
Design, Synthesis, and Biological Evaluation of Novel PARP-1 Inhibitors Based on a 1H-Thieno[3,4-d] Imidazole-4-Carboxamide Scaffold

Abstract
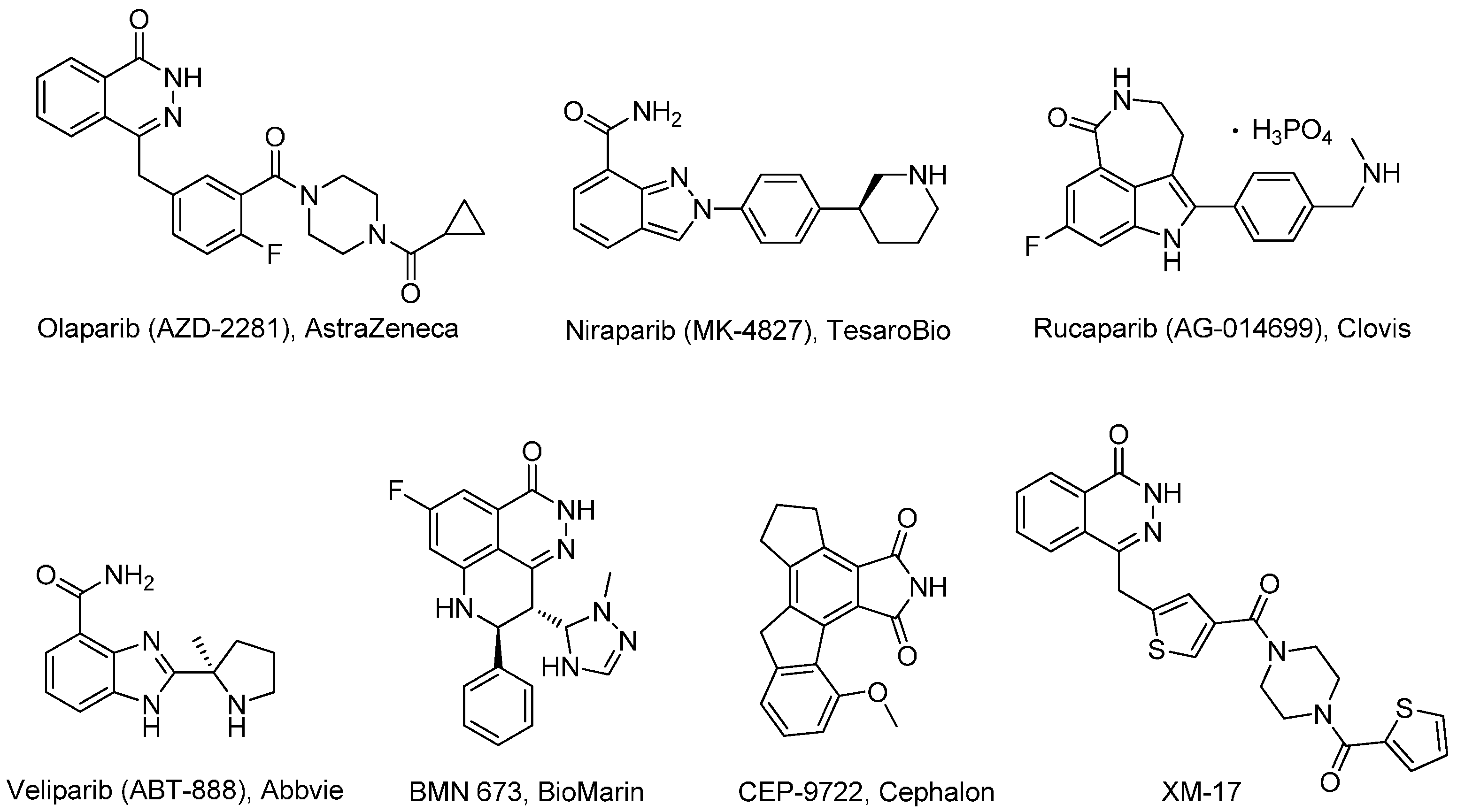
:1. Introduction
2. Results and Discussion
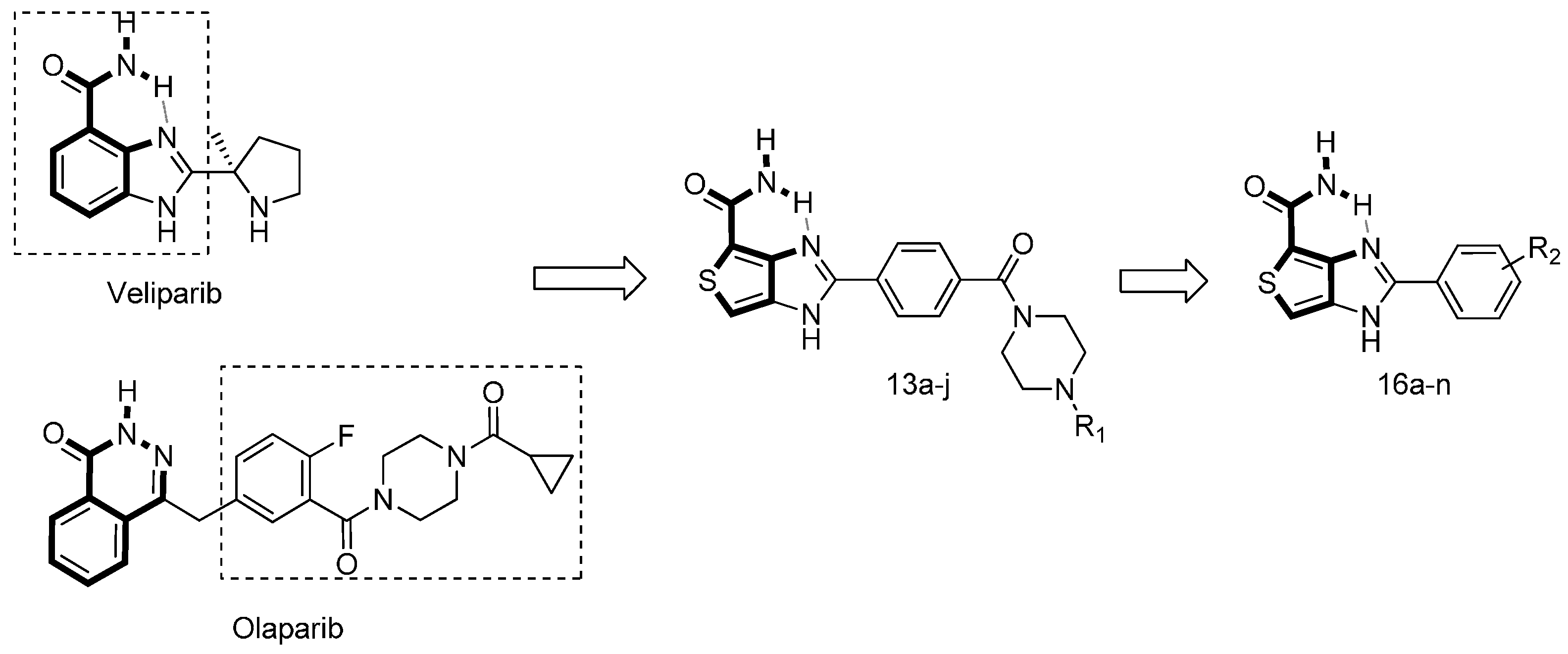
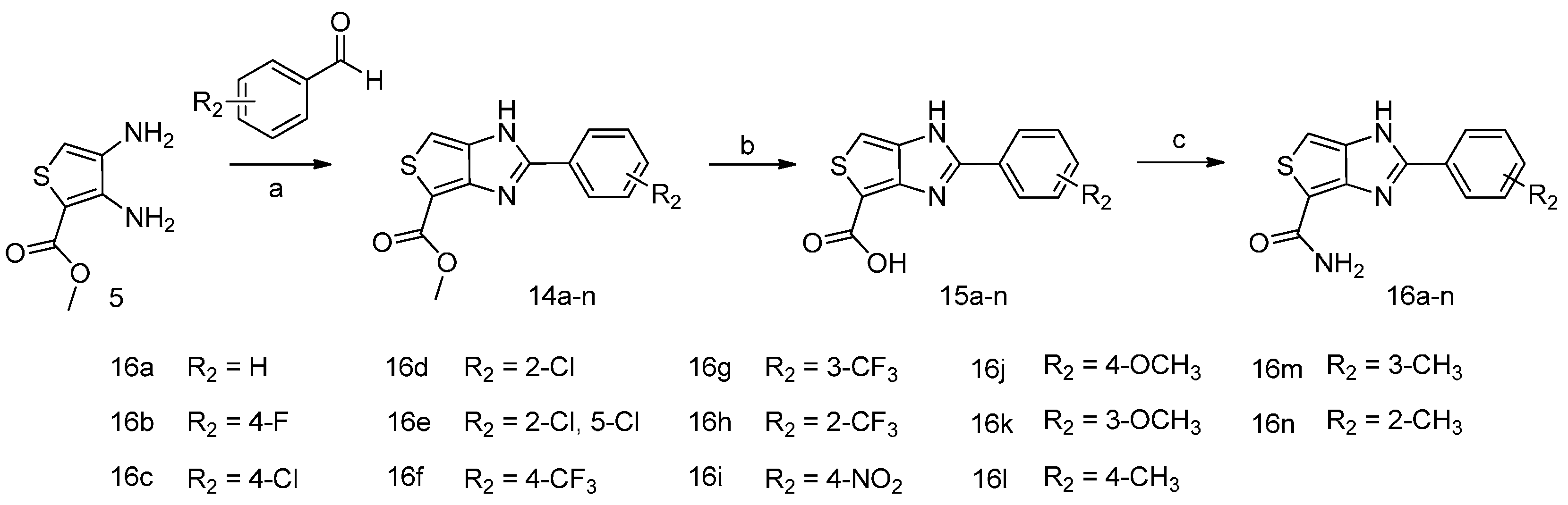
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. PARP-1 Enzyme Inhibitory Activity
2.2.2. Anti-Proliferative Activity on BRCA1/2-Deficient Cell Lines
2.2.3. Cytotoxicity Assay on Normal Human Lung Fibroblast (HLF) Cells
2.3. ADMET Prediction
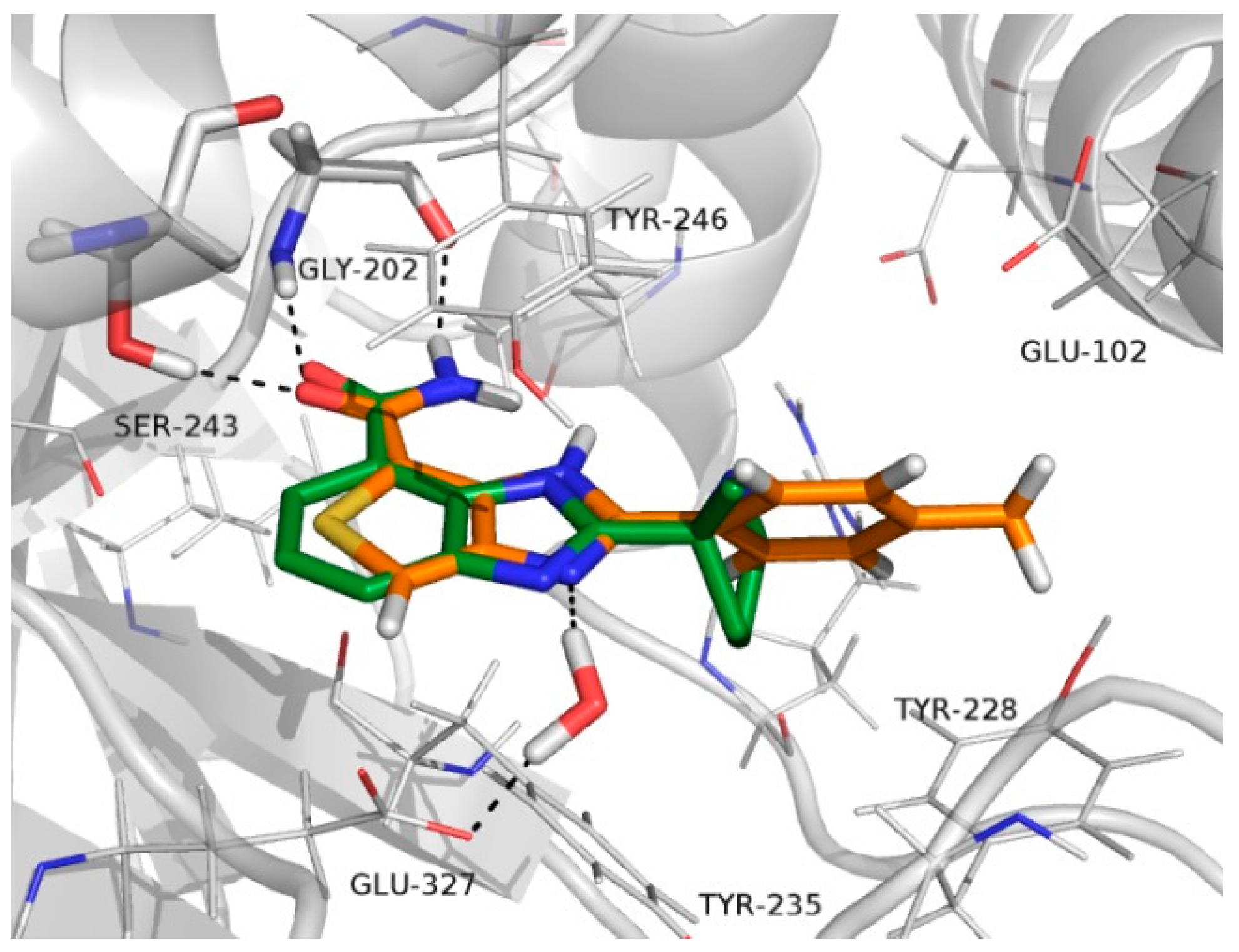
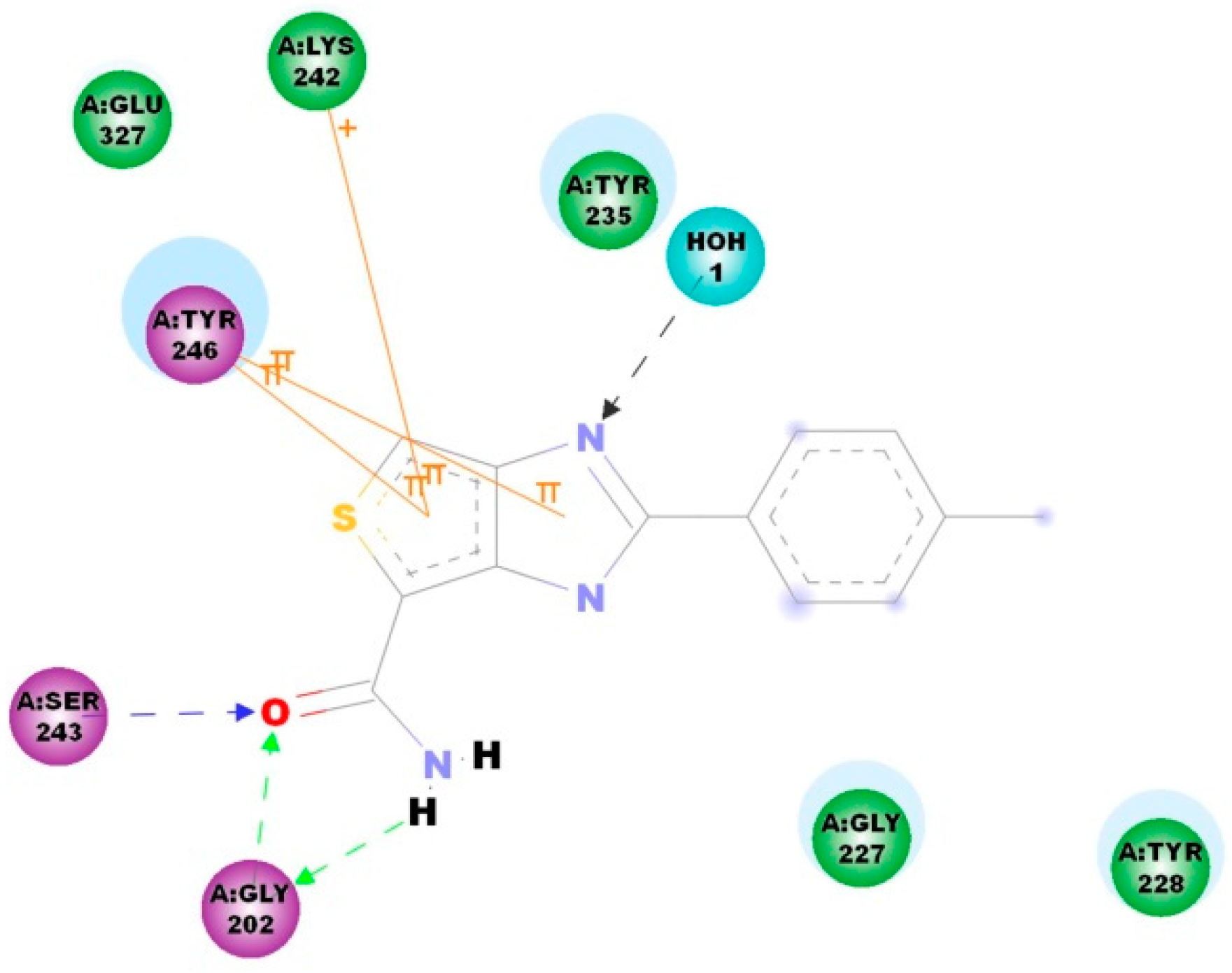
2.4. Molecular Docking
3. Experimental Section
3.1. General
3.2. Chemical Synthesis
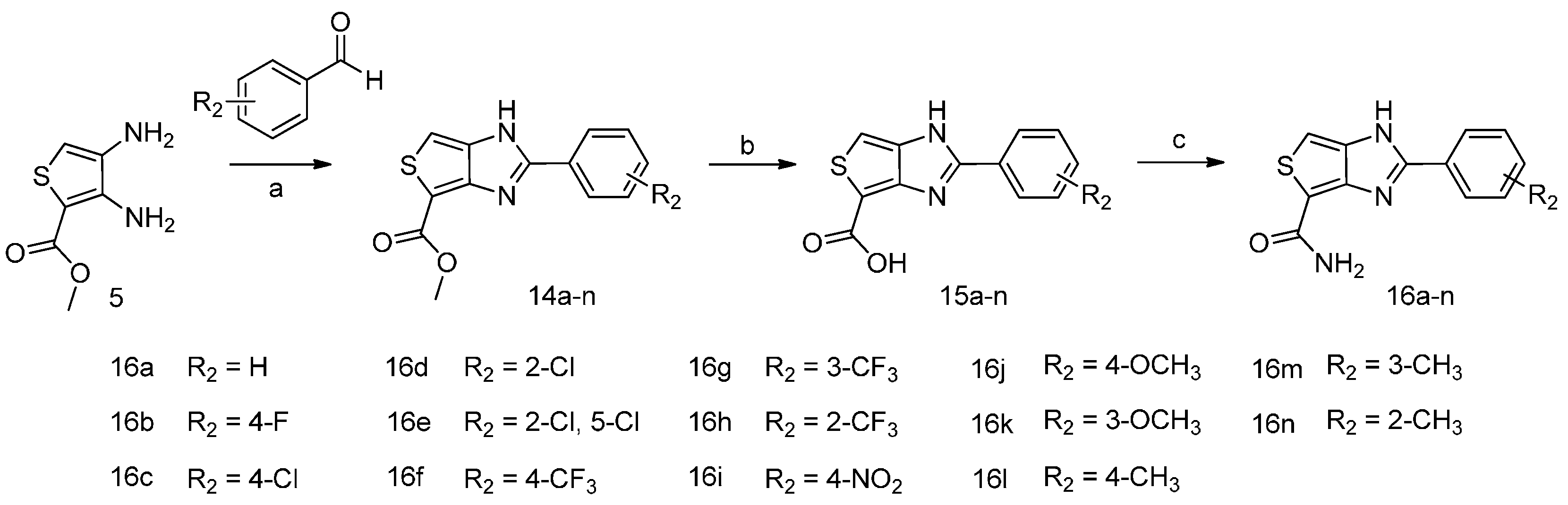
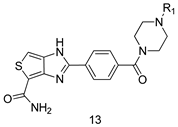
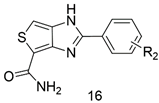
3.2.1. Synthesis of Compounds 13a–j
3.2.2. Synthesis of Compounds 16a–n
3.3. PARP-1 Enzymatic Inhibition Assay
3.4. CellTiter Glo Assay for Cell Growth Inhibition
3.5. Cytotoxicity Assay on Normal Human Lung Fibroblast (HLF) Cells
3.6. ADMET Prediction
