
An NMR-Guided Screening Method for Selective Fragment Docking and Synthesis of a Warhead Inhibitor

 ,
, 
Abstract
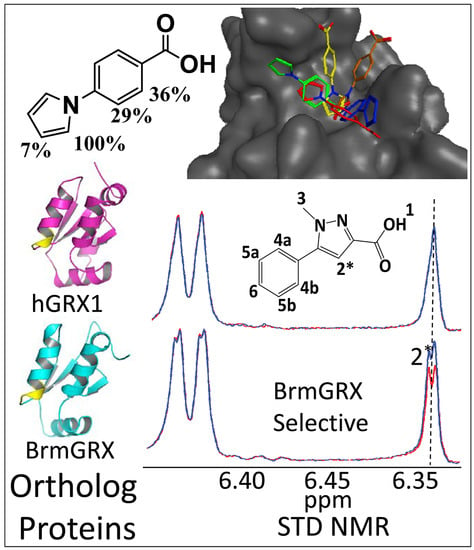
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Library
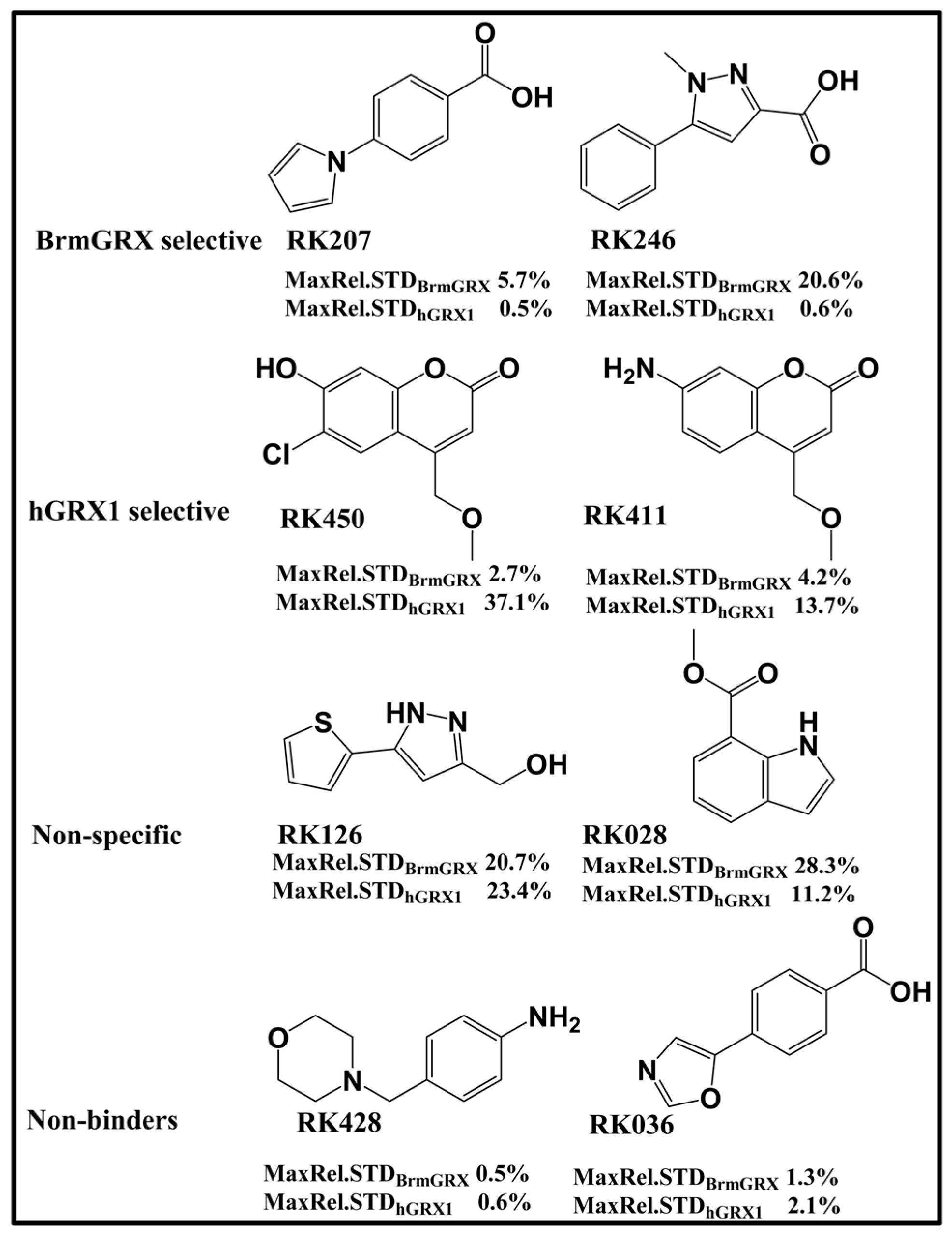
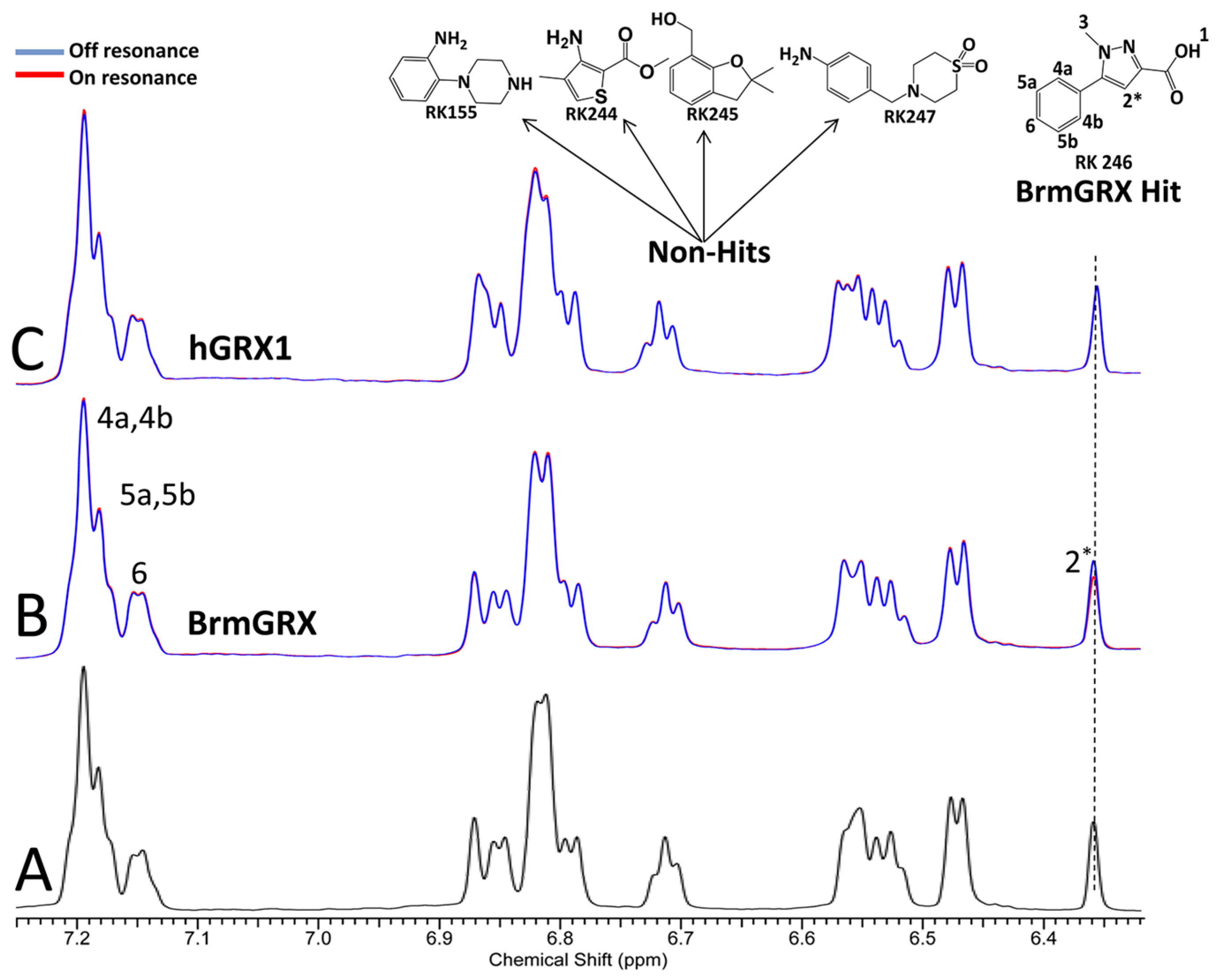
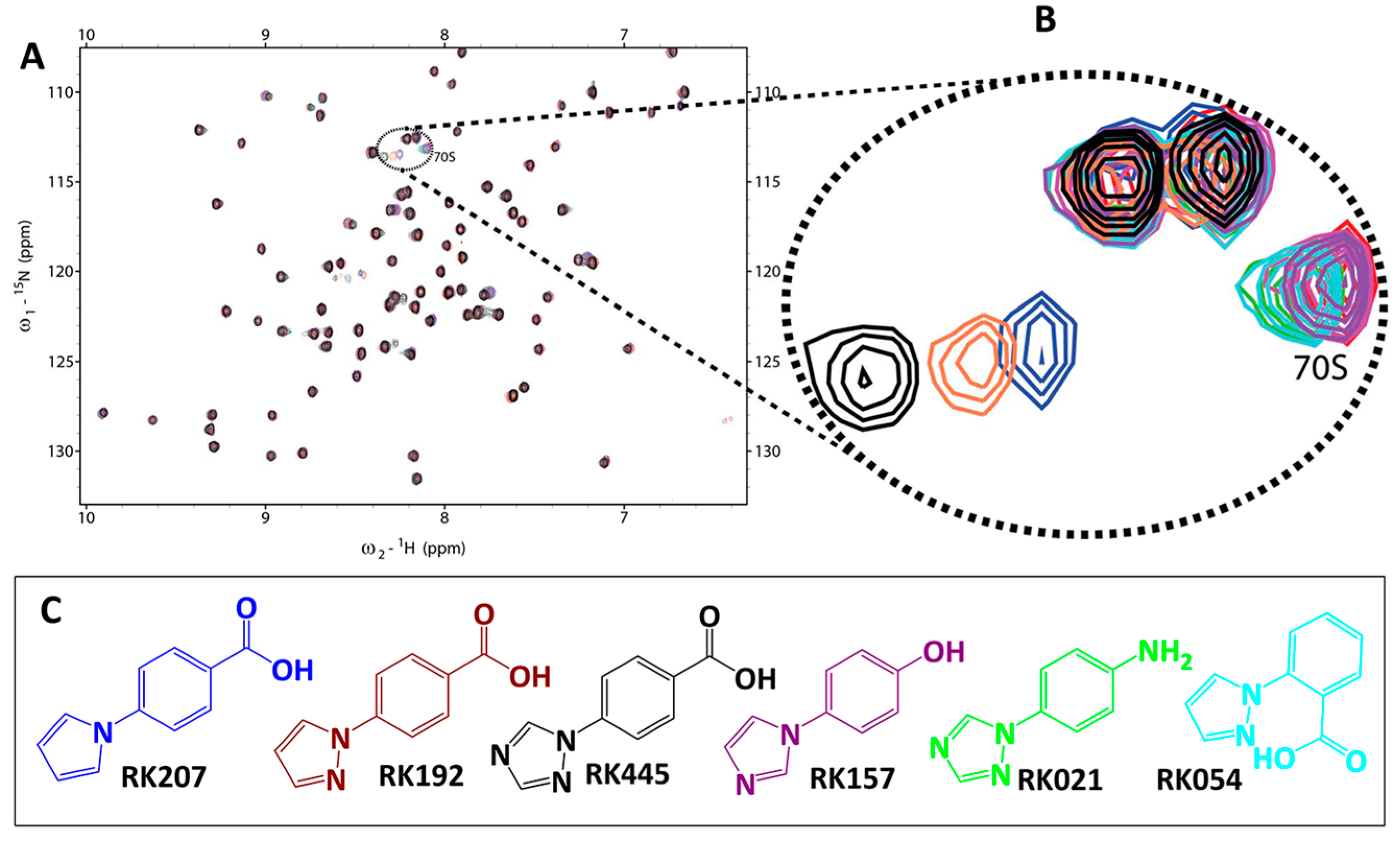
2.2. Library Screening and Hit Validation
2.3. Selection of Hit for Optimization
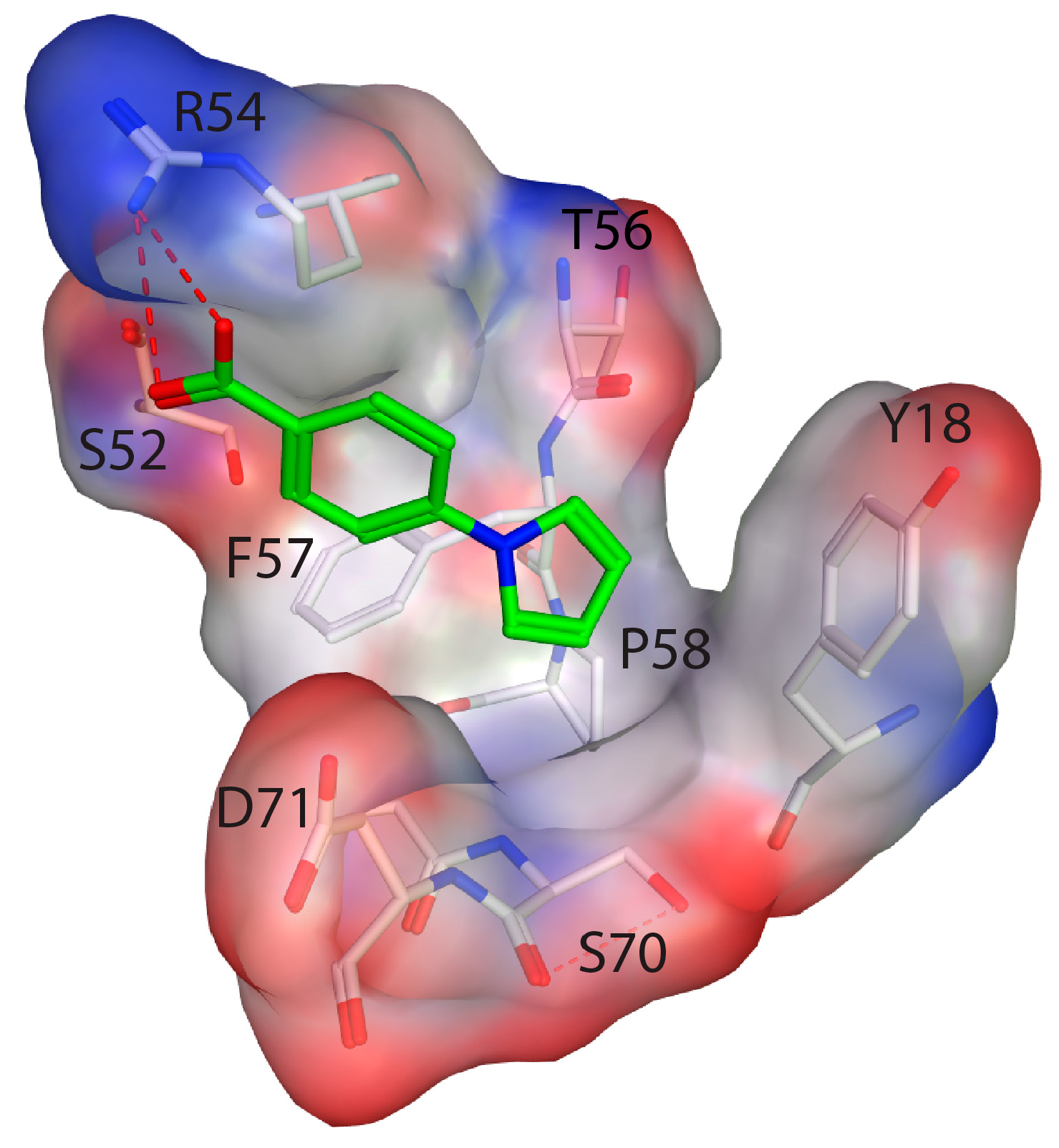
2.4. Fragment Docking for Lead Development
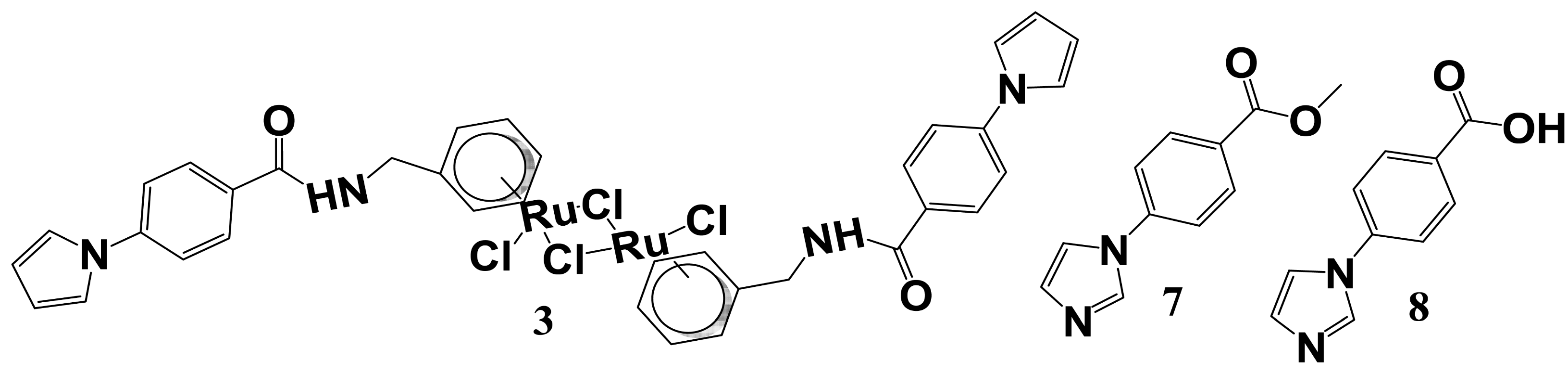
2.5. Initial Warhead-Fragment Strategy: Ruthenium Arene Derivatives
2.6. Subsequent Warhead-Fragment Strategy: Acrylamide Derivatives of a Modified Fragment
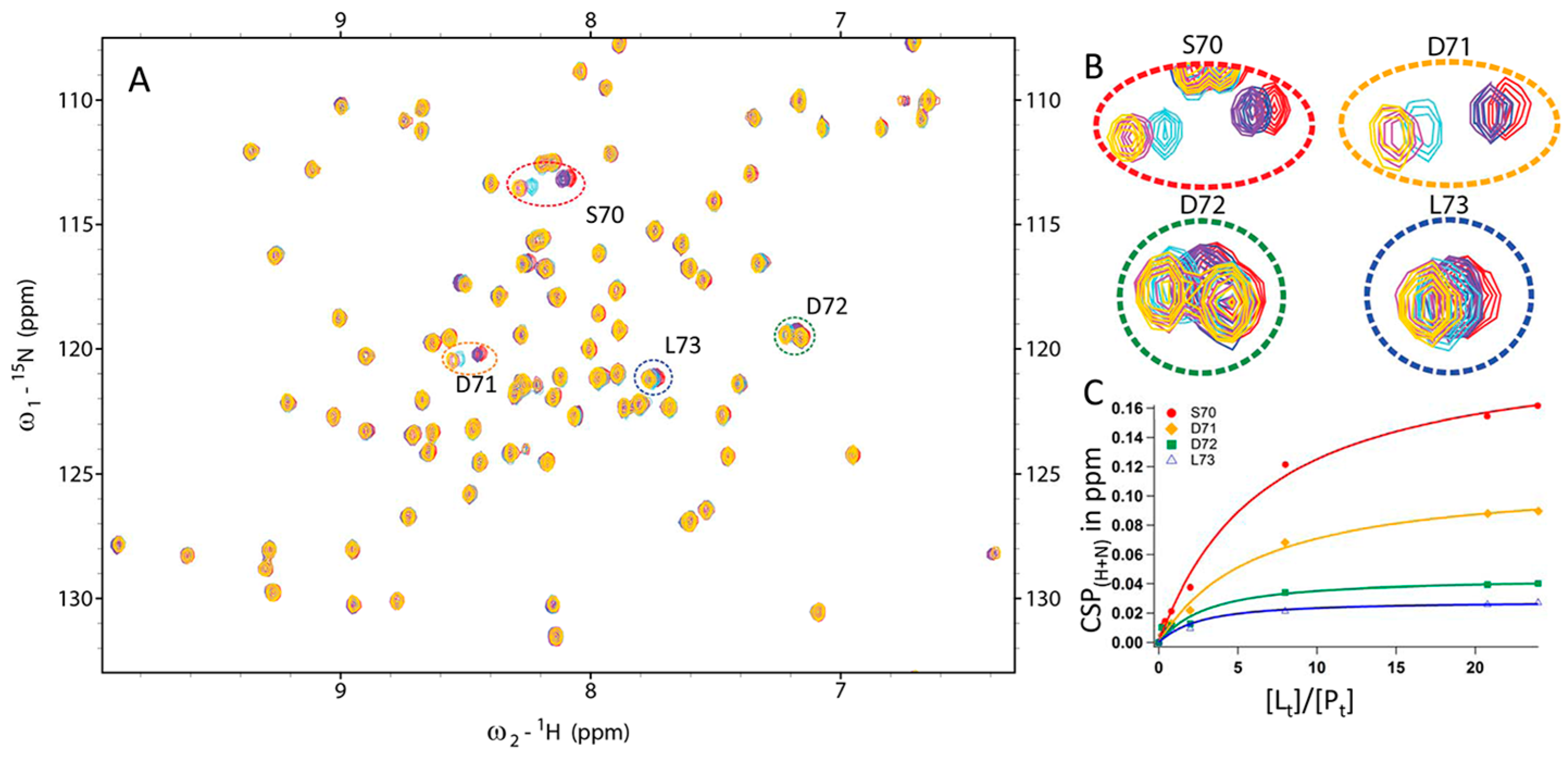
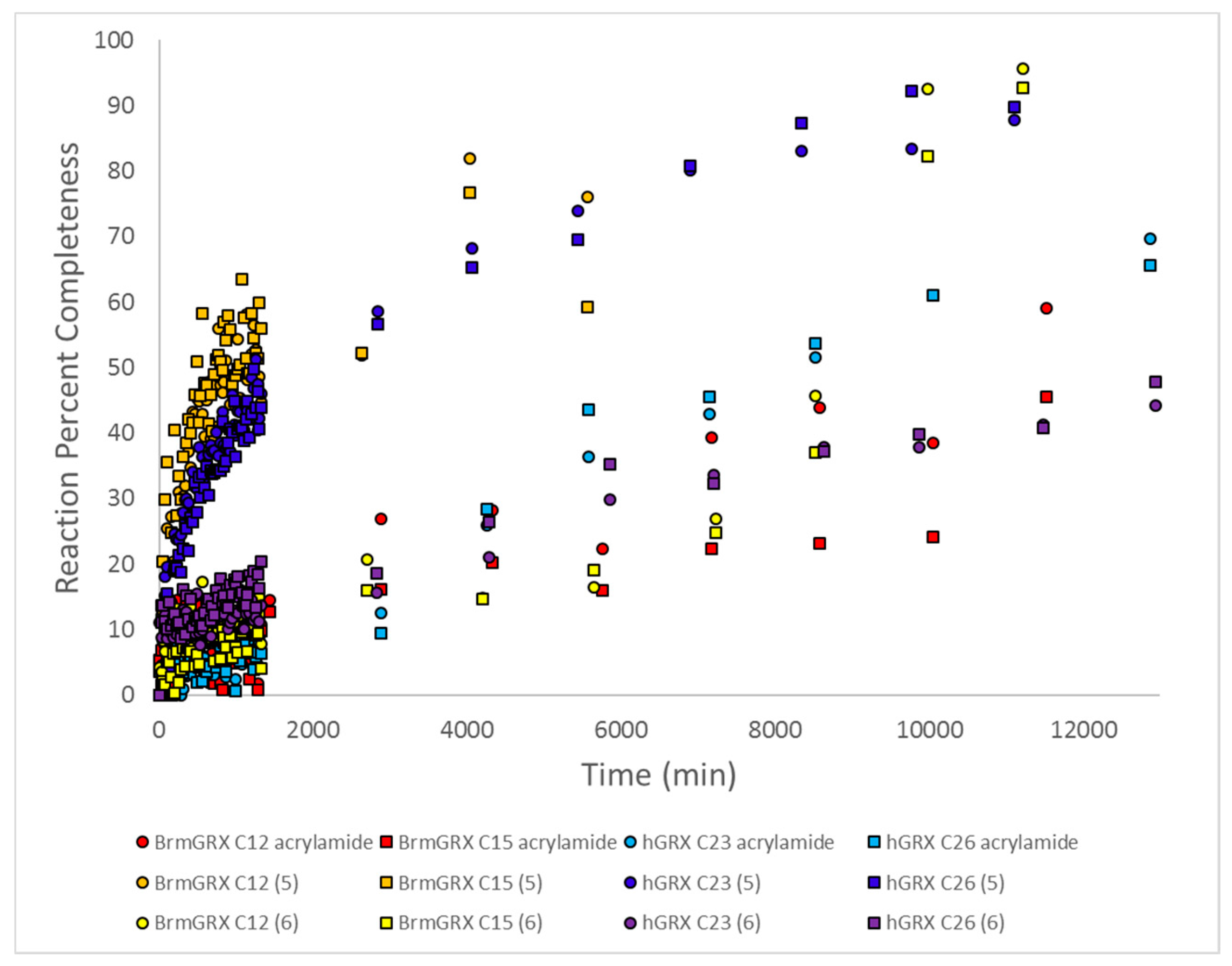
2.7. Kinetic Studies and Binding Sites Preferred by Acrylamide Warhead Containing Inhibitor
3. Materials and Methods
3.1. General Information and Library Construction
3.2. Glutaredoxin Orthologs: Expression and Purification
3.3. NMR Experiments
3.3.1. STD Screening and trNOE NMR Experiments
3.3.2. CSP Analysis of Fragment Binding
3.4. Computational Studies
3.4.1. Ligand Docking
3.4.2. SHIFTS Simulations for Computational CSPs
3.5. Synthesis
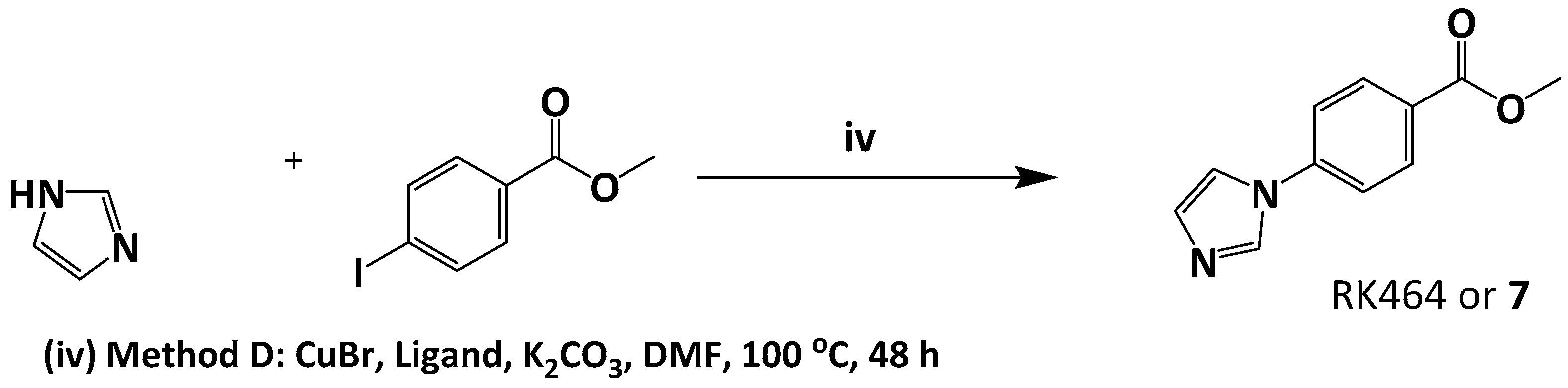
3.5.1. Preparation of Methyl 4-(1H-imidazol-1-yl) Benzoate or RK464
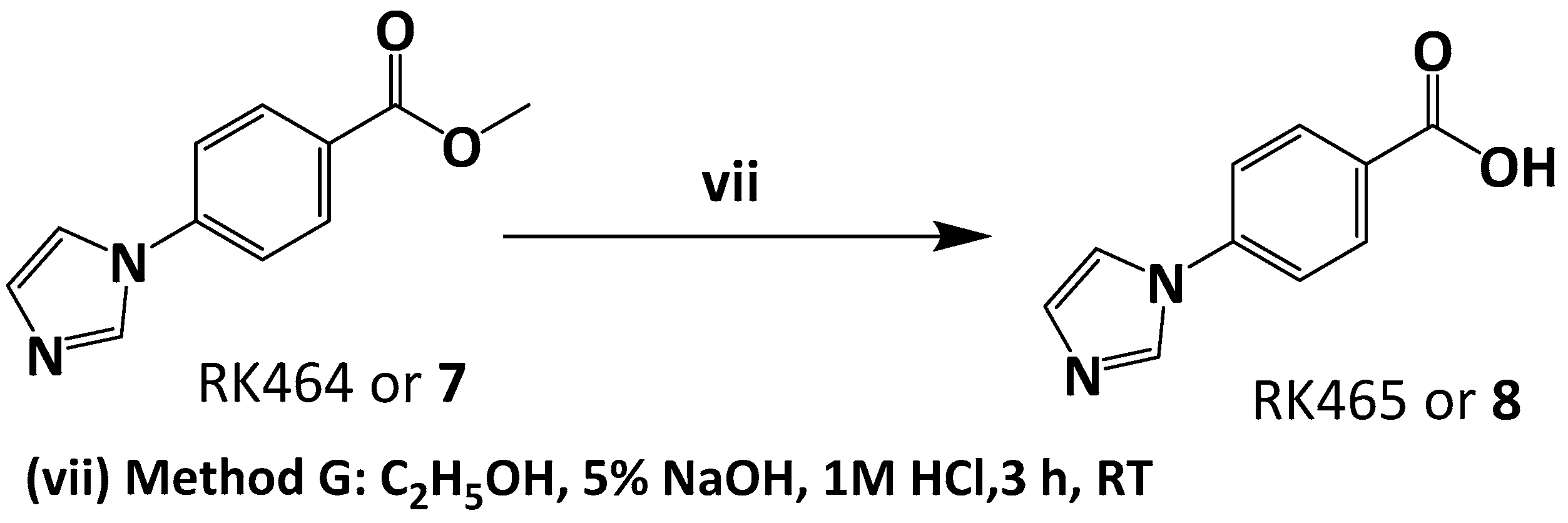
3.5.2. Preparation of 4-(1H-imidazol-1-yl)benzoic acid or RK465
3.5.3. Preparation of 4-(3-(Acrylamidomethyl)-1H-pyrrol-1-yl)benzoate (4)
3.5.4. Preparation of Methyl 4-(3-(Acrylamidomethyl)-1H-pyrrol-1-yl)benzoate (5)
3.5.5. Preparation of 4-(3-(Acrylamidomethyl)-1H-pyrrol-1-yl)benzoic acid (6)
3.6. HED Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FBDD | fragment-based drug discovery |
| STD | saturation transfer difference |
| Relative STD% | relative saturation transfer difference in percentage |
| NMR | nuclear magnetic resonance |
| HSQC | heteronuclear single quantum correlation |
| SAR | structure activity relationship |
| BrmGRX | Brucella melitensis glutaredoxin |
