
In Silico Mining for Antimalarial Structure-Activity Knowledge and Discovery of Novel Antimalarial Curcuminoids


 ,
, 

Abstract
:
1. Introduction
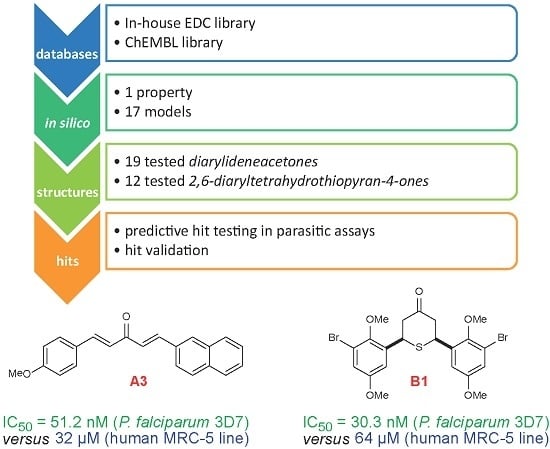
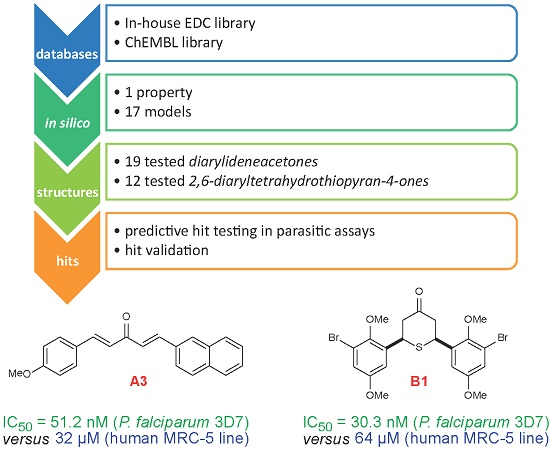
- (a)
- Collection and curation of experimental data
- (b)
- Knowledge extraction, by construction of structure-activity relationship models
- (c)
- Virtual screening of candidate collections and selection of candidates with best predicted properties using the above built models
- (d)
- Experimental in vitro testing of selected candidates
2. Results
3. Discussion
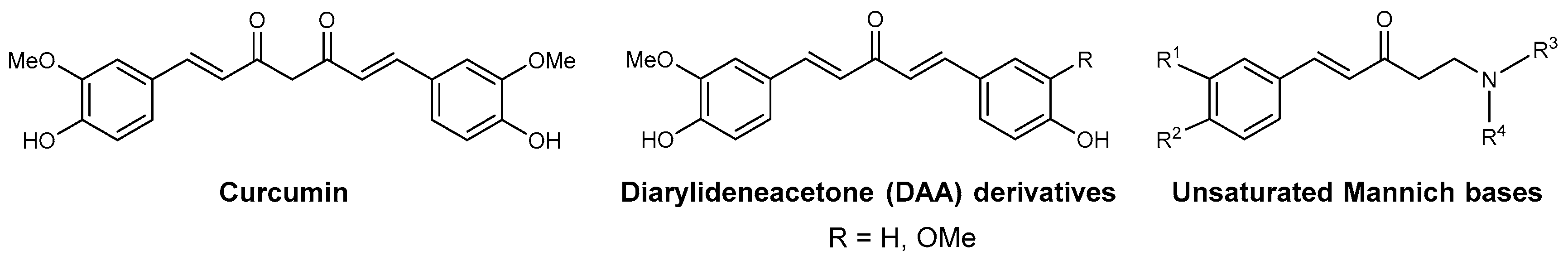
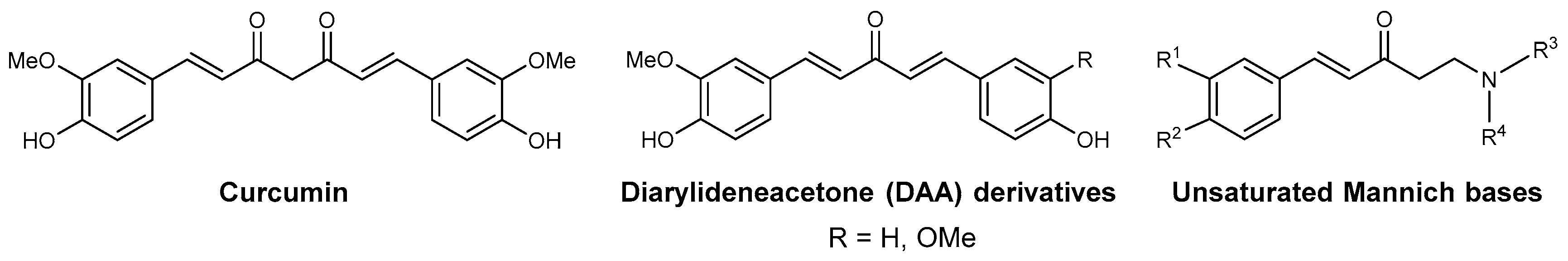
3.1. Known Antimalarial Curcuminoids and Unsaturated/Phenolic Mannich Bases
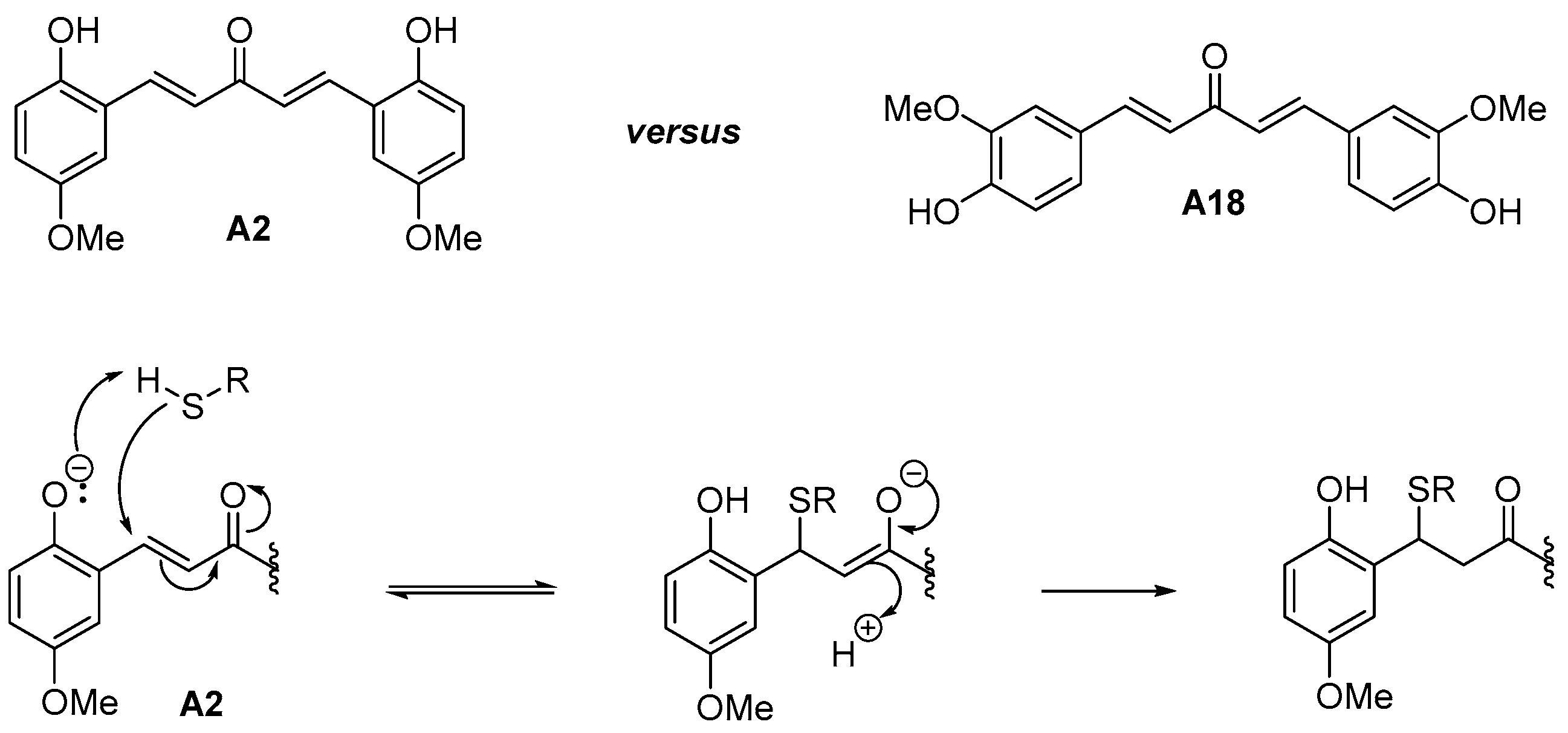
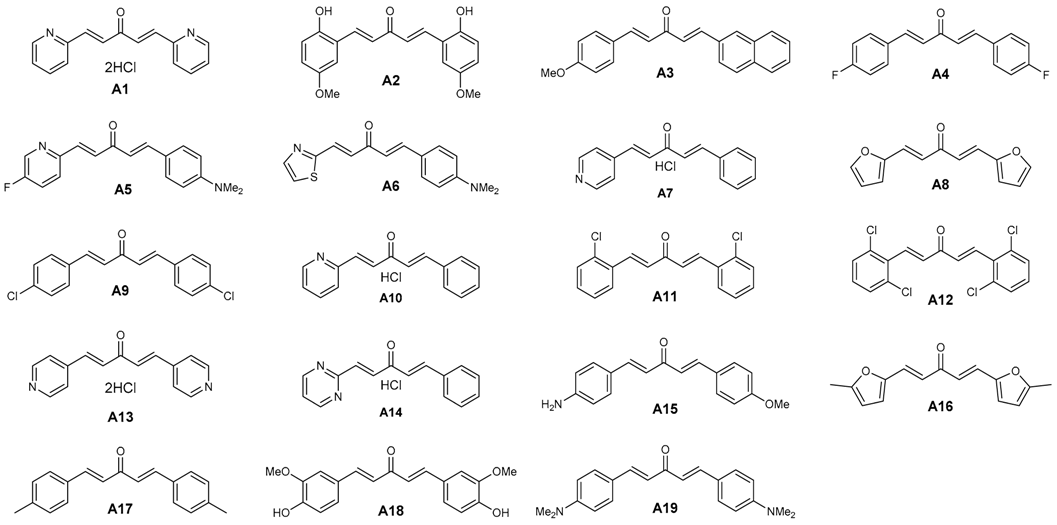
3.2. Compound Class Selection and Organic Synthesis
3.3. Virtual Screening of Candidate Collections Using the SVM Classification Consensus Models and the Selection of Candidates with the Best Predicted Properties
3.4. Experimental in Vitro Testing of the Selected Candidates and Structure-Activity Relationships
4. Materials and Methods
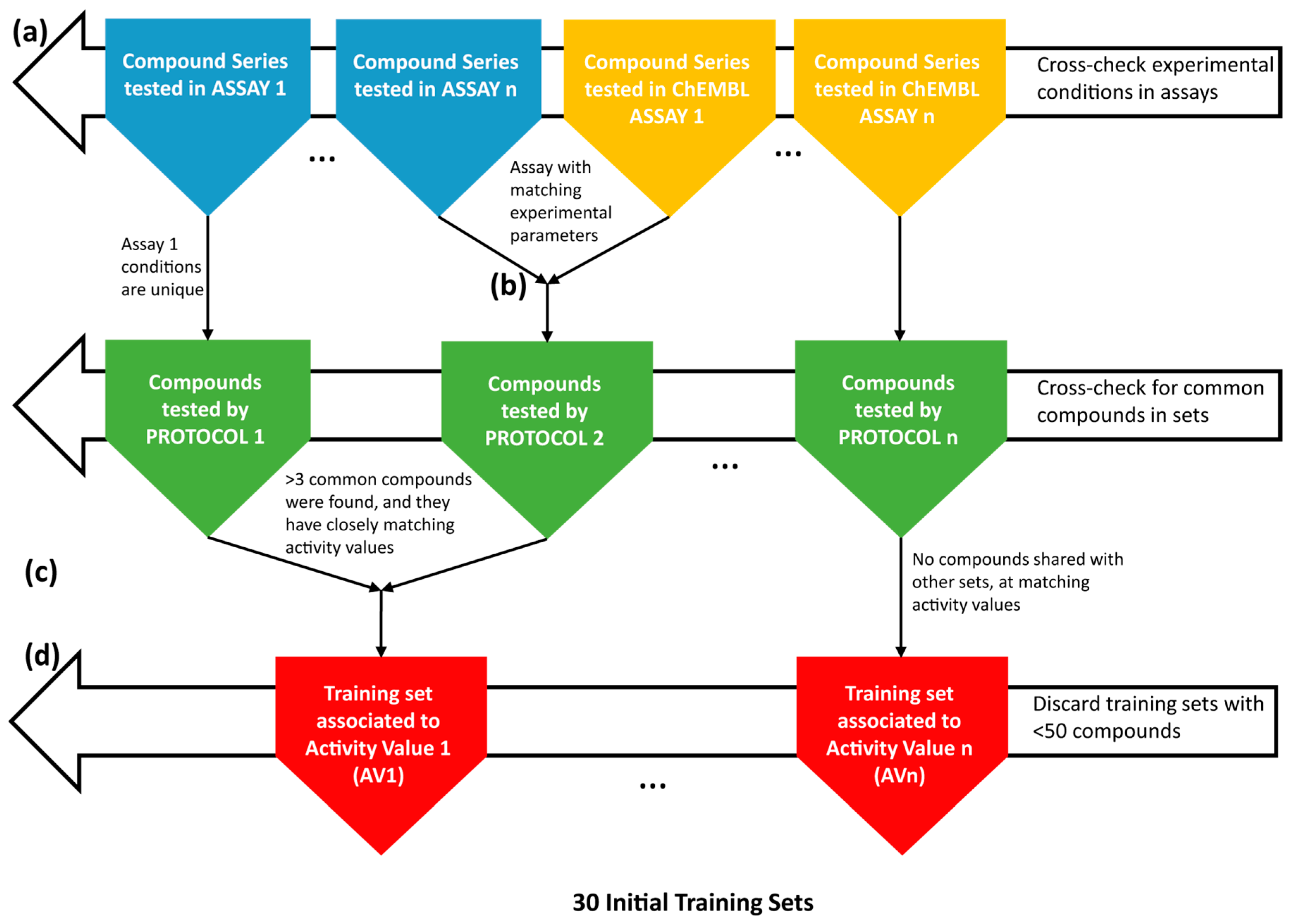
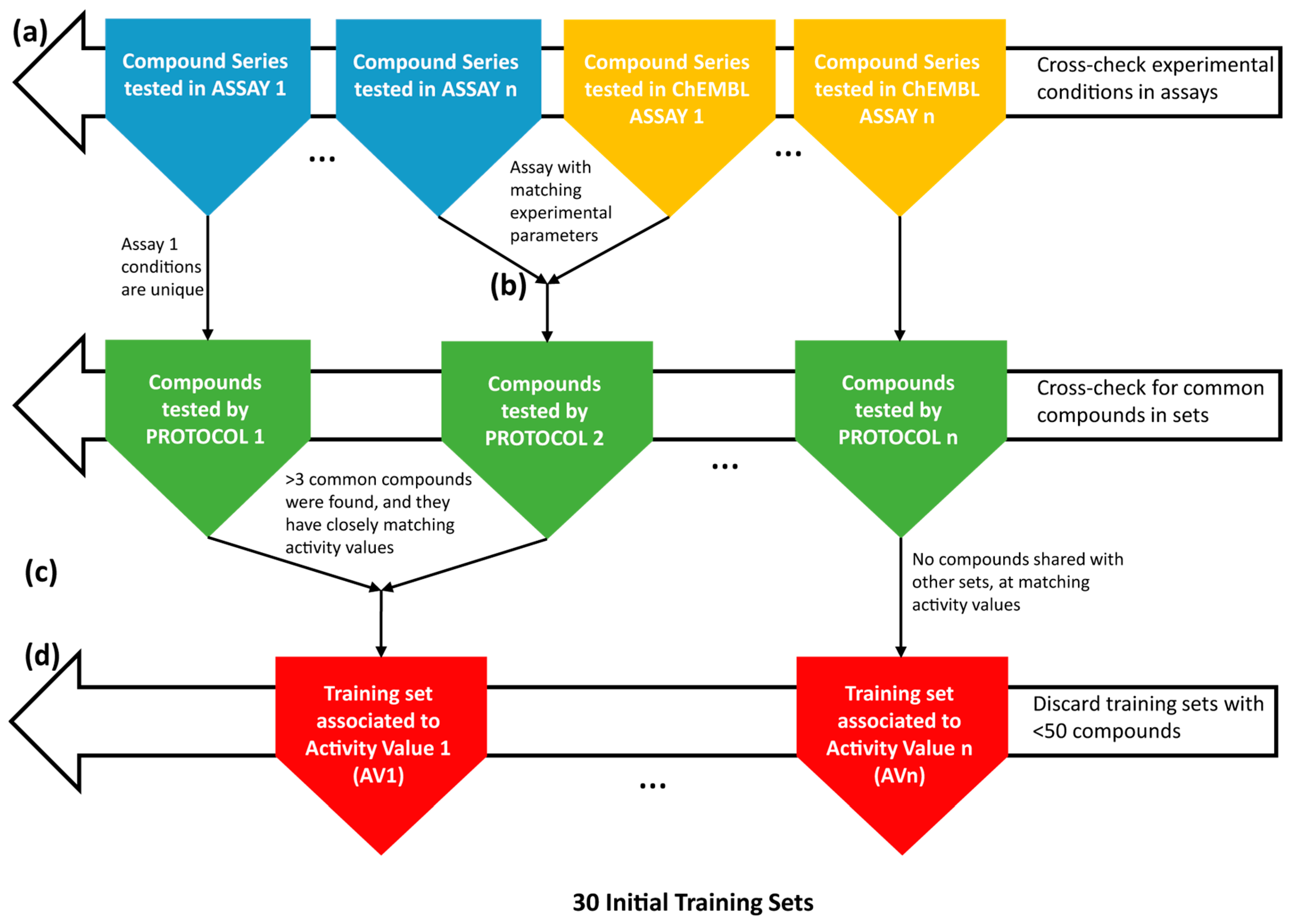
4.1. Collection and Curation of the Experimental Data
4.2. Knowledge Extraction by the Construction of Structure-Activity Models
4.3. Virtual Screening of Candidate Collections and the Selection of Candidates with the Best Predicted Properties Using the Above-Built Models
4.4. Experimental Testing of Selected Candidates
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACT | artemisinin-based combination therapy |
| AV | activity value |
| DAA | diarylideneacetones |
| DMSO | dimethylsulfoxide |
| 2,6-DATHTP | 2,6-diaryltetrahydrothiopyran-4-ones |
| GR | glutathione reductase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| ROS | reactive oxygen species |
| SI | selectivity index |
| SVM | support vector machine |
| TrxR | thioredoxin reductase |
| TrxS2 | oxidized thioredoxin |
References
- Egan, T.J. Physico-chemical aspects of hemozoin (malaria pigment) structure and formation. J. Inorg. Biochem. 2002, 91, 19–26. [Google Scholar] [CrossRef]
- Hempelmann, E.; Egan, T.J. Pigment biocrystallization in Plasmodium falciparum. Trends. Parasitol. 2002, 18. [Google Scholar] [CrossRef]