3.7. Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ame, J.C.; Spenlehauer, C.; De Murcia, G. The PARP superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.D.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Ricks, T.K.; Chiu, H.J.; Ison, G.; Kim, G.; McKee, A.E.; Kluetz, P.; Pazdur, R. Successes and Challenges of PARP Inhibitors in Cancer Therapy. Front. Oncol. 2015, 5, 222. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.; Mikropoulos, C.; Kaye, S.B.; Nutting, C.M.; Bhide, S.A.; Newbold, K.; Harrington, K. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. J. Cancer Treat Rev. 2010, 36, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Wilson, S.H. Strategic combination of DNA-damaging agent and PARP inhibitor results in enhanced cytotoxicity. Front. Oncol. 2013, 3, 257. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R. Poly(ADP-ribose)polymerase (PARP) inhibitors: From bench to bedside. Clin. Oncol. 2014, 26, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Eskander, R.N.; Tewari, K.S. PARP inhibition and synthetic lethality in ovarian cancer. Expert Rev. Clin. Pharmacol. 2014, 7, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther. 2015, 8, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Konstantinopoulos, P.A.; Matulonis, U.A. PARP inhibitors in ovarian cancer: Current status and future promise. Gynecol. Oncol. 2014, 133, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Ji, M.; Zhou, J.; Jin, J.; Xue, N.; Chen, J.; Xu, B.; Chen, X. Poly(ADP-ribose)polymerases inhibitor, Zj6413, as a potential therapeutic agent against breast cancer. Biochem. Parmacol. 2016, 107, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Lacobuzio-Donahue, C.A.; Kelsen, D.P.; O’Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Khemlina, G.; Ikeda, S.; Kurzrock, R. Molecular landscape of prostate cancer: Implications for current clinical trials. Cancer Treat. Rev. 2015, 41, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Michalarea, V.; Lopez, J.; Lorente, D.; Yap, T.A. 343 Translational phase I trial combining the AKT inhibitor AZD5363 (AZD) and PARP inhibitor Olaparib (Ola) in advanced cancer patients (pts). Eur. J. Cancer 2015, 51, S68–S68. [Google Scholar] [CrossRef]
- Du, Y.; Yamaguchi, H.; Wei, Y.K.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.; et al. Blocking