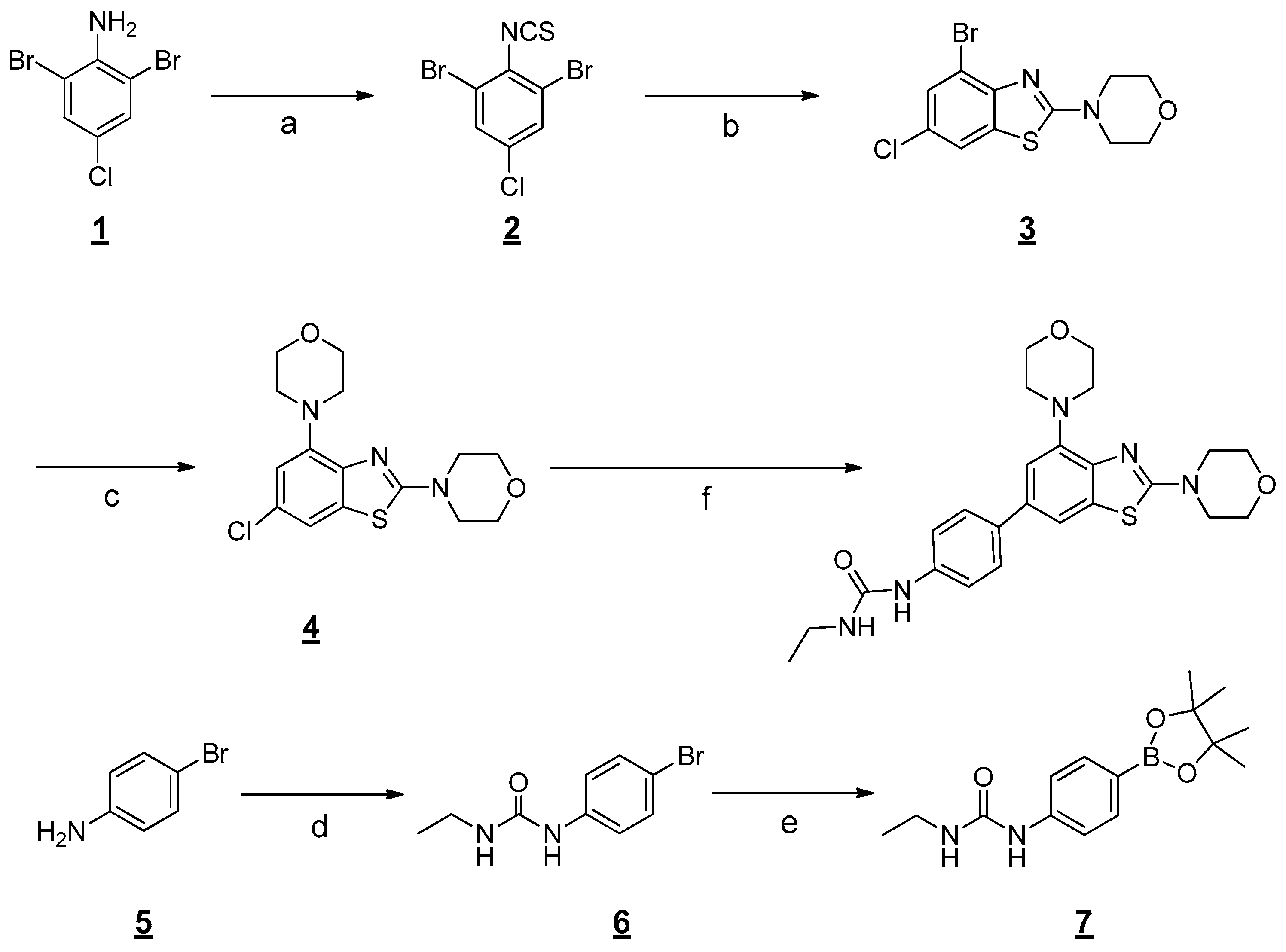
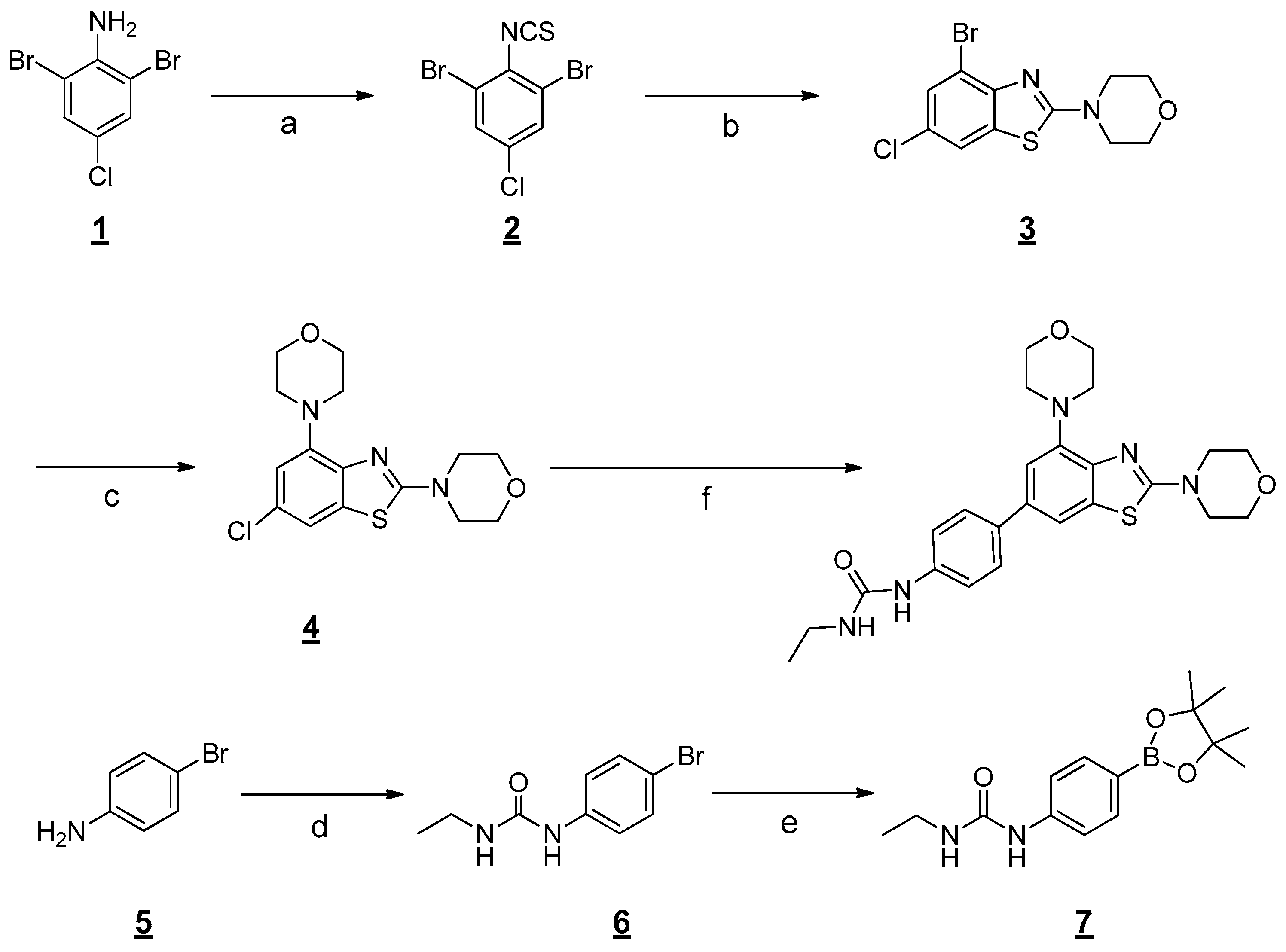
2.1. Chemistry
2.2. Biological Discussion
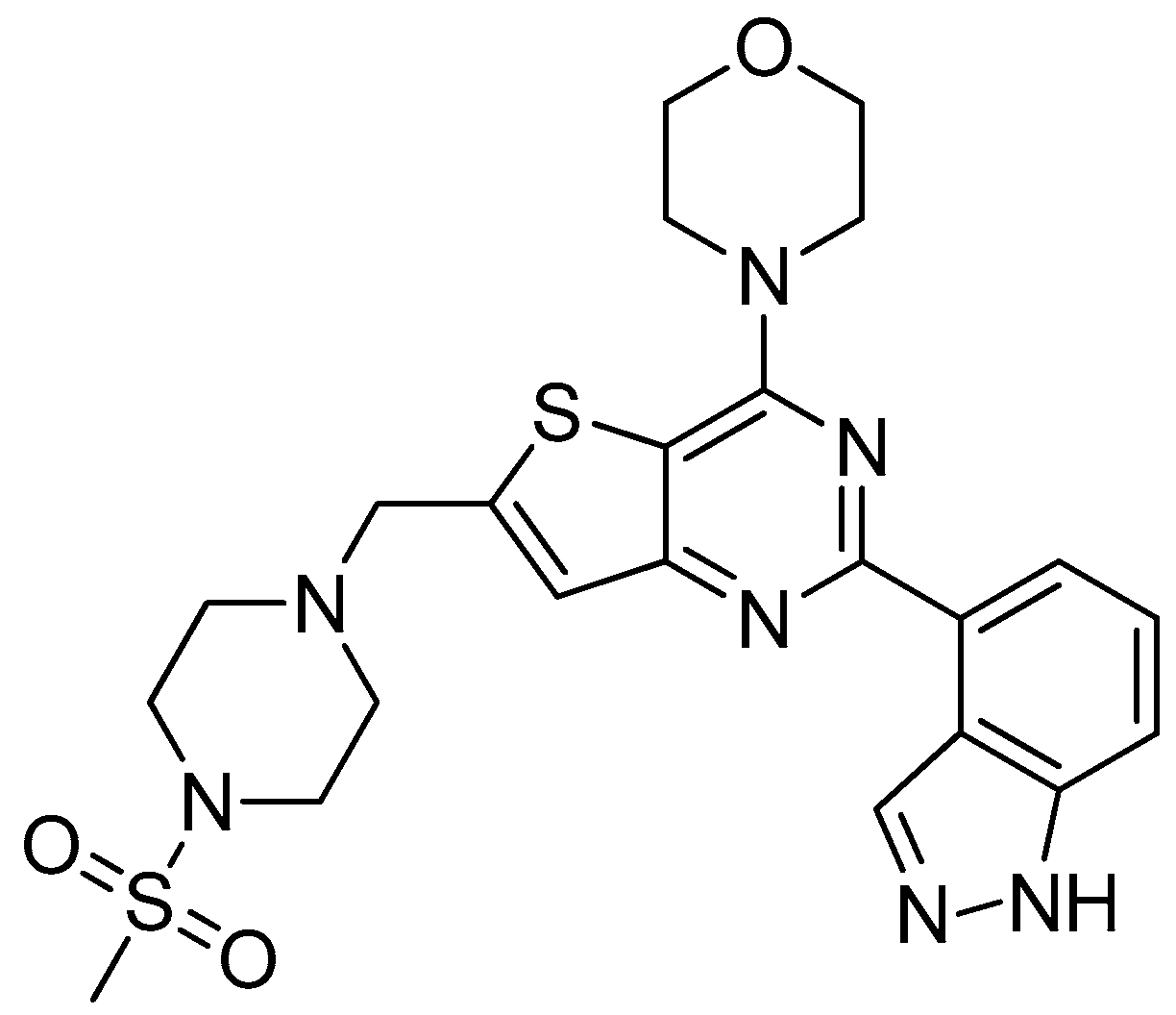
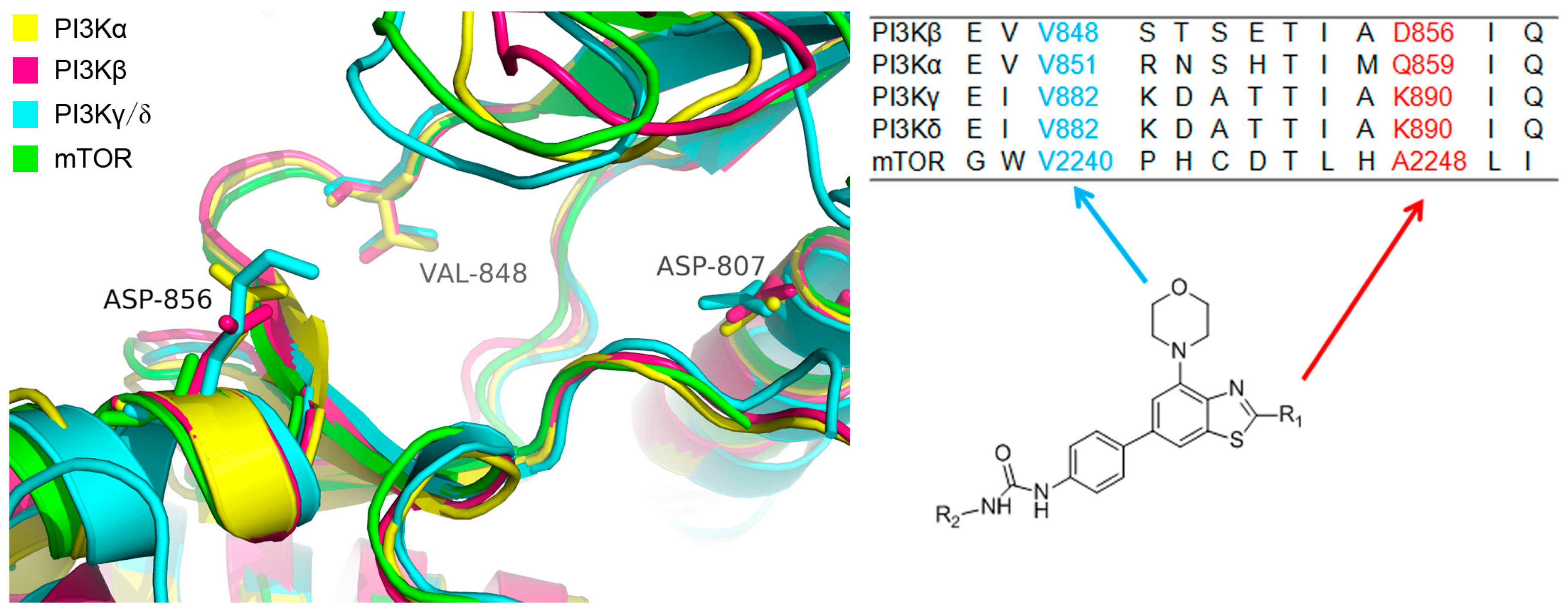
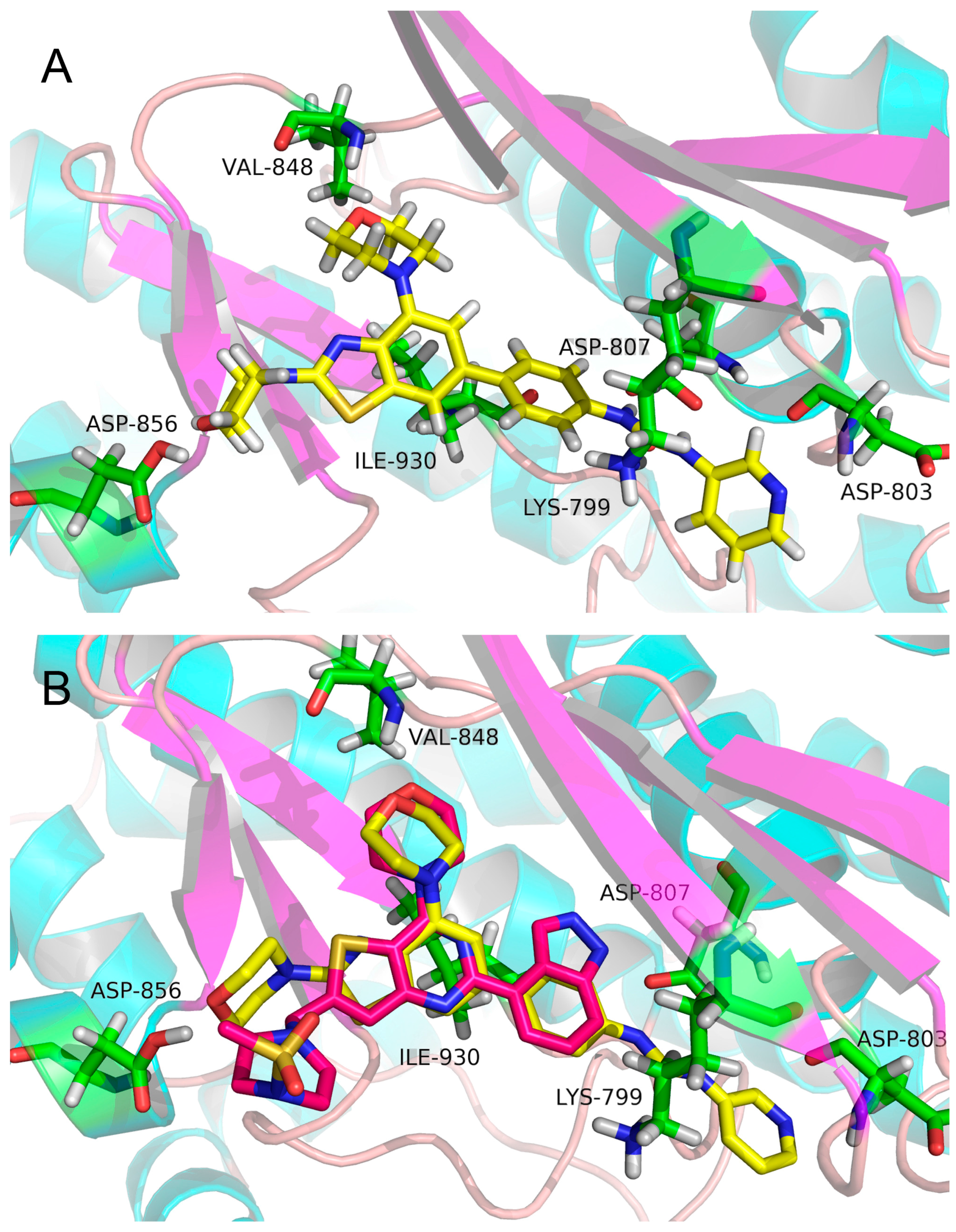
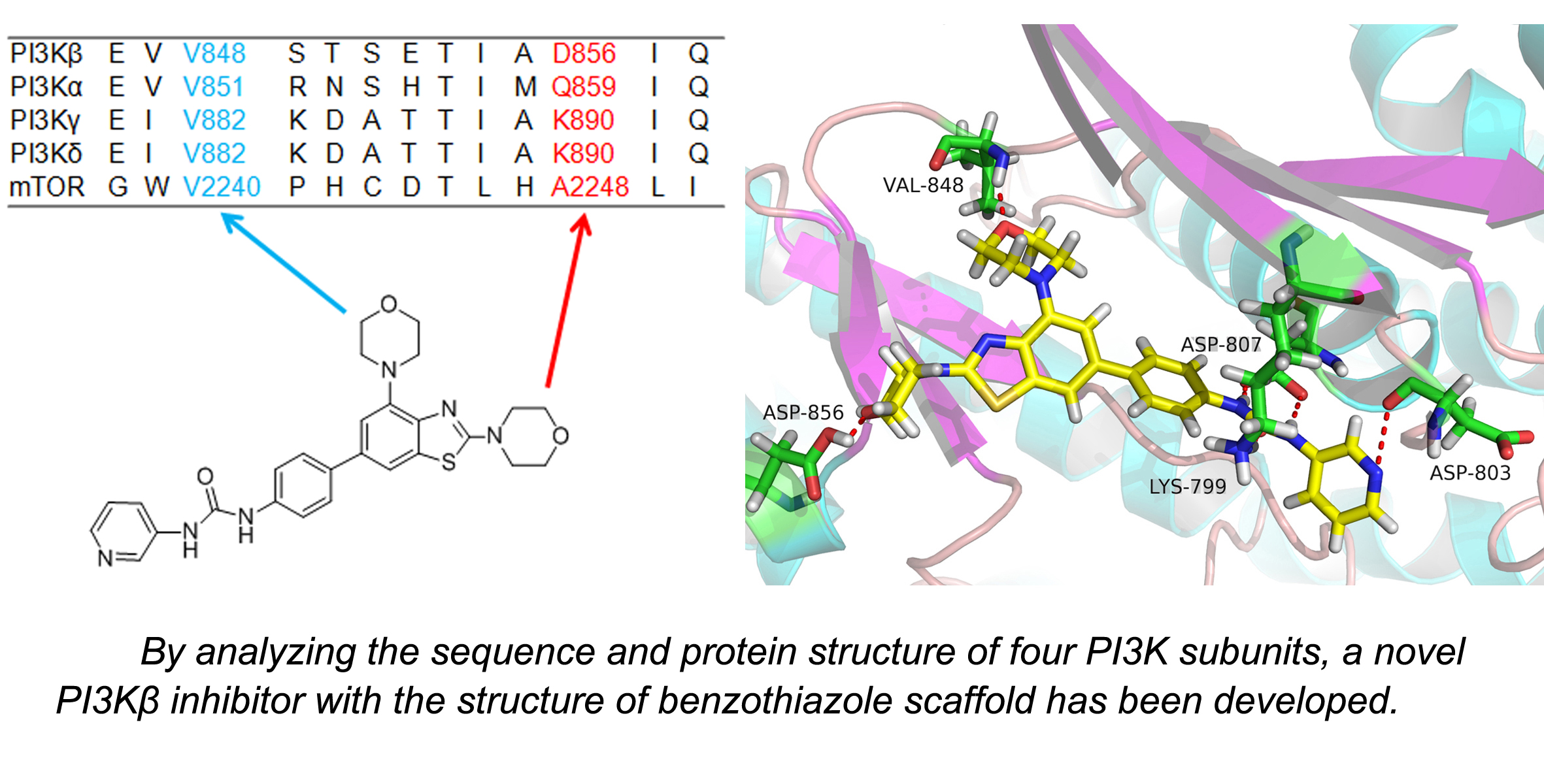
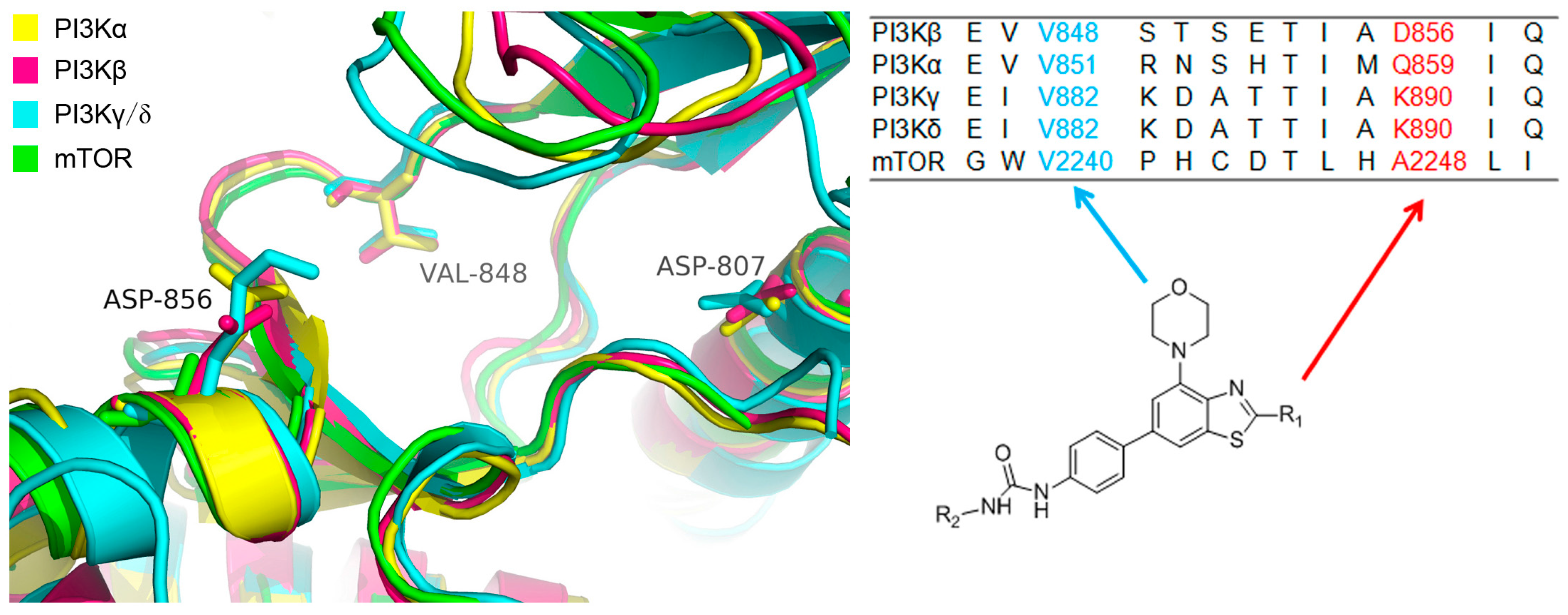
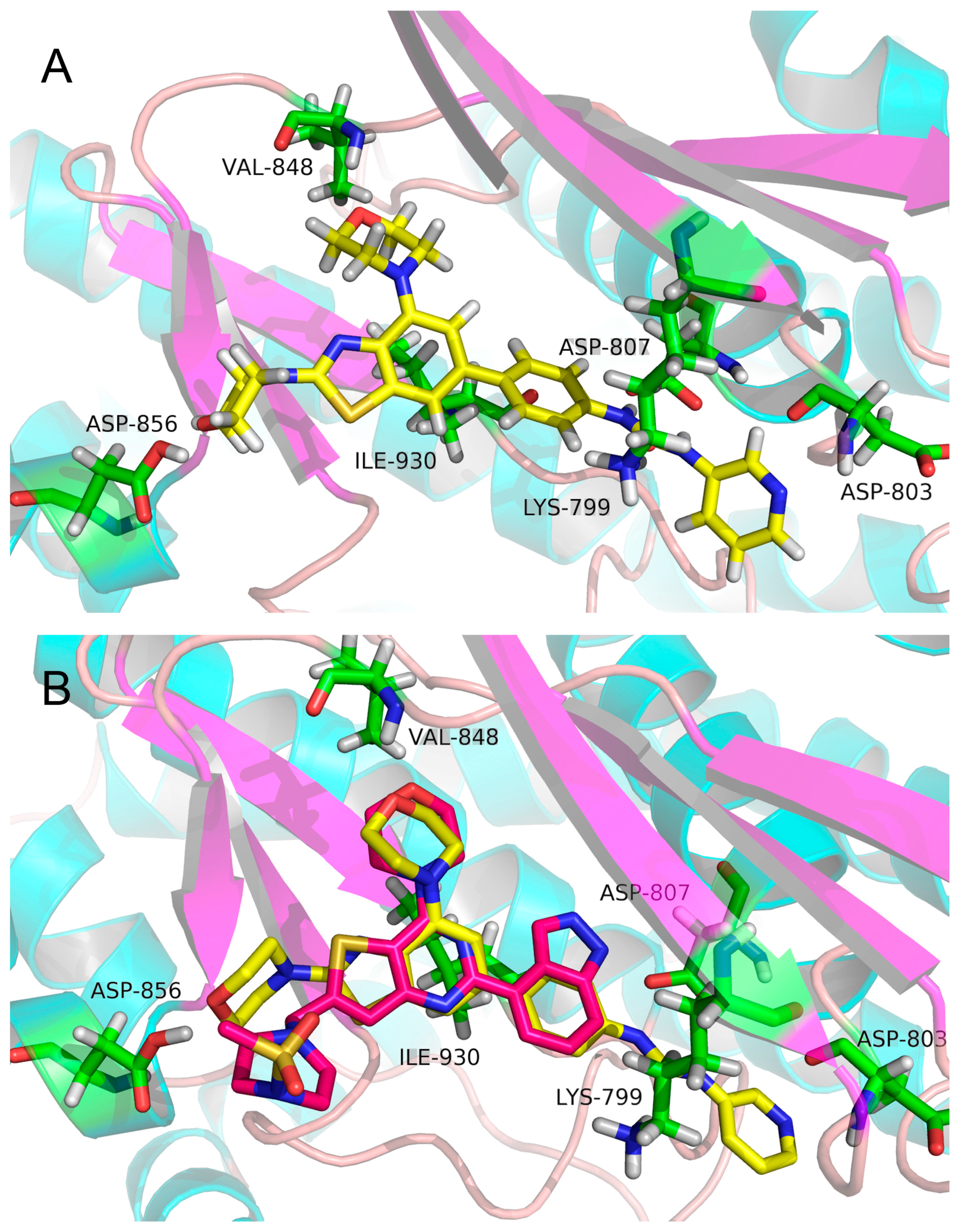
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
General procedure for the preparation of 1-(4-(2,4-Dimorpholinobenzo[d]thiazol-6-yl)phenyl)-3-ethylurea (1)
Step 1: Preparation of 1,3-Dibromo-5-chloro-2-isothiocyanatobenzene
Step 2: Preparation of 4-(4-Bromo-6-chlorobenzo[d]thiazol-2-yl)morpholine
Step 3: Preparation of 4,4′-(6-Chlorobenzo[d]thiazole-2,4-diyl)dimorpholine.
Step 4: Preparation of 1-(4-(2,4-Dimorpholinobenzo[d]thiazol-6-yl)phenyl)-3-ethylurea
3.2. Pharmacology
3.2.1. Class I PI3Ks Enzyme Assay
3.2.2. mTOR Enzyme Assay
3.2.3. Anti-Proliferative Assay
3.3. Molecular Docking Study
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PI3K | Phosphatidylinositol-3-kinases |
| mTOR | Mammalian target of rapamycin |
| PTEN | Phosphatase and tensin homolog deleted on chromosome ten |