- De Villiers, K.A.; Egan, T.J. Recent advances in the discovery of haem-targeting drugs for malaria and schistosomiasis. Molecules. 2009, 14, 2868–2887. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Rahlfs, S.; Nickel, C.; Schirmer, R.H. Glutathione—Functions and metabolism in the malarial parasite Plasmodium falciparum. Biol. Chem. 2003, 384, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Krauth-Siegel, R.L.; Leroux, A.E. Low-molecular-mass antioxidants in parasites. Antioxid. Redox. Signal. 2012, 17, 583–607. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, M.; Diederich, K.; Becker, K.; Krauth-Siegel, R.L.; Jortzik, E. Detection of thiol-based redox switch processes in parasites—Facts and future. Biol. Chem. 2015, 396, 445–463. [Google Scholar] [CrossRef] [PubMed]
- Mohring, F.; Pretzel, J.; Jortzik, E.; Becker, K. The redox systems of Plasmodium falciparum and Plasmodium vivax: Comparison, in silico analyses and inhibitor studies. Curr Med Chem. 2014, 21, 1728–1756. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, H.; Famin, O.; Zhang, J.; Krugliak, M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 1998, 56, 1305–1313. [Google Scholar] [CrossRef]
- Ginsburg, H.; Golenser, J. Glutathione is involved in the antimalarial action of chloroquine and its modulation affects drug sensitivity of human and murine species of Plasmodium. Redox Rep. 2003, 8, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Patzewitz, E.M.; Salcedo-Sora, J.E.; Wong, E.H.; Sethia, S.; Stocks, P.A.; Maughan, S.C.; Murray, J.A.; Krishna, S.; Bray, P.G.; Ward, S.A.; et al. Glutathione transport: A new role for PfCRT in chloroquine resistance. Antioxid. Redox. Signal. 2013, 19, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Müller, S. Role and Regulation of Glutathione Metabolism in Plasmodium falciparum. Molecules 2015, 20, 10511–10534. [Google Scholar] [CrossRef] [PubMed]
- Kanzok, S.M.; Schirmer, R.H.; Turbachova, I.; Iozef, R.; Becker, K. The thioredoxin system of the malaria parasite Plasmodium falciparum—Glutathione reduction revisited. J. Biol. Chem. 2000, 275, 40180–40186. [Google Scholar] [CrossRef] [PubMed]
- Sturm, N.; Jortzik, E.; Mailu, B.M.; Koncarevic, S.; Deponte, M.; Forchhammer, K.; Rahlfs, S.; Becker, K. Identification of proteins targeted by the thioredoxin superfamily in Plasmodium falciparum. PLoS. Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Krnajski, Z.; Gilberger, T.W.; Walter, R.D.; Cowman, A.F.; Muller, S. Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J. Biol. Chem. 2002, 277, 25970–25975. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Akoachere, M.B.; Krogh, R.; Nickel, C.; McLeish, M.J.; Kenyon, G.L.; Arscott, L.D.; Williams, C.H., Jr.; Davioud-Charvet, E.; Becker, K. Specific inhibitors of Plasmodium falciparum thioredoxin reductase as potential antimalarial agents. Bioorg Med Chem Lett. 2006, 16, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Davioud-Charvet, E.; McLeish, M.J.; Veine, D.; Giegel, D.; Andricopulo, A.D.; Becker, K.; Müller, S.; Schirmer, R.H.; Williams, C.H., Jr.; Kenyon, G.L. Mechanism-based inactivation of thioredoxin reductase from Plasmodium falciparum by Mannich Bases. Implications of drug design. In Flavins and Flavoproteins 2002; Chapman, S.K., Perham, R.N., Scrutton, N.S., Eds.; Agency for Scientific Publications: Berlin, Germany, 2002; pp. 845–851. [Google Scholar]