c-Met–mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Sun, Y.; Cartson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K- AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA–ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Olaparib: First global approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Bixel, K.; Hays, J.L. Olaparib in the management of ovarian cancer. Pharmgenomics Pers. Med. 2015, 8, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Courley, C.; Karlan, B.Y.; Amit, A.; Bell-McGuinn, K.M.; Chen, L.M.; Friedlander, M.; et al. Phase II, open-Label, randomized, multicenter study comparing the efficacy and safety of Olaparib, a poly(ADP-Ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2011, 30, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, A. PARP inhibitors: Its role in treatment of cancer. Chin. J. Cancer 2011, 30, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.D.; Gong, J.C.; Gandhi, V.B.; Liu, X.S.; Shi, Y.; Johnson, E.F.; Donawho, C.K.; Ellis, P.A.; Bouska, J.J.; Osterling, D.J.; et al. Discovery and SAR of orally efficacious tetrahydropyri dopyridazinone PARP inhibitors for the treatment of cancer. Bioorg. Med. Chem. 2012, 20, 4635–4645. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.V. Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From concept to clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Zhou, X.B.; Xiao, M.L.; Jiang, N.; Liu, F.; Zhou, W.X.; Wang, X.K.; Zheng, Z.Z.; Li, S. Synthesis and biological evaluation of substituted 4-(thiophen-2-ylmethyl)-2H-phthalazin-1-ones as potent PARP-1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3739–3743. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T.; et al. Specific killing of BRCA-2 deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N. PARP inhibitors for anticancer therapy. Biochem. Soc. Trans. 2014, 42, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Cai, Z.; Hu, G.; Li, Q. Identification of ALK5 inhibitor via structure-based virtual screening and ADMET prediction. J. Recept. Signal Transduct. Res. 2015, 35, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 12, 13a–j and 16a–n are available from the authors.


| Compd. | R1 | IC50 (μM) | Compd. | R1 | IC50 (μM) |
|---|---|---|---|---|---|
| Olaparib | 0.018 | 13f | 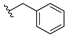 | 3.864 | |
| Veliparib | 0.005 | 13g |  | ND | |
| 13a | 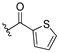 | 2.569 | 13h |  | 3.724 |