| DNA-PK | DNA-dependent protein kinase |
| ATM | Ataxia-telangiectasia mutated |
| ATR | ATM- and Rad3-related |
| R-BINAP | (R)-(+)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| EGTA | Ethylene glycol tetraacetic acid |
| EDTA | Ethylene diamine tetraacetic acid |
| DTT | Dithiothreitol |
| TR-FRET | Time-resolved fluorescence resonance energy transfer |
| RLU | relative light unit |
References
- Fruman, D.A.; Rommel, C. Pi3k and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sun, A.; Jiang, W.; Thrasher, J.B.; Terranova, P. Pi-3 kinase p110beta: A therapeutic target in advanced prostate cancers. Am. J. Clin. Exp. Urol. 2014, 2, 188–198. [Google Scholar] [PubMed]
- Giordanetto, F.; Wållberg, A.; Ghosal, S.; Iliefski, T.; Cassel, J.; Yuan, Z.-Q.; von Wachenfeldt, H.; Andersen, S.M.; Inghardt, T.; Tunek, A.; et al. Discovery of phosphoinositide 3-kinases (pi3k) p110β isoform inhibitor 4-[2-hydroxyethyl(1-naphthylmethyl)amino]-6-[(2S)-2-methylmorpholin-4-yl]-1H-pyrimidin-2-one, an effective antithrombotic agent without associated bleeding and insulin resistance. Bioorg. Med. Chem. Lett. 2012, 22, 6671–6676. [Google Scholar] [CrossRef] [PubMed]