- Davioud-Charvet, E.; McLeish, M.J.; Veine, D.; Giegel, D.; Arscott, L.D.; Adricopulo, A.D.; Becker, K.; Müller, S.; Schirmer, R.H.; Williams, C.H., Jr.; et al. Mechanism-based inactivation of thioredoxin reductase from Plasmodium falciparum by Mannich bases. Implication for cytotoxicity. Biochemistry 2003, 42, 13319–13330. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Lu, J.; Holmgren, A. Thioredoxin reductase is irreversibly modified by curcumin: A novel molecular mechanism for its anticances activity. J. Biol. Chem. 2005, 280, 25284–25290. [Google Scholar] [CrossRef] [PubMed]
- Morin, C.; Besset, T.; Moutet, J.C.; Fayolle, M.; Brückner, M.; Limosin, D.; Becker, K.; Davioud-Charvet, E. The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioreoxin and glutathione reductases and of human erythrocyte glutathione reductase. Org. Biomol. Chem. 2008, 6, 2731–2742. [Google Scholar] [CrossRef] [PubMed]
- Urig, S.; Fritz-Wolf, K.; Réau, R.; Herold-Mende, C.; Tóth, K.; Davioud-Charvet, E.; Becker, K. Undressing of phosphine gold(I) complexes as irreversible inhibitors of human disulfide reductases. Angew. Chem. Int. Ed. Engl. 2006, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Viry, E.; Battaglia, E.; Deborde, V.; Müller, T.; Réau, R.; Davioud-Charvet, E.; Bagrel, D. A sugar-modified phosphole fold complex with antipoliferative properties acting as a thioredoxin reductase inhibitor in MCF-7 cells. Chem. Med. Chem. 2008, 3, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Gandhy, S.U.; Kim, K.; Larsen, L.; Rosengren, R.J.; Safe, S. Curcumin and synthetic analogs induce reactive oxygen species and decreases specificity protein (Sp) transcription factors by targeting microRNAs. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Noratto, G.D.; Jutooru, I.; Safe, S.; Angel-Morales, G.; Mertens-Talcott, S.U. The drug resistance suppression induced by curcuminoids in colon cancer SW-480 cells is mediated by reactive oxygen species-induced disruption of the microRNA-27a-ZBTB10-Sp axis. Mol. Nutr. Food. Res. 2013, 57, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Munigunti, R.; Gathiaka, S.; Acevedo, O.; Sahu, R.; Tekwani, B.; Calderón, A.I. Determination of antiplasmodial activity and binding affinity of curcumin and demethoxycurcumin towards PfTrxR. Nat. Prod. Res. 2014, 28, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Mimche, P.N.; Thompson, E.; Taramelli, D.; Vivas, L. Curcumin enhances non-opsonic phagocytosis of Plasmodium falciparum through up-regulation of CD36 surface expression on monocytes/macrophages. J. Antimicrob. Chemother. 