| 13b | 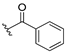 | 3.763 | 13i | 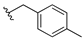 | ND |
| 13c |  | 1.874 | 13j | 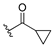 | 1.525 |
| 13d | 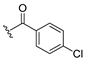 | ND b | 12 | H | 0.723 |
| 13e | 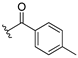 | ND |
| Compd. | R2 | IC50 (μM) | Compd. | R2 | IC50 (μM) |
|---|---|---|---|---|---|
| Olaparib | 0.013 | 16g | 3-CF3 | 0.151 | |
| Veliparib | 0.014 | 16h | 2-CF3 | ND | |
| 16a | H | 0.827 | 16i | 4-NO2 | 0.136 |
| 16b | 4-F | 0.738 | 16j | 4-OCH3 | 0.102 |
| 16c | 4-Cl | 0.354 | 16k | 3-OCH3 | 1.491 |
| 16d | 2-Cl | 1.488 | 16l | 4-CH3 | 0.043 |
| 16e | 2-Cl, 5-Cl | ND b | 16m | 3-CH3 | 5.787 |
| 16f | 4-CF3 | 0.548 | 16n | 2-CH3 | 0.328 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | IC50 (μM) (BRCA-1, HCC1937) | IC50 (μM) (BRCA-2, CAPAN-1) |
|---|---|---|
| Olaparib | 132.92 ± 14.39 | 13.36 ± 1.16 |
| Veliparib | 127.04 ± 18.10 | 57.67 ± 12.39 |
| 16g | 31.34 ± 0.35 | 38.63 ± 6.75 |
| 16i | 32.87 ± 4.21 | 26.79 ± 1.55 |
| 16j | 55.14 ± 0.24 | 46.60 ± 0.62 |
| 16l | 49.25 ± 5.43 | 38.77 ± 1.60 |
| ADMET Properties | AZD-2281 | 16g | 16i | 16j | 16l |
|---|---|---|---|---|---|