- Certal, V.; Halley, F.; Virone-Oddos, A.; Delorme, C.; Karlsson, A.; Rak, A.; Thompson, F.; Filoche-Romme, B.; El-Ahmad, Y.; Carry, J.C.; et al. Discovery and optimization of new benzimidazole- and benzoxazole-pyrimidone selective pi3kbeta inhibitors for the treatment of phosphatase and tensin homologue (pten)-deficient cancers. J. Med. Chem. 2012, 55, 4788–4805. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Kull, B.; Bjorkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skarby, T.; Inghardt, T.; Fjellstrom, O.; et al. Human target validation of phosphoinositide 3-kinase (pi3k)beta: Effects on platelets and insulin sensitivity, using azd6482 a novel pi3kbeta inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Poulogiannis, G.; Pyne, S.; Jia, S.; Zou, L.; Signoretti, S.; Loda, M.; Cantley, L.C.; Roberts, T.M. A constitutively activated form of the p110beta isoform of pi3-kinase induces prostatic intraepithelial neoplasia in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 11002–11007. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Giordanetto, F.; Green, S.; Inghardt, T.; Hennequin, L.; Hancox, U.; Lambert-van der Brempt, C.; et al. Discovery of 9-(1-anilinoethyl)-2-morpholino-4-oxo-pyrido[1,2-a]pyrimidine-7-carboxamides