2012, 67, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, D.N.; Nagaraj, V.A.; Vathsala, P.G.; Rangarajan, P.; Padmanaban, G. Curcumin-artemisin combination therapy for malaria. Antimicrob. Agents. Chemother. 2006, 50, 1859–1860. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.; Davioud-Charvet, E.; Thomas, J.J.M. Versatile Synthesis of Dissymetric Diarylideneacetones via a Palladium-Catalyzed Coupling-Isomeration Reaction. Synthesis 2012, 44, 3829–3835, Erratum: Synthesis 2013, 45, 1270. [Google Scholar]
- Gendron, T.; Kessedjian, H.; Davioud-Charvet, E.; Lanfranchi, D.A. Diastereoselective synthesis of 2,6-diaryltetrahydrothiopyran-4-ones by phase-transfer catalysis. Eur. J. Org. Chem. 2015, 8, 1790–1796. [Google Scholar] [CrossRef]
- Davioud-Charvet, E.; Wenzel, I.N.; Müller, T.J.J.; Hanquet, G.; Lanfranchi, D.A.; Leroux, F.; Gendron, T. Dibenzylidene- and heteroarylideneacetone derivatives as kinetoplastideae parasiticides and their preparation, pharmaceutical compositions and use in the treatment of trypanosomiasis and leishmaniasis. PCT Int. Appl. WO 2011033115 A2, 24 March 2011. [Google Scholar]
- Wenzel, N. Synthesis and Mechanism of Antiparasitic Mannich Base Derivatives Affecting the Redox Equilibrium of Trypanosomes and Malaria Parasites. Ph.D. Thesis, Heidelberg University, Heidelberg, Germamy, 21 September 2009. [Google Scholar]
- Gendron, T. Synthesis and Evaluation of the Antiparasitic Activity of Diarylideneacetones and Their Related Thiopyranone and S-Oxide Prodrugs. Ph.D. Thesis, Strasbourg University, Strasbourg, 23 November 2012. [Google Scholar]
- Aher, R.B.; Wanare, G.; Kawathekar, N.; Kumar, R.R.; Kaushik, N.K.; Sahal, D.; Chauhan, V.S. Dibenzylideneacetone analogues as novel Plasmodium falciparum inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3034–3036. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D; Marcou, G.; Varnek, A. Predicting the Predictability: A Unified Approach to the Applicability Domain Problem of QSAR Models. J. Chem. Inf. Model. 2009, 49, 1762–1776. [Google Scholar]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.A.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin-from molecule to biological function. Angew. Chem. Int. Ed. Engl. 2012, 51, 5308–5332. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Jitoe, A.; Isobe, J.; Nakatani, N.; Yonemori, S. Anti-oxidative and anti-inflammatory curcumin-related phenolics from rhizomes of Curcruma domestica. Phytochem. 