| S + logP | 2.26 | 2.6 | 1.7 | 1.98 | 1.3 |
| S + Sw (mg/mL) | 2.0 × 10−3 | 7.86 × 10−3 | 4.69 × 10−3 | 2.16 × 10−2 | 1.03 × 10−2 |
| S + FaSSGF (mg/mL) | 1.40 × 10−2 | 3.50 × 10−1 | 8.85 × 10−2 | 3.68 × 10−1 | 4.19 × 10−1 |
| S + Peff (×104 cm/s) | 3.26 | 3.18 | 1.59 | 1.86 | 2.32 |
| S + BBB_Filter | Low | High | High | High | High |
| Absn_risk | 0.17 | 0.31 | 0.76 | 0.0 | 0.0 |
| CYP3A4_substr | Yes | NO | NO | NO | NO |
| CYP_risk | 1.95 | 0 | 0 | 0 | 0 |
| Rat acute toxicity (LD50; mg/kg) | 380.03 | 240.10 | 1550.48 | 1741.25 | 1520.52 |
| Tox_risk | 2.73 | 2.67 | 3 | 1 | 1 |
| ADMET_risk | 4.85 | 3.02 | 3.76 | 1 | 1 |
| Compd. | Inhibition Rate (%) | IC50 (µM) | ||||||
|---|---|---|---|---|---|---|---|---|
| 0.3 µM | 1 µM | 3 µM | 10 µM | 30 µM | 100 µM | 300 µM | ||
| Olaparib | 1.88 | 1.83 | 1.10 | 1.37 | 1.04 | 29.60 | 91.45 | 134.30 ± 3.14 |
| Veliparib | 1.00 | −0.28 | −1.20 | −1.35 | −0.06 | −0.45 | 93.64 | ~200 |
| 16g | −1.65 | −1.02 | −0.63 | 0.76 | 4.13 | 69.88 | 99.40 | 77.63 ± 1.50 |
| 16i | −0.05 | 1.33 | 0.99 | 3.37 | 2.56 | 55.26 | 99.63 | 94.11 ± 2.20 |
| 16j | 0.68 | 2.85 | 2.68 | 6.95 | 9.43 | 12.63 | 32.37 | >300 |