| hGRX1 | human glutaredoxin 1 |
| E. coli | Escherichia coli |
| HTS | high throughput screening |
| log P | log of partition-coefficient |
| Kd | dissociation constant |
| L.E. | ligand efficiency |
| CSPs | chemical shift perturbations |
| trNOE | transferred nuclear Overhauser effect |
| DMSO | dimethyl sulfoxide |
| D2O | deuterium oxide |
| DTT | dithiothreitol |
| NaCl | sodium chloride |
| Et3N | triethyl amine |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate |
References
- Congreve, M.; Chessari, G.; Tisi, D.; Woodhead, A.J. Recent developments in fragment-based drug discovery. J. Med. Chem. 2008, 51, 3661–3680. [Google Scholar] [CrossRef] [PubMed]
- Prime, M.E.; Brookfield, F.A.; Courtney, S.M.; Gaines, S.; Marston, R.W.; Ichihara, O.; Li, M.; Vaidya, D.; Williams, H.; Pedret-Dunn, A.; et al. Irreversible 4-aminopiperidine transglutaminase 2 inhibitors for Huntington’s disease. ACS Med. Chem. Lett. 2012, 3, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Carmi, C.; Cavazzoni, A.; Vezzosi, S.; Bordi, F.; Vacondio, F.; Silva, C.; Rivara, S.; Lodola, A.; Alfieri, R.R.; la Monica, S.; et al. Novel irreversible epidermal growth factor receptor inhibitors by chemical modulation of the cysteine-trap portion. J. Med. Chem. 2010, 53, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Develop ment Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Wityak, J.; Prime, M.E.; Brookfield, F.A.; Courtney, S.M.; Erfan, S.; Johnsen, S.; Johnson, P.D.; Li, M.; Marston, R.W.; Reed, L.; et al. SAR development of lysine-based irreversible inhibitors of transglutaminase 2 for huntington’s disease. ACS Med. Chem. Lett. 2012, 3, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Carmi, C.; Galvani, E.; Vacondio, F.; Rivara, S.; Lodola, A.; Russo, S.; Aiello, S.; Bordi, F.; Costantino, G.; Cavazzoni, A.; et al. Irreversible inhibition of epidermal growth factor receptor activity by 3-aminopropanamides. J. Med. Chem. 2012, 55, 2251–2264. [Google Scholar] [CrossRef] [PubMed]