as pi3kbeta/delta inhibitors for the treatment of pten-deficient tumours. Bioorg. Med. Chem. Lett. 2014, 24, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Certal, V.; Carry, J.C.; Halley, F.; Virone-Oddos, A.; Thompson, F.; Filoche-Romme, B.; El-Ahmad, Y.; Karlsson, A.; Charrier, V.; Delorme, C.; et al. Discovery and optimization of pyrimidone indoline amide pi3kbeta inhibitors for the treatment of phosphatase and tensin homologue (pten)-deficient cancers. J. Med. Chem. 2014, 57, 903–920. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Liu, Q.; Xie, S.; Carlson, C.; Von, T.; Vogel, K.; Riddle, S.; Benes, C.; Eck, M.; Roberts, T.; et al. Functional characterization of an isoform-selective inhibitor of pi3k-p110beta as a potential anticancer agent. Cancer Discov. 2012, 2, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Inhibitors of pi3kbeta as potential treatment for cancer. ACS Med. Chem. Lett. 2013, 4, 815–816. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, G.; Cao, X.; Luo, X.; Li, Z.; Deng, Y.; Li, X.; Wang, S.; Liu, M.; Hu, J.; et al. Down-regulation of p110beta expression increases chemosensitivity of colon cancer cell lines to oxaliplatin. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Kim, K.S.; Moon, J.S.; Song, J.A.; Choi, S.H.; Kim, K.I.; Kim, T.H.; An, H.J. Targeted inhibition of phosphatidyl inositol-3-kinase p110beta, but not p110alpha, enhances apoptosis and sensitivity to paclitaxel in chemoresistant ovarian cancers. Apoptosis 2013, 18, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (gdc-0941) as a potent, selective, orally bioavailable inhibitor of class i pi3 kinase for the treatment of cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef] [PubMed]
- Verheijen, J.C.; Richard, D.J.; Curran, K.; Kaplan, J.; Lefever, M.; Nowak, P.; Malwitz, D.J.; Brooijmans, N.; Toral-Barza, L.; Zhang, W.-G.; et al. Discovery of 4-morpholino-6-aryl-1H-pyrazolo[3,4-d]pyrimidines as highly potent and selective atp-competitive inhibitors of the mammalian target of rapamycin (mtor): Optimization of the 6-aryl substituent. J. Med. Chem. 2009, 52, 8010–8024. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Chen, C.; Sun, C.; Xu, S.; Wu, C.; Lei, F.; Xia, H.; Tu, Q.; Zheng, P. Design, synthesis and docking studies of novel thienopyrimidine derivatives bearing chromone moiety as