| 16l | 0.42 | 3.80 | 7.12 | 10.16 | 13.65 | 23.20 | 27.94 | >300 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Liu, F.; Jiang, N.; Zhou, W.; Zhou, X.; Zheng, Z. Design, Synthesis, and Biological Evaluation of Novel PARP-1 Inhibitors Based on a 1H-Thieno[3,4-d] Imidazole-4-Carboxamide Scaffold. Molecules 2016, 21, 772. https://doi.org/10.3390/molecules21060772
Wang L, Liu F, Jiang N, Zhou W, Zhou X, Zheng Z. Design, Synthesis, and Biological Evaluation of Novel PARP-1 Inhibitors Based on a 1H-Thieno[3,4-d] Imidazole-4-Carboxamide Scaffold. Molecules. 2016; 21(6):772. https://doi.org/10.3390/molecules21060772
Chicago/Turabian StyleWang, Lingxiao, Feng Liu, Ning Jiang, Wenxia Zhou, Xinbo Zhou, and Zhibing Zheng. 2016. "Design, Synthesis, and Biological Evaluation of Novel PARP-1 Inhibitors Based on a 1H-Thieno[3,4-d] Imidazole-4-Carboxamide Scaffold" Molecules 21, no. 6: 772. https://doi.org/10.3390/molecules21060772
APA StyleWang, L., Liu, F., Jiang, N., Zhou, W., Zhou, X., & Zheng, Z. (2016). Design, Synthesis, and Biological Evaluation of Novel PARP-1 Inhibitors Based on a 1H-Thieno[3,4-d] Imidazole-4-Carboxamide Scaffold. Molecules, 21(6), 772. https://doi.org/10.3390/molecules21060772