- Leproult, E.; Barluenga, S.; Moras, D.; Wurtz, J.M.; Winssinger, N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: Application to the design of selective covalent inhibitors. J. Med. Chem. 2011, 54, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Cocco, M.; Garella, D.; di Stilo, A.; Borretto, E.; Stevanato, L.; Giorgis, M.; Marini, E.; Fantozzi, R.; Miglio, G.; Bertinaria, M. Electrophilic warhead-based design of compounds preventing NLRP3 inflammasome-dependent pyroptosis. J. Med. Chem. 2014, 57, 10366–10382. [Google Scholar] [CrossRef] [PubMed]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Prog. Polym. Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.R.; Mamuya, N.; Johnson, B.D.; Reich, M.F.; Gruber, B.C.; Ye, F.; Nilakantan, R.; Shen, R.; Discafani, C.; DeBlanc, R.; et al. 6-Substituted-4-(3-bromophenylamino)quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J. Med. Chem. 2001, 44, 2719–2734. [Google Scholar] [CrossRef] [PubMed]
- Slichenmyer, W.J.; Elliott, W.L.; Fry, D.W. CI-1033, a pan-erbB tyrosine kinase inhibitor. Semin. Oncol. 2001, 28, 80–85. [Google Scholar] [CrossRef]
- Minkovsky, N.; Berezov, A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr. Opin. Investig. Drugs 2008, 9, 1336–1346. [Google Scholar] [PubMed]
- Hevener, K.E.; Mehboob, S.; Su, P.C.; Truong, K.; Boci, T.; Deng, J.; Ghassemi, M.; Cook, J.L.; Johnson, M.E. Discovery of a novel and potent class of F. tularensis enoyl-reductase (FabI) inhibitors by molecular shape and electrostatic matching. J. Med. Chem. 2012, 55, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Halling, S.M.; Peterson-Burch, B.D.; Bricker, B.J.; Zuerner, R.L.; Qing, Z.; Li, L.-L.; Kapur, V.; Alt, D.P.; Olsen, S.C. Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J. Bacteriol. 2005, 187, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Pappas, G. The changing Brucella ecology: Novel reservoirs, new threats. Int. J. Antimicrob. Agents 2010, 36, S8–S11. [Google Scholar] [CrossRef] [PubMed]