mtor/pi3kalpha inhibitors. Eur. J. Med. Chem. 2015, 93, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ma, H.; Sun, Z.; Luo, L.; Tian, S.; Zheng, J.; Zhang, X. Discovery of a 6-(pyridin-3-yl)benzo[d]thiazole template for optimization of hedgehog and pi3k/akt/mtor dual inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3665–3670. [Google Scholar] [CrossRef] [PubMed]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of molecular lipophilicity: State-of-the-art and comparison of log p methods on more than 96,000 compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Barlaam, B.; Berglund, S.; Edman, K.; Karlsson, O.; Lindberg, J.; Nylander, S.; Inghardt, T. Discovery of 9-(1-phenoxyethyl)-2-morpholino-4-oxo-pyrido[1,2-a]pyrimidine-7-carboxamides as oral pi3kβ inhibitors, useful as antiplatelet agents. Bioorg. Med. Chem. Lett. 2014, 24, 3936–3943. [Google Scholar] [CrossRef] [PubMed]
- Cushing, T.D.; Hao, X.; Shin, Y.; Andrews, K.; Brown, M.; Cardozo, M.; Chen, Y.; Duquette, J.; Fisher, B.; Gonzalez-Lopez de Turiso, F.; et al. Discovery and in vivo evaluation of (S)-N-(1-(7-fluoro-2-(pyridin-2-yl)quinolin-3-yl)ethyl)-9H-purin-6-amine (amg319) and related pi3kdelta inhibitors for inflammation and autoimmune disease. J. Med. Chem. 2015, 58, 480–511. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Li, B.; Shao, J.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. A general and facile one-pot process of isothiocyanates from amines under aqueous conditions. Beilstein J. Org. Chem. 2012, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Banerjee, A.; Chakraborty, S.; Patel, B.K. Regioselective intramolecular arylthiolations by ligand free cu and pd catalyzed reaction. ACS Catal. 2012, 2, 544–551. [Google Scholar] [CrossRef]
- Verheijen, J.C.; Zask, A.; Verheijen, J.C.; Curran, K.; Kaplan, J.; Richard, D.J.; Nowak, P.; Malwitz, D.J.; Brooijmans, N.; Bard, J.; et al. Atp-competitive inhibitors of the mammalian target of rapamycin: Design and synthesis of highly potent and selective pyrazolopyrimidines. J. Med. Chem. 2009, 52, 5013–5016. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Ying, H.; Ma, X.; Qiu, N.; Wu, P.; Yang, B.; Hu, Y. Design, synthesis and biological evaluation of novel 4-alkynyl-quinoline derivatives as pi3k/mtor dual inhibitors. Eur. J. Med. Chem. 2015, 99, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.