1993, 32, 1557–1560. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Massiah, M.A.; Bozak, R.E.; Hicks, R.J.; Talalay, P. Potency of Micheal reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulhydryl groups. Proc. Natl. Acad. Sci. USA 2001, 98, 3404–3409. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.M.; Hunsaker, L.A.; Abcouwer, S.F.; Deck, L.M.; vander Jagt, D.L. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005, 13, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.E.; Benya, R.V.; Turgeon, D.K.; Vareed, S.; Neuman, M.; Rodriguez, L.; Kakarala, M.; Carpenter, P.M.; McLaren, C.; Meyskens, F.L.; et al. Phase IIa Clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer. Prev. Res. 2011, 4, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Zhou, H.; Yang, S.; Wu, X.; Jiang, C.; Zhao, Y.; Liang, D.; Li, X.; Liang, G. A novel monocharbonyl analogue of curcumin, (1E,4E)-1,5-Bis(2,3-dimethoxyphenyl)penta-1,4-dien-3-one, induced cancer cell H460 apoptosis via activation of endoplasmic reticulum stress signalling pathway. J. Med. Chem. 2011, 54, 3768–3778. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.C.; Vatsala, P.G.; Keshamouni, V.G.; Padmanab, G.; Rangarajan, P.N. Curcumin for malaria therapy. Biochem. Biophys. Res. Commun. 2005, 326, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Karmodiya, K.; Surolia, A. Synthesis and exploration of novel curcumin analogues as anti-malarial agents. Bioorg. Med. Chem. 2008, 16, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Singh, A.; Pathak, L.P.; Shrivastava, N.; Tripathi, P.K.; Singh, M.P.; Singh, K. Inhibition of P. falciparum PFATP6 by curcumin and its derivatives: A bioinformatics study. Cell. Mol. Biol. 2012, 58, 182–186. [Google Scholar] [PubMed]
- Manohar, S.; Khan, S.I.; Kandi, S.K.; Raj, K.; Sun, G.; Yang, X.; Calderon Molina, A.D.; Ni, N.; Wang, B.; Rawat, D.S. Synthesis, antimalarial activity and cytotoxic potential of new monocarbonyl analogues of curcumin. Bioorg. Med. Chem. Lett. 2013, 23, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.; Lanfranchi, D.A.; Davioud-Charvet, E. Redox-active agents in reactions involving the trypanothione/trypanothione reductase-based system to combat kinetoplastidal parasites. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery; Jäger, T., Koch, O., Flohė, L., Eds.; Wiley-Blackwell VCH, Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; Volume 4, pp. 405–428. [Google Scholar]