- Pappas, G.; Papadimitriou, P.; Akritidis, N.; Christou, L.; Tsianos, E.V. The new global map of human brucellosis. Lancet Infect. Dis. 2006, 6, 91–99. [Google Scholar] [CrossRef]
- Leeper, T.; Zhang, S.; Van Voorhis, W.C.; Myler, P.J. Comparative analysis of glutaredoxin domains from bacterial opportunistic pathogens. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chemie Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef] [PubMed]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Barelier, S.; Pons, J.; Marcillat, O.; Lancelin, J.M.; Krimm, I. Fragment-based deconstruction of Bcl-xL inhibitors. J. Med. Chem. 2010, 53, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Klages, J.; Coles, M.; Kessler, H. NMR-based screening: A powerful tool in fragment-based drug discovery. Analyst 2007, 132, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, H.; Ye, C.; Liu, M. Structure-based drug design: NMR-based approach for ligand-protein interactions. Drug Discov. Today Technol. 2006, 3, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Barelier, S.; Pons, J.; Gehring, K.; Lancelin, J.-M.; Krimm, I. Ligand specificity in fragment-based drug design. J. Med. Chem. 2010, 53, 5256–5266. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Bures, M.; Praestgaard, J.; Fesik, S.W. Privileged molecules for protein binding identified from NMR-based screening. J. Med. Chem. 2000, 43, 3443–3447. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.A.; Strange, P.G. Evidence for the importance of a carboxyl group in the binding of ligands to the D2 dopamine receptor. J. Neurochem. 1990, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.
- Moon, S.; Case, D.A. A new model for chemical shifts of amide hydrogens in proteins. J. Biomol. NMR 2007, 38, 139–150. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.A.; Wyss, D.F. Alignment of weakly interacting molecules to protein surfaces using simulations of chemical shift perturbations. J. Biomol. NMR 2000, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.A.; Wyss, D.F. Spatial Localization of Ligand Binding Sites from Electron Current Density Surfaces Calculated from NMR Chemical Shift Perturbations. J. Am. Chem. Soc. 2002, 124, 11758–11763. [Google Scholar] [CrossRef] [PubMed]