| Compd. | R1 | R2 | % Inhib. 1 μM |
|---|---|---|---|
| 1 | 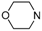 | ethyl | 52.1% |
| 2 | 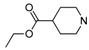 | ethyl | 23.0% |
| 3 | 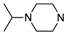 | ethyl | 11.7% |
| 4 | 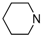 | ethyl | 17.6% |
| GDC-0941 | / | / | 74.5% |
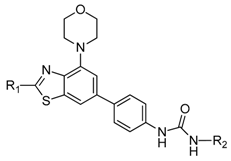
| Compd. | R1 | R2 | PI3Kβ, 1 μM, % |
|---|---|---|---|
| 1 | |  | 52.1% |
| 5 | | 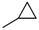 | 10.2% |
| 6 | | 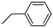 | 15.0% |
| 7 | | 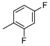 | 37.7% |
| 8 | |  | 27.2% |
| 9 | | 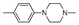 | 22.5% |
| 10 | | 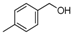 | 75.9% |
| 11 | | 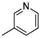 | 88.3% |
| GDC-0941 | / | / | 74.5% |
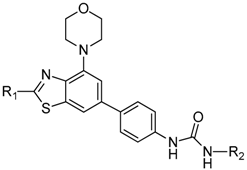
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50, μM | ||||
|---|---|---|---|---|---|
| PI3Kβ | PI3Kα | PI3Kγ | PI3Kδ | mTOR | |
| 10 | 0.12 | >50 | >50 | 15.31 | >50 |
| 11 | 0.02 | 5.43 | 7.52 | 4.01 | 39.85 |
| GDC-0941 | 0.15 | 0.01 | 0.27 | 0.01 | 1.70 |
| Compd. | IC50, μM | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A549 | MCF7 | SKOV3 | AGS | MES-SA | HepG2 | SW-620 | PC-3 | DU145 | MRC5 | |
| 1 | >100 | 4.06 | 21.23 | 9.69 | 12.31 | >100 | 8.31 | 15.32 | 9.50 | 6.06 |
| 2 | >100 | 4.04 | >100 | >100 | >100 | >100 | >100 | 85.00 | >100 | >100 |
| 3 | 2.27 | 6.53 | 3.76 | 15.35 | 3.92 | 6.20 | 10.93 | 25.23 | 71.69 | 3.53 |
| 4 | 14.77 | 6.58 | 5.59 | 12.16 | 21.85 | >100 | >100 | 33.50 | >100 | 83.47 |
| 5 | 14.39 | 40.78 | 26.86 | 64.94 | 33.15 | >100 | 30.78 | >100 | >100 | 38.50 |
| 6 | 5.67 | 19.94 | 8.85 | 46.97 | 24.90 | >100 | 41.25 | >100 | >100 | 16.52 |
| 7 | 3.72 | 14.17 | 18.92 | 36.60 | 6.49 | 15.83 | >100 | 14.17 | 22.98 | 28.26 |
| 8 | 7.53 | 11.55 | 20.43 | >100 | >100 | 9.83 | >100 | 25.19 | 17.53 | 35.69 |
| 9 | 32.69 | >100 | 35.20 | 37.92 | 75.29 | >100 | 82.65 | 42.40 | 38.55 | >100 |
| 10 | 84.96 | 0.75 | 0.63 | 1.78 | 0.72 | 2.70 | 5.39 | 5.74 | 2.95 | 1.19 |
| 11 | 3.48 | 0.36 | 2.64 | 0.40 | 0.55 | 3.43 | 1.17 | 0.35 | 0.62 | 33.11 |
| GDC-0941 | 6.91 | 0.89 | 0.30 | 3.62 | 0.23 | 1.10 | 3.27 | 5.92 | 1.41 | 20.72 |
| Compd. | Property of Absorption and Metabolism | ||
|---|---|---|---|
| MlogP a | Absn Risk b | CYP Risk c | |
| 11 | 1.8 | 2.8 | 1.5 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, S.; Cao, R.; Liu, X.; Luo, X.; Zhong, W. Design, Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Selective PI3Kβ Inhibitors. Molecules 2016, 21, 876. https://doi.org/10.3390/molecules21070876
Cao S, Cao R, Liu X, Luo X, Zhong W. Design, Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Selective PI3Kβ Inhibitors. Molecules. 2016; 21(7):876. https://doi.org/10.3390/molecules21070876
Chicago/Turabian StyleCao, Shuang, Ruiyuan Cao, Xialing Liu, Xiang Luo, and Wu Zhong. 2016. "Design, Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Selective PI3Kβ Inhibitors" Molecules 21, no. 7: 876. https://doi.org/10.3390/molecules21070876
APA StyleCao, S., Cao, R., Liu, X., Luo, X., & Zhong, W. (2016). Design, Synthesis and Biological Evaluation of Novel Benzothiazole Derivatives as Selective PI3Kβ Inhibitors. Molecules, 21(7), 876. https://doi.org/10.3390/molecules21070876