- Burckhalter, J.H.; Tendick, F.H.; Jones, E.M.; Jones, P.A.; Holcomb, W.F.; Rawlins, A.L. Aminoalkylphenols as antimalarials (heterocyclicamino)-α-amino-O-cresols; the synthesis of camoquin. J. Am. Chem. Soc. 1948, 70, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.; Irare, S.G.; Ellis, D.S.; Warhurst, D.C.; Robinson, B.L. The chemotherapy of rodent malaria, XXXVIII. Studies on the activity of three new antimalarials (WR 194,965, WR 228,258 and WR 225,448) against rodent and human malaria parasites (Plasmodium berghei and P. falciparum). Ann. Trop. Med. Parasitol. 1984, 78, 567–579. [Google Scholar] [PubMed]
- Thompson, P.E.; Weston, K.; Glazko, A.J.; Fisken, R.A.; Reutner, T.F.; Bayles, A.; Weston, J.K. Laboratory studies on amopyroquin (propoquin), an antimalarial compound. Antibiot. Chemother. (Northfield) 1958, 8, 450–460. [Google Scholar] [PubMed]
- Zheng, X.Y.; Xia, Y.; Gao, F.H.; Chen, C. Synthesis of 7351, a new antimalarial drug. Yao Xue Xue Bao 1979, 14, 736–737. [Google Scholar] [PubMed]
- Friebolin, W.; Jannack, B.; Wenzel, N.; Furrer, J.; Oeser, T.; Sanchez, C.P.; Lanzer, M.; Yardley, V.; Becker, K.; Davioud-Charvet, E. Antimalarial dual drugs based on potent inhibitors of glutathione reductase from Plasmodium falciparum. J. Med. Chem. 2008, 51, 1260–1277. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, I.N.; Wong, P.E.; Maes, L.; Müller, T.J.J.; Krauth-Siegel, L.R.; Barrett, M.; Davioud-Charvet, E. Unsaturated Mannich bases active against multidrug-resistant T. brucei brucei strains. Chem. Med. Chem. 2009, 4, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Ciana, C.L.; Siegrist, R.; Aissaoui, H.; Marx, L.; Racine, S.; Meyer, S.; Binkert, C.; de Kanter, R.; Fischli, C.; Wittlin, S.; et al. Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Bioorg. Med. Chem. Lett. 2013, 23, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, N.I.; Chavain, N.; Wang, Y.; Friebolin, W.; Maes, L.; Pradines, B.; Lanzer, M.; Yardley, V.; Brun, R.; Herold-Mende, C.; et al. Antimalarial versus cytotoxic properties of dual drugs derived from 4-aminoquinolines and Mannich bases: Interaction with DNA. J. Med. Chem. 2010, 53, 3214–3226. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Ueno, M.; Suzuki, R.; Ishitani, H.; Kim, H.S.; Wataya, Y. Catalytic Asymmetric Synthesis of Antimalarial Alkaloids Febrifugine and Isofebrifugine and Their Biological Activity. J. Org. Chem. 1999, 64, 6833–6841. [Google Scholar] [CrossRef] [PubMed]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef] [PubMed]
- Gamo, F.J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, L.; Vanderwall, D.E.; Green, D.V.S.; Kumar, V.; Hasa, S.; et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, K.; Abdelaziz, A.; Rybacka, A.; Roncaglioni, A.; Tropsha, A.; Varnek, A.; Zakharov, A.; Worth, A.; Richard, A.M.; Grulke, C.M.; et al. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect. 2016. [Google Scholar] [CrossRef] [PubMed]