- Wyss, D.F.; Arasappan, A.; Senior, M.M.; Wang, Y.-S.; Beyer, B.M.; Njoroge, F.G.; McCoy, M.A. Non-peptidic small-molecule inhibitors of the single-chain hepatitis C virus NS3 protease/NS4A cofactor complex discovered by structure-based NMR screening. J. Med. Chem. 2004, 47, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; ten Brink, T.; Guichou, J.-F.; Cala, O.; Krimm, I. Comparing binding modes of analogous fragments using NMR in fragment-based drug design: Application to PRDX5. PLoS ONE 2014, 9, e102300. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.C.; Ten Brink, T.; Cala, O.; Guichou, J.-F.F.; Krimm, I. Protein-ligand structure guided by backbone and side-chain proton chemical shift perturbations. J. Biomol. NMR 2014, 60, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; Cala, O.; Krimm, I. Overview of Probing Protein-Ligand Interactions Using NMR. Curr. Protoc. Protein Sci. 2015, 81. [Google Scholar] [CrossRef]
- Herrick, R.S.; Ziegler, C.J.; Leeper, T.C. Structure and function in organometallic•protein complexes. J. Organomet. Chem. 2014, 751, 90–110. [Google Scholar] [CrossRef]
- Petruk, A.A.; Vergara, A.; Marasco, D.; Bikiel, D.; Doctorovich, F.; Estrin, D.A.; Merlino, A. Interaction between proteins and Ir based CO releasing molecules: Mechanism of adduct formation and CO release. Inorg. Chem. 2014, 53, 10456–10462. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Stjernschantz, E.; Oostenbrink, C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys. J. 2010, 98, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Tummino, P.J.; Copeland, R.A. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry 2008, 47, 5481–5492. [Google Scholar] [CrossRef] [PubMed]