- ChEMBL Version 20; European Bioinformatics Institute (EMBL-EBI): Cambridge, UK, 2016.
- ChemAxon. Available online: http://www.chemaxon.com/products/standardizer (accessed on 15 May 2016).
- Horvath, D.; Brown, J.B.; Marcou, G.; Varnek, A. An Evolutionary Optimiser of libsvm Models. Challenges 2014, 5, 450–472. [Google Scholar] [CrossRef]
- Solov’ev, V.P.; Varnek, A.; Wipff, G. Modeling of Ion Complexation and Extraction Using Substructural Molecular Fragments. J. Chem. Inf. Comput. Sci. 2000, 40, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, P.; Gaspar, G.; Marcou, G.; Varnek, A.; Horvath, D. Mappability of drug-like space: Towards a polypharmacologically competent map of drug-relevant compounds. J. Comput. Aided. Mol. Des. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ruggiu, F.; Marcou, G.; Varnek, A.; Horvath, D. ISIDA Property-Labelled Fragment Descriptors. Mol. Inf. 2010, 29, 855–868. [Google Scholar] [CrossRef]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. 2001, 2. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef] [PubMed]
- ICE Stimator Software Version 1.2; Institut Claude Bernard AP-HP: Paris, France, 2006.
- Sample Availability: Samples of the compounds A1–A19, B1–B12 are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAA Code | Plasmodium falciparum 3D7 Strain a | Cytotoxicity MRC-5 b | SI | In Silico Status Plasmodium falciparum | Ref. |
|---|---|---|---|---|---|
| IC50 ± SD (nM) | CC50 (µM) | CC50/IC50 | A/I | ||
| A1 | 30.1 ± 8.5 | 1.91 | 63.4 | A | [29,30] |
| A2 | 30.2 ± 6.3 | 1.88 | 62.2 | A | [29,30] |
| A3 | 51.2 ± 10.7 | 32 | 625 | A | [29,31] |
| A4 | 70.6 ± 9.7 | nd | nd | A | [28,32] |
| A5 | 110.2 ± 20.3 | 32.22 | 292.4 | A | [27] |
| A6 | 110.8 ± 12.5 | >64.00 | >577.6 | A | [27] |
| A7 | 120.2 ± 17.3 | 1.99 | 16.5 | A | [27] |
| A8 | 120.7 ± 10.5 | >64.00 | >530.2 | A | [29,31] |
| A9 | 120.7 ± 12.4 | 42.44 | 351.6 | A | [29,31,32] |
| A10 | 160.8 ± 20.5 | 1.53 | 9.5 | A | [27] |
| A11 | 180.9 ± 29.6 | 29.37 | 162.3 | A | [29,31,32] |
| A12 | 240.1 ± 20.4 | >64.00 | >266.5 | A | [28] |
| A13 | >500 | 1.13 | <2.3 | A | [29,30] |
| A14 | >500 | 1.56 | <3.1 | A | [27] |
| A15 | >500 | nd | nd | A | [29,31] |
| A16 | >500 | 5.36 | <10.7 | A | [29,31] |
| A17 | >500 | 32.46 | <64.9 | A | [28,32] |
| A18 | >500 | 1.06 | <2.1 | I | [29,30] |
| A19 | >500 | 62.28 | <124.5 | I | [29,31,32] |
| chloroquine | 20.3 ± 5.2 | >64.00 | >3153 | A |
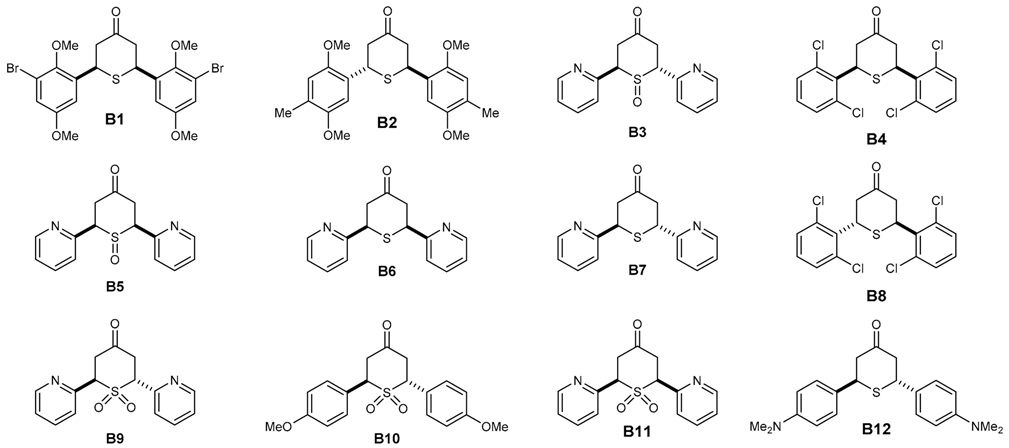
| 2,6-DATHTP Code | P. falciparum 3D7 Strain a | Cytotoxicity MRC-5 b | SI | In Silico Status P. falciparum | Ref. |
|---|---|---|---|---|---|
| IC50 ± SD (nM) | CC50 (µM) | CC50/IC50 | A/I | ||
| B1 | 30.3 ± 6.1 | >64.00 | >2112.2 | A | [28] |
| B2 | 30.6 ± 9.2 | nd | nd | A | [28] |
| B3 | 40.1 ± 7.3 | 8 | 199.5 | A | [29,31] |
| B4 | 60.4 ± 10.2 | nd | nd | A | [28] |
| B5 | 240.1 ± 48.1 | 8.06 | 33.6 | A | [29,31] |
| B6 | >500 | >64.00 | <128 | A | [29,31] |
| B7 | >500 | >64.00 | <128 | A | [29,31] |
| B8 | >500 | >64.00 | <128 | A | [28] |
| B9 | >500 | 7.89 | <15.8 | A | [29,31] |
| B10 | >500 | >64.00 | <128 | A | [29,31] |
| B11 | >500 | 7.47 | <14.9 | A | [29,31] |
| B12 | >500 | 4.96 | <9.9 | I | [29,31] |
| chloroquine | 20.3 ± 5.2 | >64.00 | >3158 | A |
| Training Set | Activity Threshold (log Unit) | Size | No. of Active | Cross-Validated Model Fitness Score |
|---|---|---|---|---|
| FS53 | 7.0 | 94 | 25 | 0.953 |
| FS39+52 | 6.0 | 107 | 31 | 0.912 |
| FS31 | 7.5 | 65 | 19 | 0.910 |
| FS33+67 | 6.5 | 70 | 17 | 0.905 |
| FS78 | 7.5 | 66 | 27 | 0.870 |
| FS61 | 7.0 | 143 | 58 | 0.865 |
| FS15 | 7.0 | 116 | 35 | 0.849 |
| FS34 | 7.0 | 123 | 33 | 0.825 |
| CHEMBL896244 | 7.0 | 225 | 74 | 0.819 |
| FS10 | 7.0 | 59 | 14 | 0.796 |
| FS76 | 7.5 | 160 | 57 | 0.787 |
| CHEMBL1038869 | 6.5 | 159 | 43 | 0.730 |
| CHEMBL730080 | 6.0 | 977 | 273 | 0.728 |
| CHEMBL896245 | 7.0 | 113 | 37 | 0.721 |
| CHEMBL730081 | 6.5 | 164 | 41 | 0.711 |
| CHEMBL730641 | 6.5 | 158 | 51 | 0.702 |
| CHEMBL1038870 | 6.5 | 156 | 36 | 0.701 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viira, B.; Gendron, T.; Lanfranchi, D.A.; Cojean, S.; Horvath, D.; Marcou, G.; Varnek, A.; Maes, L.; Maran, U.; Loiseau, P.M.; et al. In Silico Mining for Antimalarial Structure-Activity Knowledge and Discovery of Novel Antimalarial Curcuminoids. Molecules 2016, 21, 853. https://doi.org/10.3390/molecules21070853
Viira B, Gendron T, Lanfranchi DA, Cojean S, Horvath D, Marcou G, Varnek A, Maes L, Maran U, Loiseau PM, et al. In Silico Mining for Antimalarial Structure-Activity Knowledge and Discovery of Novel Antimalarial Curcuminoids. Molecules. 2016; 21(7):853. https://doi.org/10.3390/molecules21070853
Chicago/Turabian StyleViira, Birgit, Thibault Gendron, Don Antoine Lanfranchi, Sandrine Cojean, Dragos Horvath, Gilles Marcou, Alexandre Varnek, Louis Maes, Uko Maran, Philippe M. Loiseau, and et al. 2016. "In Silico Mining for Antimalarial Structure-Activity Knowledge and Discovery of Novel Antimalarial Curcuminoids" Molecules 21, no. 7: 853. https://doi.org/10.3390/molecules21070853
APA StyleViira, B., Gendron, T., Lanfranchi, D. A., Cojean, S., Horvath, D., Marcou, G., Varnek, A., Maes, L., Maran, U., Loiseau, P. M., & Davioud-Charvet, E. (2016). In Silico Mining for Antimalarial Structure-Activity Knowledge and Discovery of Novel Antimalarial Curcuminoids. Molecules, 21(7), 853. https://doi.org/10.3390/molecules21070853