- Engel-Andreasen, J.; Shimpukade, B.; Ulven, T. Selective copper catalysed aromatic N-arylation in water. Green Chem. 2013, 15, 336–340. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Kong, J.S.; Venkatraman, S.; Furness, K.; Nimkar, S.; Shepherd, T.A.; Wang, Q.M.; Aubé, J.; Hanzlik, R.P. Synthesis and evaluation of peptidyl Michael acceptors that inactivate human rhinovirus 3C protease and inhibit virus replication. J. Med. Chem. 1998, 41, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hanzlik, R.P. Structure-activity relationships for inhibition of papain by peptide Michael acceptors. J. Med. Chem. 1992, 35, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Kathman, S.G.; Xu, Z.; Statsyuk, A.V. A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem. 2014, 57, 4969–4974. [Google Scholar] [CrossRef] [PubMed]
- Mah, R.; Thomas, J.R.; Shafer, C.M. Drug discovery considerations in the development of covalent inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Luthman, M.; Holmgren, A. Glutaredoxin from calf thymus. Purification to homogeneity. J. Biol. Chem. 1982, 257, 6686–6690. [Google Scholar] [PubMed]
- Jao, S.-C.; English Ospina, S.M.; Berdis, A.J.; Starke, D.W.; Post, C.B.; Mieyal, J.J. Computational and mutational analysis of human glutaredoxin (thioltransferase): Probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry 2006, 45, 4785–4796. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; Vlamis-Gardikas, A.; Nilsson, L. The -Cys-X1-X2-Cys- motif of reduced glutaredoxins adopts a consensus structure that explains the low pK(a) of its catalytic cysteine. Biochemistry 2012, 51, 8189–8207. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Butina, D. Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Model. 1998, 38, 983–996. [Google Scholar] [CrossRef]
- Bruker APEX II. APEX II 2013; Bruker AXS Inc.: Madison, WI, USA, 2013. [Google Scholar]
- Sheldrick, G.M. SADABS. In Program for Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Madden, T.L.; Tatusov, R.L.; Zhang, J. Applications of network BLAST server. Methods Enzymol 1996, 266, 131–141. [Google Scholar] [PubMed]
- Myler, P.J.; Stacy, R.; Stewart, L.; Staker, B.L.; van Voorhis, W.C.; Varani, G.; Buchko, G.W. The Seattle Structural Genomics Center for Infectious Disease (SSGCID). Infect. Disord. Drug Targets 2009, 9, 493–506. [Google Scholar] [CrossRef] [PubMed]
- ACD/Structure Elucidator, version 15.01; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2015; Available online: www.acdlabs.com (accessed on 12 December 2015).
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank: A computer-based archival file for macromolecular structures. Arch. Biochem. Biophys. 1978, 185, 584–591. [Google Scholar] [CrossRef]
- Sun, C.; Berardi, M.J.; Bushweller, J.H. The NMR solution structure of human glutaredoxin in the fully reduced form. J. Mol. Biol. 1998, 280, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Fielding, L. NMR methods for the determination of protein-ligand dissociation constants. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 219–242. [Google Scholar] [CrossRef]
- Arai, M.; Ferreon, J.C.; Wright, P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: Binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012, 134, 3792–3803. [Google Scholar] [CrossRef] [PubMed]
- IGOR Pro, 6.36; WaveMetrics Inc.: Tigard, OR, USA, 2014.
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Wells, J.A.; Braisted, A.C. Tethering: Fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Janin, Y.L. Non-quinolone inhibitors of bacterial type IIA topoisomerases: A feat of bioisosterism. Chem. Rev. 2014, 114, 2313–2342. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, N.-N.; Yin, P.-D.; Cui, P.-X.; Zhou, C.-Z. Glutathionylation-triggered conformational changes of glutaredoxin Grx1 from the yeast Saccharomyces cerevisiae. Proteins 2008, 72, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Yin, S.; Dokholyan, N. V Rapid flexible docking using a stochastic rotamer library of ligands. J. Chem. Inf. Model. 2010, 50, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided. Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Oldfield, E. Correlation between 15N NMR chemical shifts in proteins and secondary structure. J. Biomol. NMR 1994, 4, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.L.; Houk, A.R.; Jensen, J.H. Cooperative hydrogen bonding effects are key determinants of backbone amide proton chemical shifts in proteins. J. Am. Chem. Soc. 2006, 128, 9863–9872. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.W.; Mallion, R.B. Ring current theories in nuclear magnetic resonance. Prog. Nucl. Magn. Reson. Spectrosc. 1979, 13, 303–344. [Google Scholar] [CrossRef]
- Nagai, S.; Black, S. A thiol-disulfide transhydrogenase from yeast. J. Biol. Chem. 1968, 243, 1942–1947. [Google Scholar] [PubMed]
- Zaffagnini, M.; Michelet, L.; Massot, V.; Trost, P.; Lemaire, S.D. Biochemical characterization of glutaredoxins from Chlamydomonas reinhardtii reveals the unique properties of a chloroplastic CGFS-type glutaredoxin. J. Biol. Chem. 2008, 283, 8868–8876. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khattri, R.B.; Morris, D.L.; Davis, C.M.; Bilinovich, S.M.; Caras, A.J.; Panzner, M.J.; Debord, M.A.; Leeper, T.C. An NMR-Guided Screening Method for Selective Fragment Docking and Synthesis of a Warhead Inhibitor. Molecules 2016, 21, 846. https://doi.org/10.3390/molecules21070846
Khattri RB, Morris DL, Davis CM, Bilinovich SM, Caras AJ, Panzner MJ, Debord MA, Leeper TC. An NMR-Guided Screening Method for Selective Fragment Docking and Synthesis of a Warhead Inhibitor. Molecules. 2016; 21(7):846. https://doi.org/10.3390/molecules21070846
Chicago/Turabian StyleKhattri, Ram B., Daniel L. Morris, Caroline M. Davis, Stephanie M. Bilinovich, Andrew J. Caras, Matthew J. Panzner, Michael A. Debord, and Thomas C. Leeper. 2016. "An NMR-Guided Screening Method for Selective Fragment Docking and Synthesis of a Warhead Inhibitor" Molecules 21, no. 7: 846. https://doi.org/10.3390/molecules21070846
APA StyleKhattri, R. B., Morris, D. L., Davis, C. M., Bilinovich, S. M., Caras, A. J., Panzner, M. J., Debord, M. A., & Leeper, T. C. (2016). An NMR-Guided Screening Method for Selective Fragment Docking and Synthesis of a Warhead Inhibitor. Molecules, 21(7), 846. https://doi.org/10.3390/molecules21070846